
HDAC Inhibitor Abrogates LTA−Induced PAI-1 Expression in Pleural Mesothelial Cells and Attenuates Experimental Pleural Fibrosis


Abstract
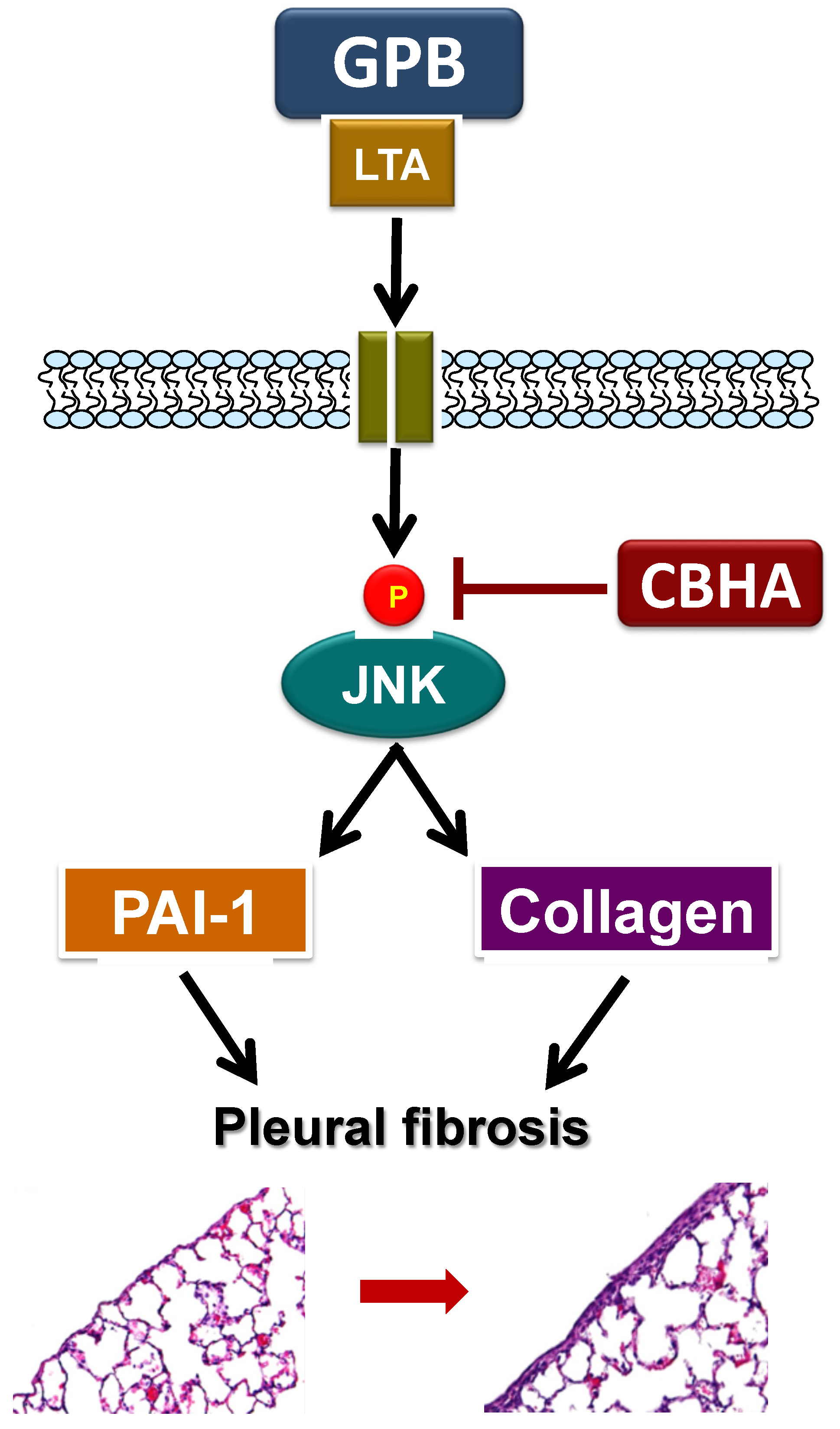
:1. Introduction
2. Results
2.1. Pleural Fluid Pamameters between Gram-Positive Bacteria (GPB) Parapneumonic Pleural Effusion (PPE) Patients with Residual Pleural Thickening (RPT) ≤ 10 mm and RPT > 10 mm
2.2. Multivariate Logistic Regression Analysis for Variables Related to RPT > 10 mm among GPB PPE Patients
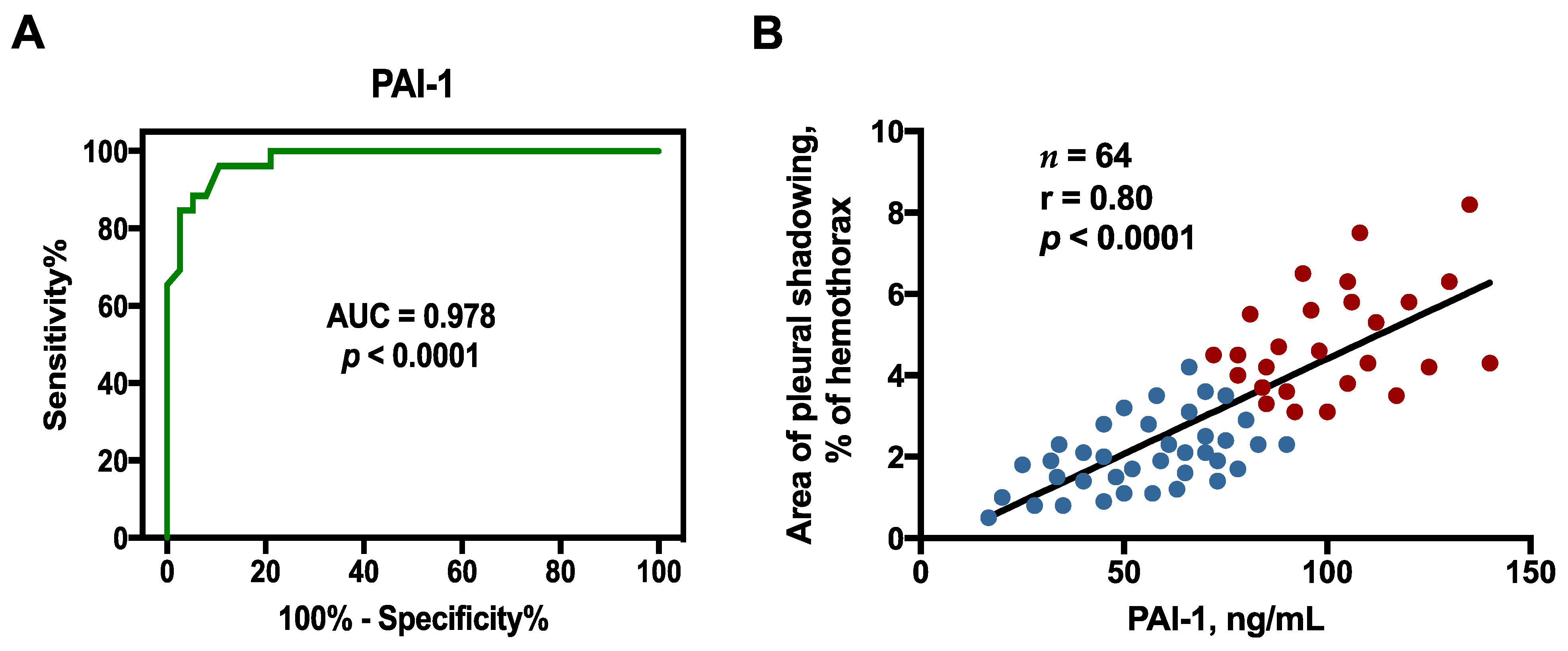
2.3. Optimal Sensitivity, Specificity, and Cutoff Value of Factors to Predict RPT > 10 mm
2.4. Correlation between Effusion Plasminogen Activator Inhibitor-1 (PAI-1) and Residual Pleural Fibrosis
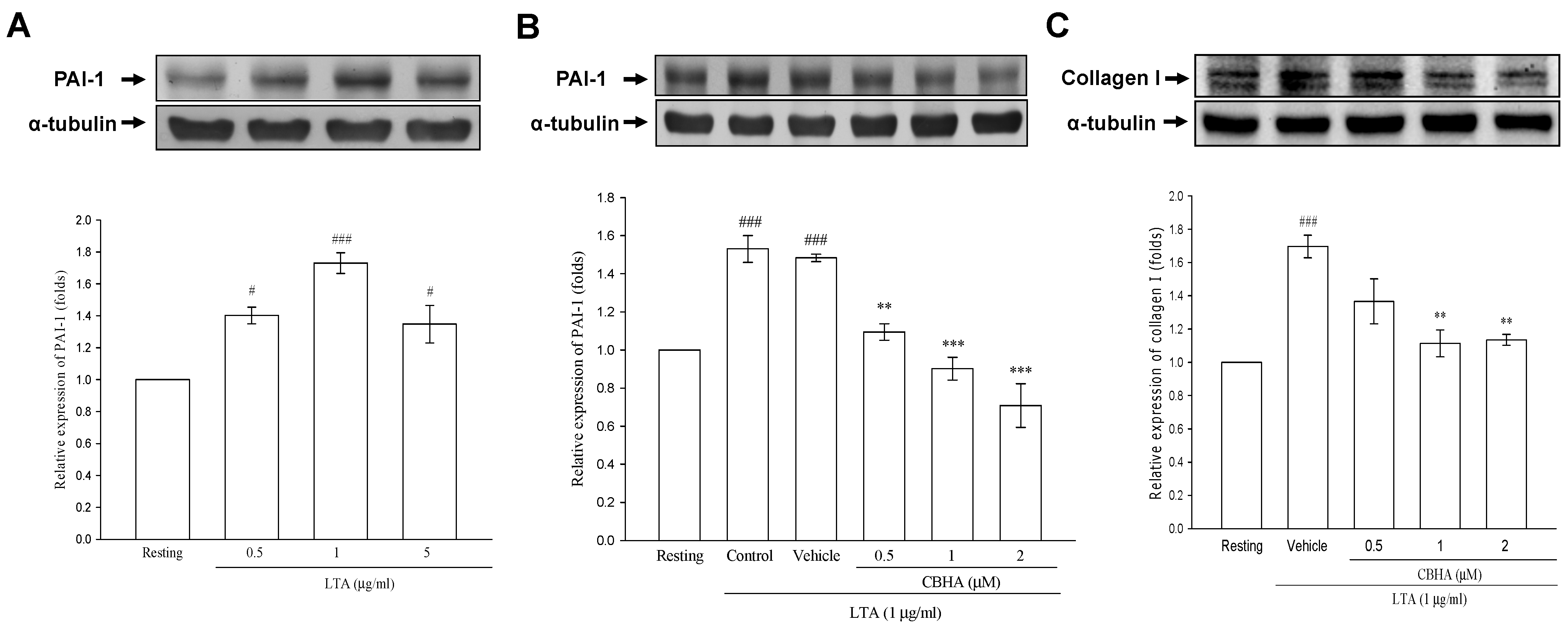
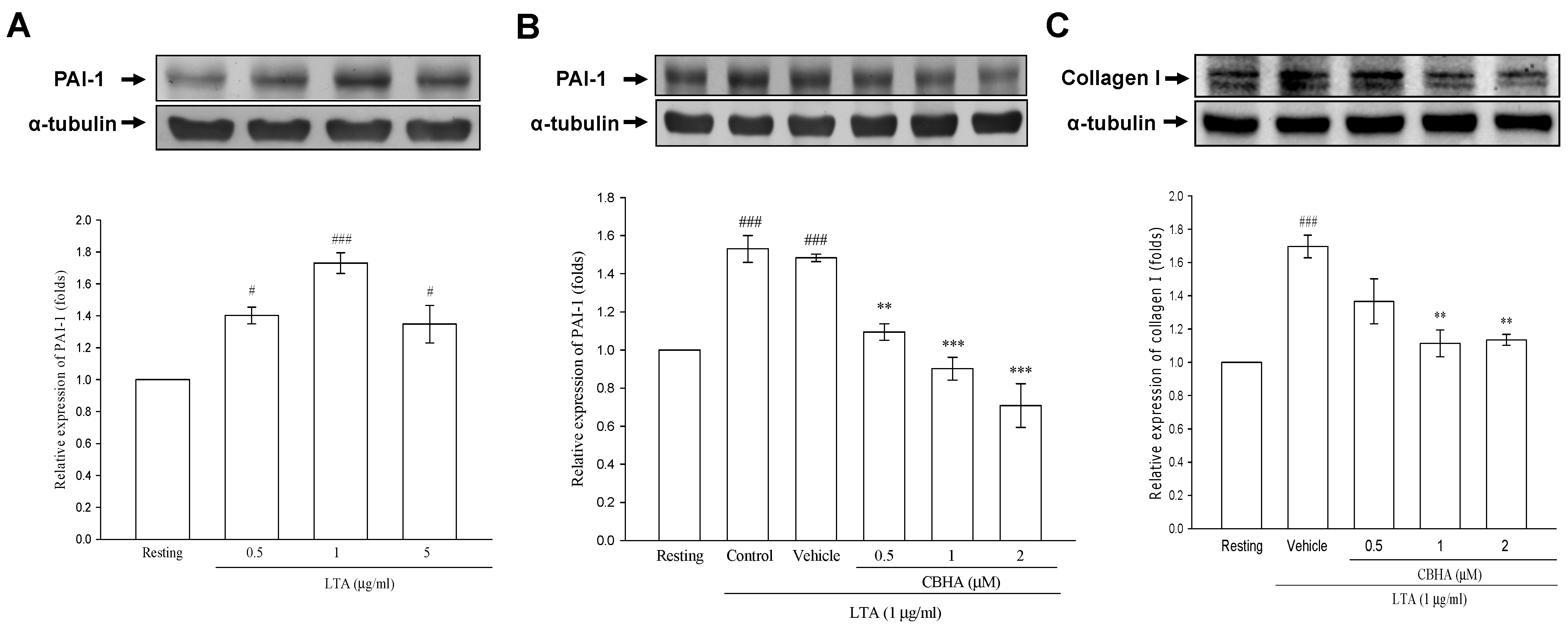
2.5. CBHA Inhibits Lipoteichoid Acid (LTA)-Induced PAI-1 and Collagen Expression in Human Pleural Mesothelial Cells (PMCs)
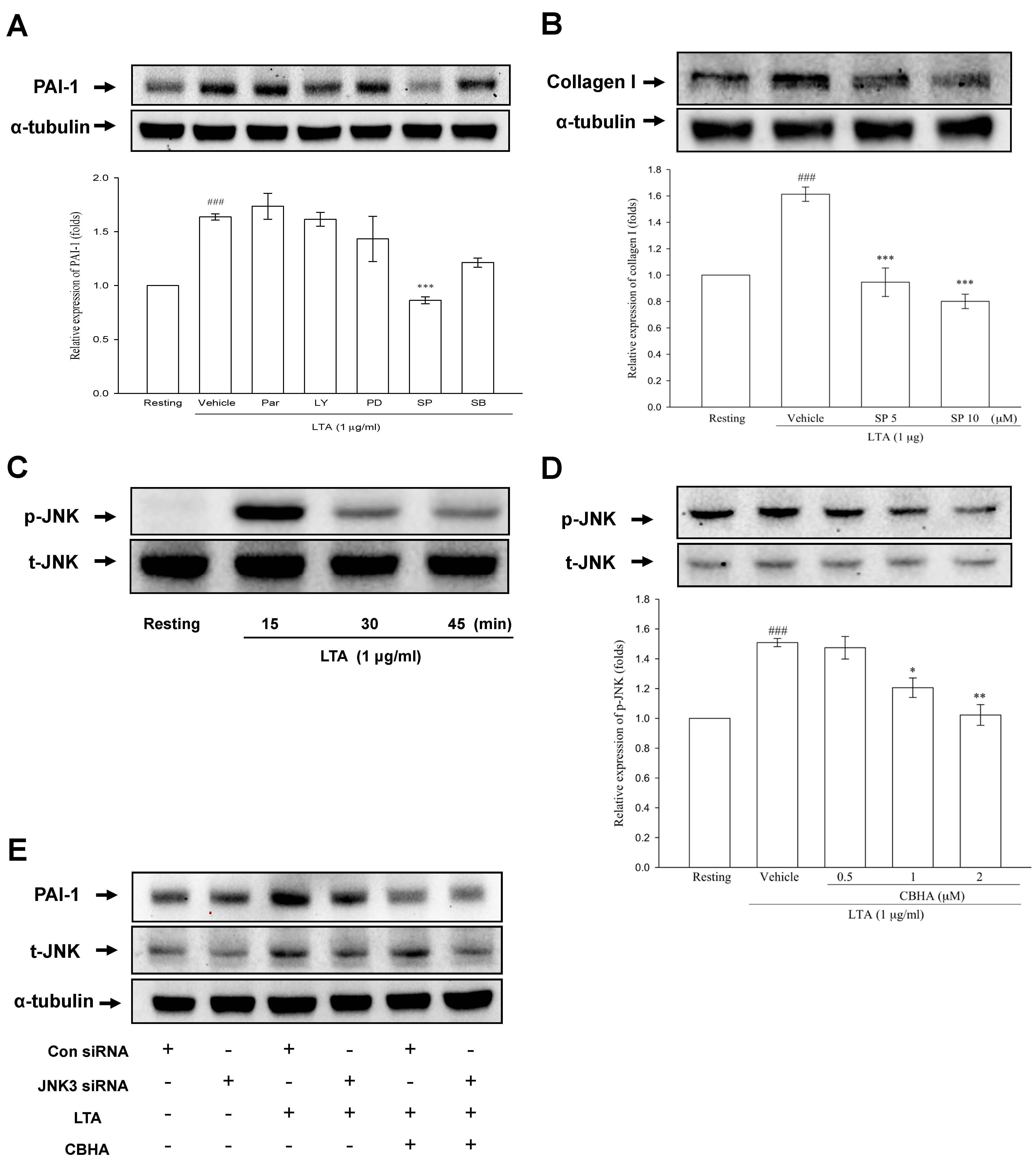
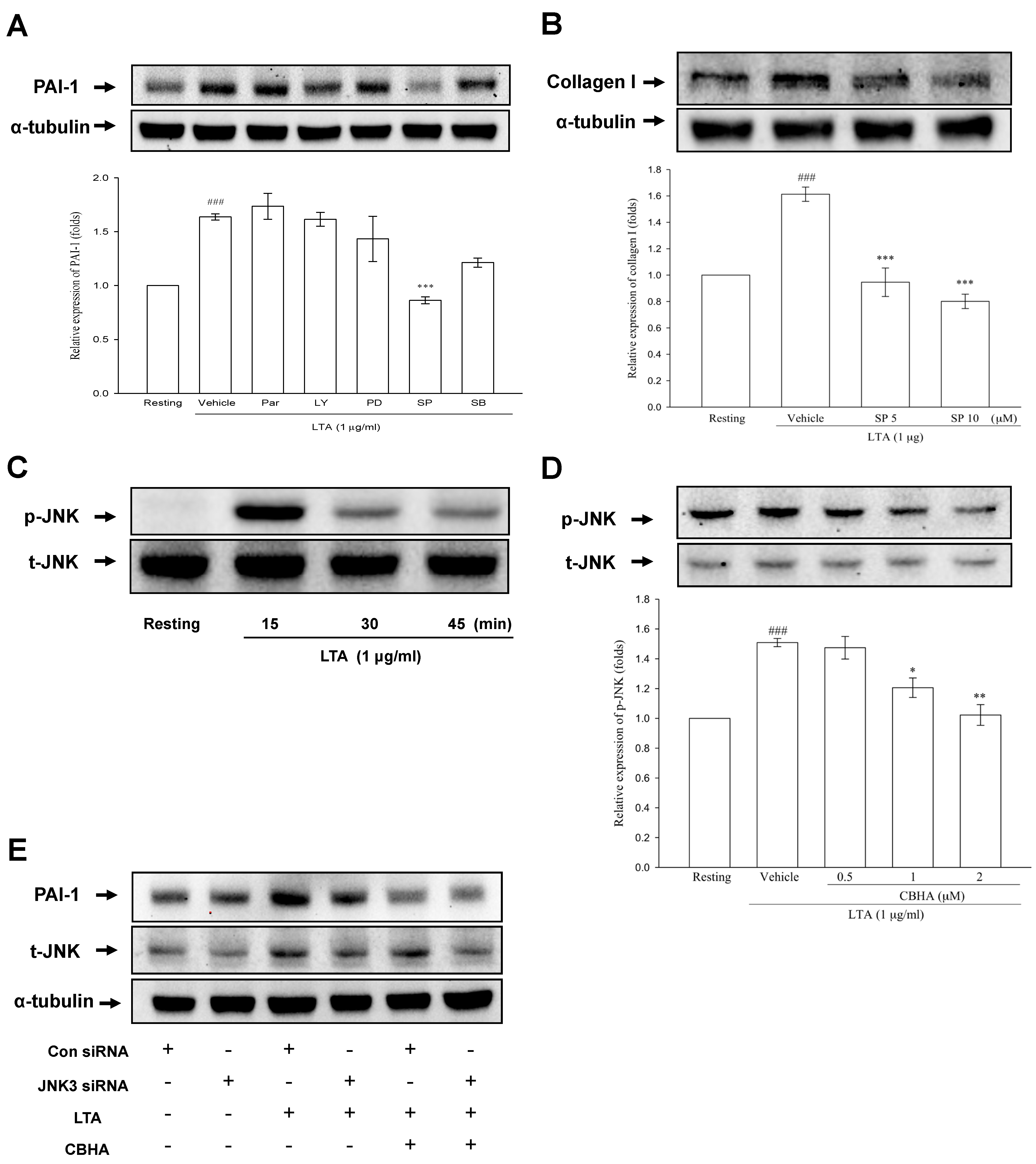
2.6. CBHA Blocks LTA-Induced JNK Phosphorylation in Human PMCs
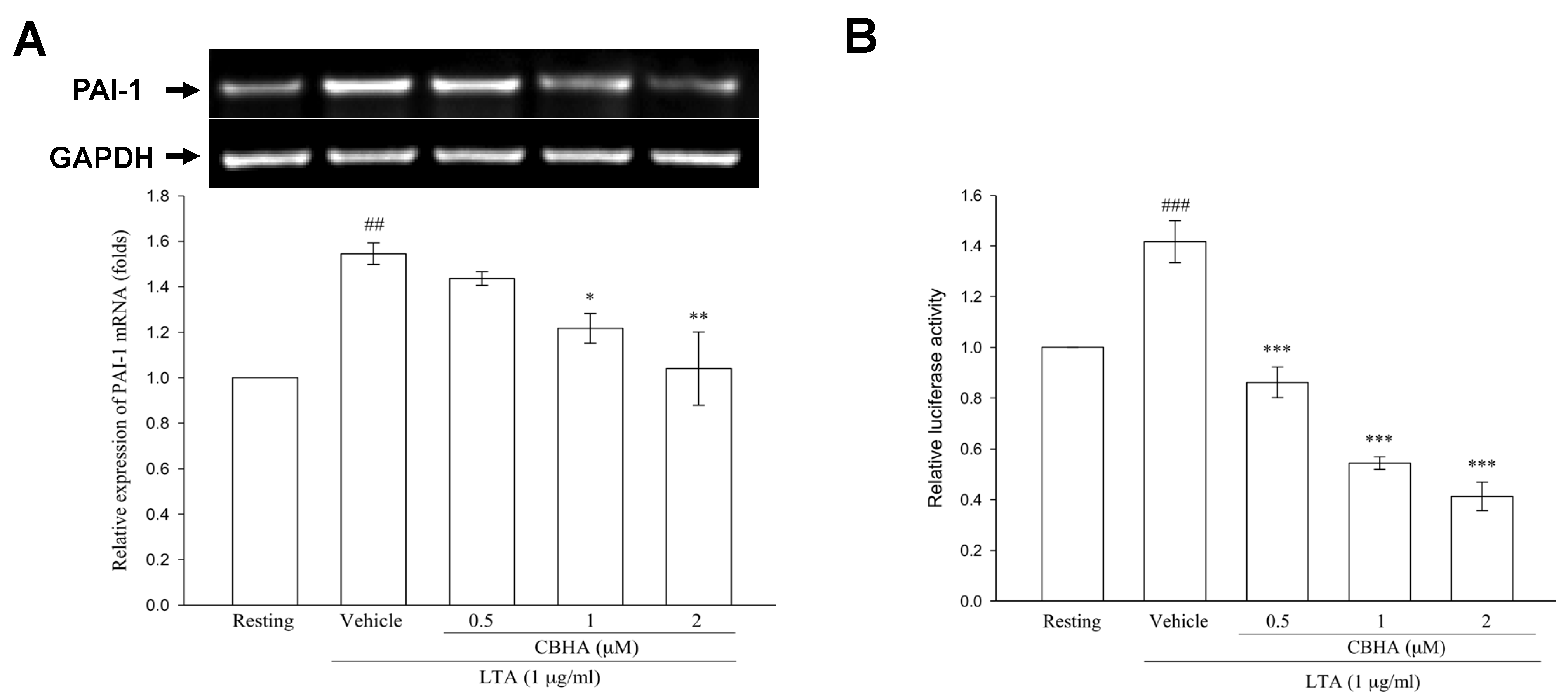
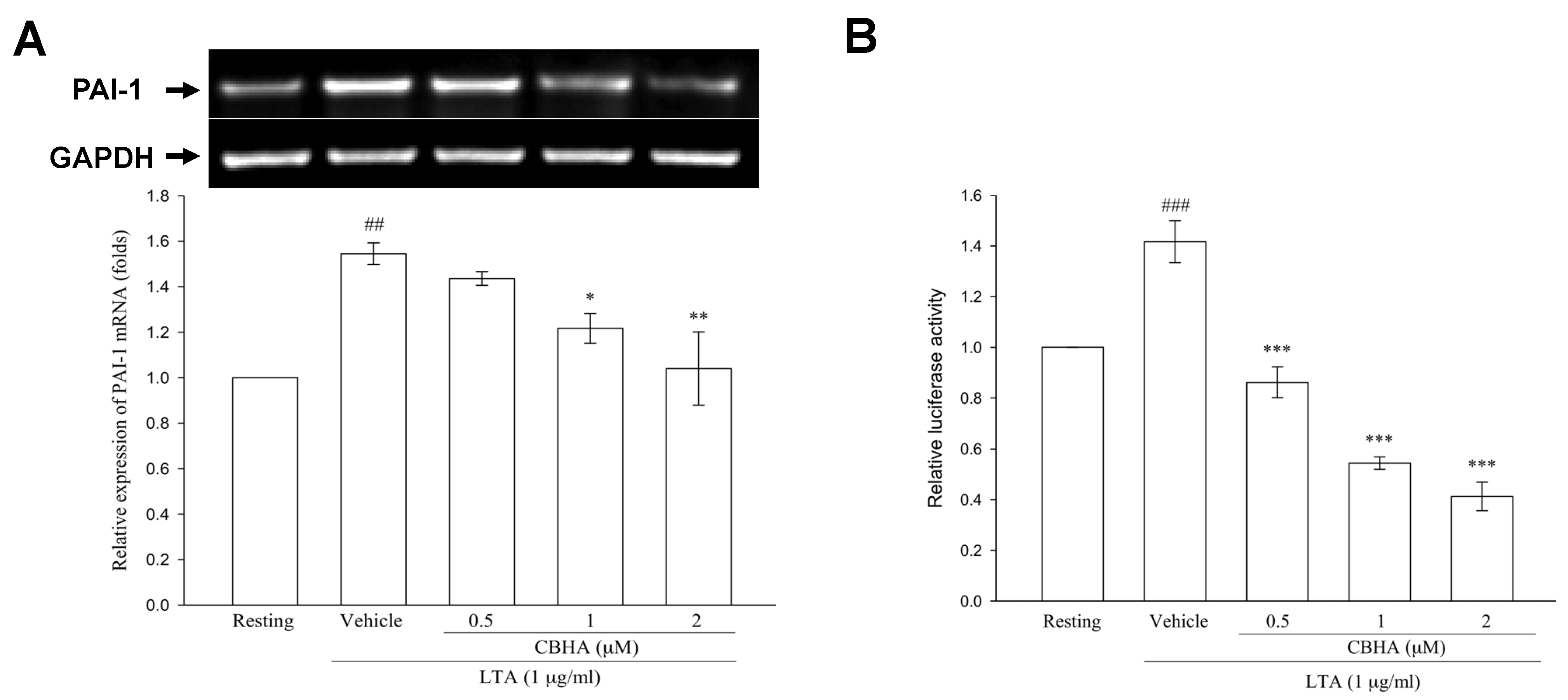
2.7. CBHA Reduces LTA-Induced PAI-1 mRNA Expression and Promoter Activity in Human PMCs
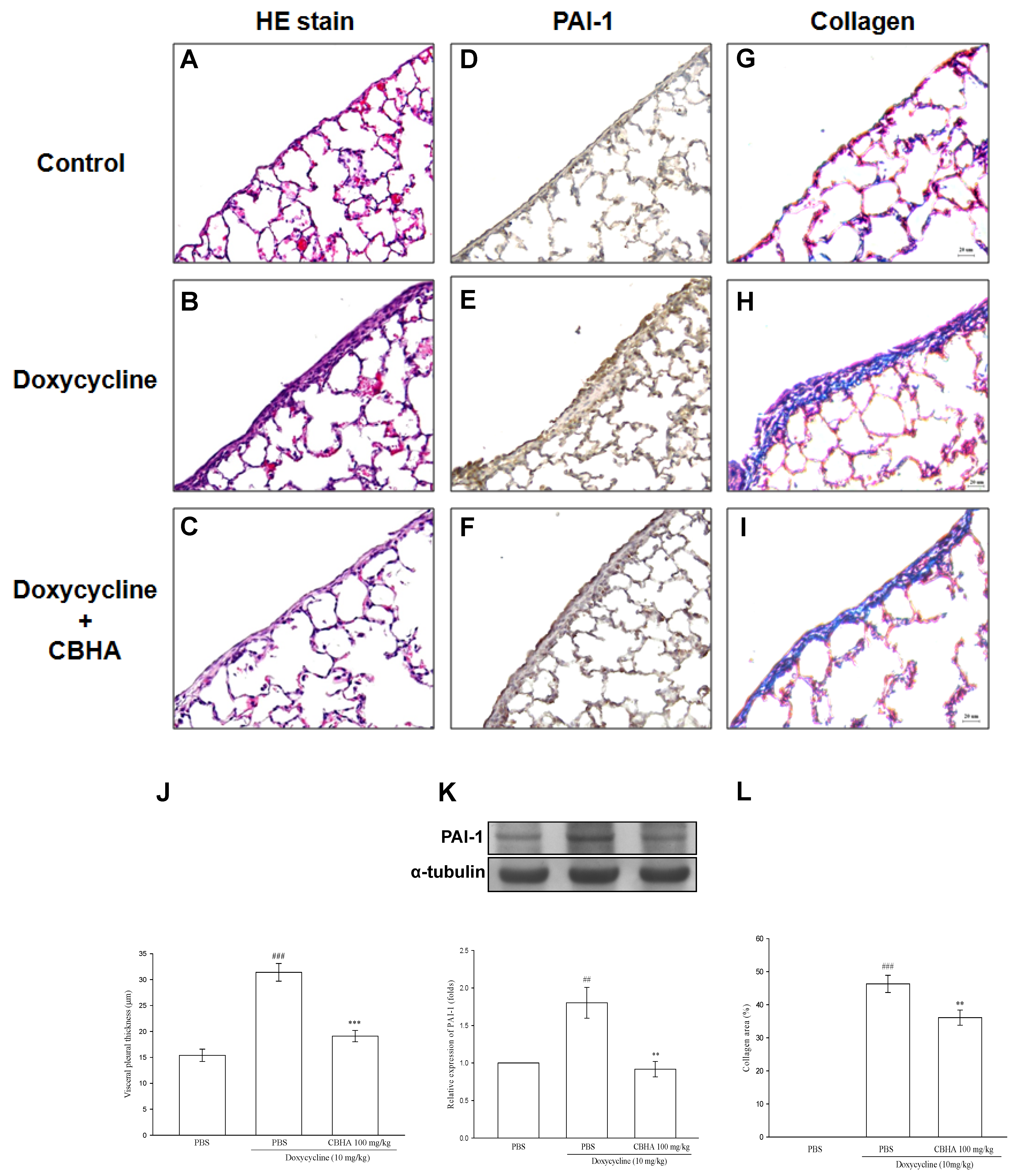
2.8. CBHA Minimizes Doxycycline–Induced PAI-1 and Collagen Expression on Visceral Pleural Mesothelium and Pleural Fibrosis in Rat Model
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Patient Enrollment
4.3. Thoracentesis and Analysis of Pleural Fluids
4.4. Chest Radiographs (CXRs) and Pulmonary Function
4.5. Measurement of Cytokines and Fibrinolytic Factors
4.6. Human Pleural Mesothelial Cell Culture
4.7. Western Blotting Assay
4.8. RNA Interference
4.9. RNA Extraction and Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
4.10. PAI-1 Luciferase Activity Assay
4.11. Pleural Fibrosis Rat Model
4.12. Histology and Immunohistochemistry
4.13. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hage, C.A.; Abdul-Mohammed, K.; Antony, V.B. Pathogenesis of pleural infection. Respirology 2004, 9, 12–15. [Google Scholar] [CrossRef]
- Jantz, M.A.; Antony, V.B. Pleural fibrosis. Clin. Chest Med. 2006, 27, 181–191. [Google Scholar] [CrossRef]
- Rabieian, R.; Boshtam, M.; Zareei, M.; Kouhpayeh, S.; Masoudifar, A.; Mirzaei, H. Plasminogen activator inhibitor type-1 as a regulator of fibrosis. J. Cell. Biochem. 2018, 119, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.C.; Chen, Y.C.; Chen, F.J.; Chang, S.C. Cytokines and fibrinolytic enzymes in tuberculous and parapneumonic effusions. Clin. Immunol. 2005, 116, 166–173. [Google Scholar] [CrossRef]
- Chung, C.L.; Hsiao, S.H.; Hsiao, G.; Sheu, J.R.; Chen, W.L.; Chang, S.C. Clinical importance of angiogenic cytokines, fibrinolytic activity and effusion size in parapneumonic effusions. PLoS ONE 2013, 8, e53169. [Google Scholar] [CrossRef]
- Maskell, N.A.; Batt, S.; Hedley, E.L.; Davies, C.W.H.; Gillespie, S.H.; Davies, R.J.O. The bacteriology of pleural infection by genetic and standard methods and its mortality significance. Am. J. Respir. Crit. Care Med. 2006, 174, 817–823. [Google Scholar] [CrossRef]
- Dessing, M.C.; Schouten, M.; Draing, C.; Levi, M.; von Aulock, S.; Van Der Poll, T. Role played by Toll-like receptors 2 and 4 in lipoteichoic acid–Induced lung inflammation and coagulation. J. Infect. Dis. 2008, 197, 245–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, N.M.; Davies, H.E.; Salzberg, M.; Truog, P.; Midgely, R.; Kerr, D.; Clelland, C.; Hedley, E.L.; Lee, Y.C.; Davies, R.J. Use of lipoteichoic acid-T for pleurodesis in malignant pleural effusion: A phase I toxicity and dose-escalation study. Lancet Oncol. 2008, 9, 946–952. [Google Scholar] [CrossRef]
- Lee, K.L.; Chen, W.L.; Chen, R.J.; LAI, K.S.; Chung, C.L. Lipoteichoic acid upregulates plasminogen activator inhibitor-1 expression in parapneumonic effusions. Respirology 2018, 23, 89–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, S.; Kang, G.; Eom, G.H. HDAC inhibitors: Therapeutic potential in fibrosis-associated human diseases. Int. J. Mol. Sci. 2019, 20, 1329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, C.L.; Sheu, J.R.; Chen, W.L.; Chou, Y.C.; Hsiao, C.J.; Hsiao, S.H.; Hsu, M.J.; Cheng, Y.W.; Hsiao, G. Histone deacetylase inhibitor m-carboxycinnamic acid bis-hydroxamide attenuates plasminogen activator inhibitor-1 expression in human pleural mesothelial cells. Am. J. Respir. Cell Mol. Biol. 2012, 46, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.L.; Sheu, J.R.; Hsiao, C.J.; Hsiao, S.H.; Chung, C.L.; Hsiao, G. Histone deacetylase inhibitor impairs plasminogen activator inhibitor-1 expression via inhibiting TNF-α-activated MAPK/AP-1 signaling cascade. Biomed. Res. Int. 2014, 2014, 231012. [Google Scholar] [CrossRef]
- Halili, M.A.; Andrews, M.R.; Labzin, L.I.; Schroder, K.; Matthias, G.; Cao, C.; Lovelace, E.; Reid, R.C.; Le, G.T.; Hume, D.A.; et al. Differential effects of selective HDAC inhibitors on macrophage inflammatory responses to the Toll-like receptor 4 agonist LPS. J. Leukoc. Biol. 2010, 87, 1103–1114. [Google Scholar] [CrossRef] [PubMed]
- Waetzig, V.; Herdegen, T. Context-specific inhibition of JNKs: Overcoming the dilemma of protection and damage. Trends Pharm. Sci. 2005, 26, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Tang, K.; Bilaceroglu, S.; Kalomenidis, I.; Salleng, K.J.; Lane, K.B.; Light, R.W. Iodopovidone is as effective as doxycycline in producing pleurodesis in rabbits. Respirology 2010, 15, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Niki, T.; Rombouts, K.; De Bleser, P.; De Smet, K.; Rogiers, V.; Schuppan, D.; Yoshida, M.; Gabbiani, G.; Geerts, A. A histone deacetylase inhibitor, trichostatin A, suppresses myofibroblastic differentiation of rat hepatic stellate cells in primary culture. Hepatology 1999, 29, 858–867. [Google Scholar] [CrossRef]
- Richon, V.M.; Emiliani, S.; Verdin, E.; Webb, Y.; Breslow, R.; Rifkind, R.A.; Marks, P.A. A class of hybrid polar inducers of transformed cell differentiation inhibits histone deacetylases. Proc. Natl. Acad. Sci. USA 1998, 95, 3003–3007. [Google Scholar] [CrossRef] [Green Version]
- Spange, S.; Wagner, T.; Heinzel, T.; Krämer, O.H. Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int. J. Biochem. Cell Biol. 2009, 41, 185–198. [Google Scholar] [CrossRef]
- Tung, C.W.; Hsu, Y.C.; Cai, C.J.; Shih, Y.H.; Wang, C.J.; Chang, P.J.; Lin, C.L. Trichostatin A ameliorates renal tubulointerstitial fibrosis through modulation of the JNK-dependent Notch-2 signaling pathway. Sci. Rep. 2017, 7, 14495. [Google Scholar] [CrossRef]
- Lyu, X.; Hu, M.; Peng, J.; Zhang, X.; Sanders, Y.Y. HDAC inhibitors as antifibrotic drugs in cardiac and pulmonary fibrosis. Ther. Adv. Chronic Dis. 2019, 10, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Sahn, S.A.; Taryle, D.A.; Good, J.T., Jr. Experimental empyema. Time course and pathogenesis of pleural fluid acidosis and low pleural fluid glucose. Am. Rev. Respir. Dis. 1979, 120, 355–361. [Google Scholar] [PubMed]
- Mavroudis, C.; Ganzel, B.L.; Katzmark, S.; Polk, H.C., Jr. Effect of hemothorax on experimental empyema thoracis in the guinea pig. J. Thorac. Cardiovasc. Surg. 1985, 89, 42–49. [Google Scholar] [CrossRef]
- Mierzejewski, M.; Korczynski, P.; Krenke, R.; Janssen, J.P. Chemical pleurodesis—a review of mechanisms involved in pleural space obliteration. Respir. Res. 2019, 20, 247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menzies, S.M.; Rahman, N.M.; Wrightson, J.M.; Davies, H.E.; Shorten, R.; Gillespie, S.H.; Davies, C.W.; Maskell, N.A.; Jeffrey, A.A.; Lee, Y.C.; et al. Blood culture bottle culture of pleural fluid in pleural infection. Thorax 2011, 66, 658–662. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, C.Y.; Sheu, J.R.; Yang, C.H.; Chen, W.L.; Tsai, J.H.; Chung, C.L. Thrombin upregulates PAI-1 and mesothelial–mesenchymal transition through PAR-1 and contributes to tuberculous pleural fibrosis. Int. J. Mol. Sci. 2019, 20, 5076. [Google Scholar] [CrossRef] [Green Version]
- De Pablo, A.; Villena, V.; Echave-Sustaeta, J.; Encuentra, A.L. Are pleural fluid parameters related to the development of residual pleural thickening in tuberculosis? Chest 1997, 112, 1293–1297. [Google Scholar] [CrossRef] [Green Version]
- Chung, C.L.; Sheu, J.R.; Liu, H.E.; Chang, S.C.; Chou, Y.C.; Chen, W.L.; Chou, D.S.; Hsiao, G. Dynasore, a dynamin inhibitor, induces PAI-1 expression in MeT-5A human pleural mesothelial cells. Am. J. Respir. Cell Mol. Biol. 2009, 40, 692–700. [Google Scholar] [CrossRef]
- Idell, S.; Na, M.J.; Liao, H.; Gazar, A.E.; Drake, W.; Lane, K.B.; Koenig, K.; Komissarov, A.; Tucker, T.; Light, R.W. Single-chain urokinase in empyema induced by Pasturella multocida. Exp. Lung Res. 2009, 35, 665–681. [Google Scholar] [CrossRef]
- Io, K.; Nishino, T.; Obata, Y.; Kitamura, M.; Koji, T.; Kohno, S. SAHA suppresses peritoneal fibrosis in mice. Perit. Dial. Int. 2015, 35, 246–258. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Yamashita, Y.; Toyokuni, S. A novel method for efficient collection of normal mesothelial cells in vivo. J. Clin. Biochem. Nutr. 2010, 46, 265–268. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | GPB PPE | p-Value * | |
|---|---|---|---|
| RPT ≤ 10 mm | RPT > 10 mm | ||
| Subject, n | 38 | 26 | |
| Age, years | 70 (42–80) | 69 (48–78) | 0.472 |
| Males, n | 25 | 17 | 0.973 |
| Symptom onset to enrollment, days | 8 (5–11) | 9 (6–12) | 0.604 |
| Pleural fluid | |||
| pH value | 7.31 (6.45–7.50) | 7.10 (6.42–7.39) | 0.126 |
| Glucose, mg/dL | 98 (15–291) | 63 (16–185) | 0.570 |
| Protein, g/L | 4.3 (2.7–7.5) | 4.7 (3.2–7.5) | 0.193 |
| LDH, IU/dL | 599 (258–1760) | 907 (263–5190) | 0.019 |
| Leukocyte count, cells/μL | 3565 (1050–23,460) | 8865 (1170–33,250) | 0.058 |
| PAI-1, ng/mL | 57.5 (16.7–90.0) | 99.0 (72.9–140.3) | <0.001 |
| tPA, ng/mL | 14.9 (3.2–21.0) | 12.8 (3.1–18.3) | 0.071 |
| TNF-α, pg/mL | 31.9 (21.8–85.9) | 42.6 (23.7.8–110.3) | 0.026 |
| IL-1β, pg/mL | 34.3 (21.0–94.5) | 50.3 (23.0–101.6) | 0.042 |
| IL-8, pg/mL | 590.6 (35.7–1906.2) | 773.4 (121.7–1572.0) | 0.036 |
| Pleural fluid bacteria culture | 0.595 | ||
| Streptococcus spp., n | 15 | 12 | |
| Staphylococcus spp., n | 23 | 14 | |
| Initial pleural effusion CXR score, % | 48 (25–65) | 54 (32–81) | 0.017 |
| Residual pleural shadowing CXR score, % | 1.9 (0.5–4.2) | 4.5 (3.1–8.2) | <0.001 |
| FVC at 12 months, % predicted | 80 (76–84) | 73 (70–76) | <0.001 |
| Variables | OR | 95% CI | p-Value |
|---|---|---|---|
| LDH, IU/dL | 1.00 | 0.99‒1.00 | 0.135 |
| PAI-1, ng/mL | 2.72 | 1.27‒5.84 | 0.006 |
| TNF-α, pg/mL | 1.08 | 1.01‒1.23 | 0.055 |
| IL-1β, pg/mL | 1.05 | 0.99‒1.15 | 0.272 |
| IL-8, pg/mL | 1.00 | 0.99‒1.01 | 0.989 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, W.-L.; Chen, M.-C.; Hsu, S.-F.; Hsiao, S.-H.; Chung, C.-L. HDAC Inhibitor Abrogates LTA−Induced PAI-1 Expression in Pleural Mesothelial Cells and Attenuates Experimental Pleural Fibrosis. Pharmaceuticals 2021, 14, 585. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14060585
Chen W-L, Chen M-C, Hsu S-F, Hsiao S-H, Chung C-L. HDAC Inhibitor Abrogates LTA−Induced PAI-1 Expression in Pleural Mesothelial Cells and Attenuates Experimental Pleural Fibrosis. Pharmaceuticals. 2021; 14(6):585. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14060585
Chicago/Turabian StyleChen, Wei-Lin, Mei-Chuan Chen, Shang-Fu Hsu, Shih-Hsin Hsiao, and Chi-Li Chung. 2021. "HDAC Inhibitor Abrogates LTA−Induced PAI-1 Expression in Pleural Mesothelial Cells and Attenuates Experimental Pleural Fibrosis" Pharmaceuticals 14, no. 6: 585. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14060585