
Kynurenines as a Novel Target for the Treatment of Malignancies


1
Department of Pharmacodynamics, Medical University of Bialystok, Mickiewicza 2c, 15-222 Bialystok, Poland
2
Department of Monitored Pharmacotherapy, Medical University of Bialystok, Mickiewicza 2c, 15-222 Bialystok, Poland
*
Author to whom correspondence should be addressed.
Pharmaceuticals 2021, 14(7), 606; https://0-doi-org.brum.beds.ac.uk/10.3390/ph14070606
Submission received: 10 May 2021
/
Revised: 21 June 2021
/
Accepted: 22 June 2021
/
Published: 23 June 2021
(This article belongs to the Special Issue Novel Therapeutic Targets in Cancer)
Abstract
:Malignancies are unquestionably a significant public health problem. Their effective treatment is still a big challenge for modern medicine. Tumors have developed a wide range of mechanisms to evade an immune and therapeutic response. As a result, there is an unmet clinical need for research on solutions aimed at overcoming this problem. An accumulation of tryptophan metabolites belonging to the kynurenine pathway can enhance neoplastic progression because it causes the suppression of immune system response against cancer cells. They are also involved in the development of the mechanisms responsible for the resistance to antitumor therapy. Kynurenine belongs to the most potent immunosuppressive metabolites of this pathway and has a significant impact on the development of malignancies. This fact prompted researchers to assess whether targeting the enzymes responsible for its synthesis could be an effective therapeutic strategy for various cancers. To date, numerous studies, both preclinical and clinical, have been conducted on this topic, especially regarding the inhibition of indoleamine 2,3-dioxygenase activity and their results can be considered noteworthy. This review gathers and systematizes the knowledge about the role of the kynurenine pathway in neoplastic progression and the findings regarding the usefulness of modulating its activity in anticancer therapy.
1. Introduction
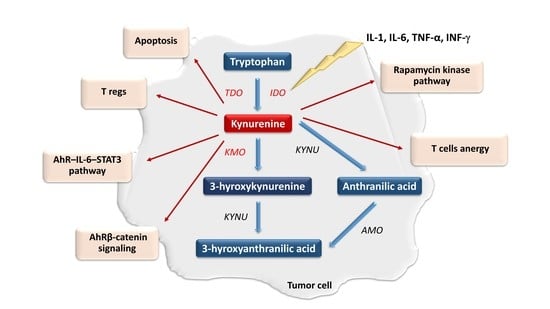
Tryptophan (TRP) belongs to the essential biogenic amino acids. It undergoes extensive metabolism through several metabolic pathways [1,2,3,4,5,6,7]. They play an important role in the regulation of numerous physiological and pathological processes. The kynurenine pathway (KP) is the main pathway responsible for the metabolism of this amino acid [2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22]. Within KP, TRP is oxidized into N-formylkynurenine using tryptophan 2,3-dioxygenase (TDO) and two indoleamine 2,3-dioxygenase isoforms (IDO-1 and IDO-2) [23,24]. Under physiological conditions, IDO and TDO catalyze the same reaction in parallel but have different tissue distributions. TDO activity is highest in the liver, while IDO occurs in almost all body tissues [25,26,27,28,29]. In the next step, N-formylokynurenine is converted to kynurenine (KYN) by the enzyme formamidase. KYN is metabolized by the three branches of KP, leading to the formation of kynurenic acid, anthranilic acid and 3-hydroxykynurenine (3-HKYN) [30,31,32,33]. The catabolism of KYN to 3-HKYN occurs with the participation of kynurenine 3-monooxygenase (KMO), while to kynurenic acid by kynurenine aminotransferase (KAT) [28,29]. In turn, 3-HKYN is converted to xanthurenic acid by the KAT and to 3-HAA after modification with the kynureninase (KYNU) (Figure 1) [34,35,36]. The above enzymes are important levels of the KP cascade because they participate in a synthesis of toxic metabolites of KP, responsible for the development of a wide range of systemic disorders, including neoplastic progression [2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22]. In 2005 Muller et al. were the first to evaluate the effectiveness of IDO inhibition in an in-vivo tumor model [37]. Research concerning this issue led the researchers to conclude that modulation of the KP activity has considerable potential as a new strategy for tumor treatment [37,38,39].
2. The Role of the Kynurenine Pathway in Neoplastic Processes
Effective cancer treatment is still a great challenge for modern medicine. Tumors have produced a broad range of mechanisms to evade an immune and therapeutic response [37,38,39,40]. An accumulation of the KP metabolites can enhance neoplastic progression because it suppresses immune processes directed against cancer cells. KYN is one of the most potent immunosuppressive metabolites belonging to this pathway. [40,41,42]. The expression and activity of IDO and TDO, responsible for its synthesis, are upregulated in most cancers. These enzymes are thought to be involved in the processes responsible for the development of immune escape of tumors and resistance to immuno-, radio- and chemotherapy [40,43,44,45,46,47].
IDO is activated in response to the release of pro-inflammatory factors such as interleukins (IL-1 and IL-6), tumor necrosis factor-α and interferon-γ (IFN-γ) [48,49,50]. It enhances immune tolerance by an increase in a KYN and its downstream metabolites levels within the tumor microenvironment [51,52]. KYN induces general control nonderepressible-2 kinase and mammalian target of rapamycin kinase pathways, which reduce the immune response [41,53,54,55,56]. Other effects of KYN include increased TH1 and effector T cells anergy and apoptosis rate and T regulatory cells (T regs) immunosuppressive activity [57,58,59,60]. In turn, IDO-activated T regs can promote IDO1 expression in dendritic cells (DCs) during the antigen presentation process, additionally enhancing the immunosuppressive mechanism [61,62]. The IDO-dependent KYN synthesis can also directly promote tumor growth and metastasis via the generation of pro-proliferative metabolites and activation of β-catenin signaling in IDO-expressing tumors [63,64]. Therefore, prolonged IDO1 overexpression in the tumor microenvironment negatively correlates with patient outcomes [46,65,66,67]. IDO-positive tumors and microenvironment tissues showed basal phosphorylation and acetylation of signal transducer and activator of transcription 3 (STAT3) [68,69]. The inhibition of IL-6 or STAT3 using small interfering RNA or pharmacological inhibitors decreases IDO activity. In turn, KYN activates the aryl hydrocarbon receptor (AhR), which upregulates the expression of IL-6 and STAT3. It indicates that IDO expression may be also upregulated by autocrine stimulation [18,19,20,70]. Inhibition of AhR signaling restores proliferation and activation of T cells in the IDO-expressing human cancers. This indicates IDO inhibitors and AhR antagonists as a new therapeutic solution in cancer therapy (Figure 2) [18,19,71,72].
The involvement of TDO in cancer biology seems to be similar to IDO. Its overexpression has been observed in several cancers [73,74,75]. As mentioned earlier, although IDO and TDO catalyze the same reaction, their tissue distribution is different [28,29,30]. Therefore, the impact of the TDO on the overall TRP metabolism cannot be ignored [73,74,75]. This is supported by the fact that isomers of 1-methyl-tryptophan (1-MT), commonly used in research as IDO inhibitors, do not fully inhibit TRP catabolism via KP. Although, preclinical studies found that targeting TDO significantly decreased the incidence of cancer progression and restored anticancer immunity [73,74,75,76], inhibition of IDO1 may enhance TDO expression and activity (Figure 2). Therefore, TDO inhibitors could be useful as auxiliary therapeutics. In summary, the inhibition of IDO, TDO, or both, impairing immune resistance of tumors, may represent a safe and efficient approach for cancer therapy, stimulating tumoral immune rejection and improving the efficacy of cancer immunotherapy [76].
3. Modulation of Kynurenine Pathway Activity in the Management of Neoplastic Diseases
3.1. Pharmacological Modulation of IDO and TDO Activity
Most approaches related to targeting KP involve pharmacological inhibition of enzymes belonging to this pathway. There is an increasing number of discovered IDO and TDO inhibitors [37,38,39,40,52,75,76,77]. Most of them are IDO1 inhibitors developed for cancer therapy [37,38,39,40,52]. Preclinical models and clinical trials showed that their administration in monotherapy slows the growth of tumors but did not eliminate or prevent them. However, their combination with chemotherapies or immunotherapies leads to beneficial results in many types of cancer, increasing the response rate compared to conventional therapies [77]. Also, IDO2 is a potential therapeutic target for diseases accompanied by immune escape, such as cancer. Compared to IDO1, its upregulation is more frequently observed in autoimmune reactions but is also found in cancer. The knowledge about IDO2 in the immune system is scarce, but evidence suggests that it supports IDO1 in mediating T regs suppressive mechanisms [78,79,80]. However, the detailed mechanisms of both isoforms in different diseases are still not fully understood.
In preclinical research 1-MT significantly reduced tumor-mediated immunosuppression, leading to the regression of systemic tumors and proving its utility in dealing with immune escape [37,38,39,40,81,82]. Additionally, its administration in combination with carboxyamidotriazole (CAI) potently increases the activity of cytotoxic CD8+ T cells and their ability to kill tumor cells, probably due to the reduced expression of programmed cell death protein-1 (PD-1) and increased IFN-γ secretion in the tumor microenvironment associated with decreased KYN level [83]. Additionally, inhibition of KYN synthesis due to blocking of IDO by 1-MT leads to an increase in T cell and NK cell proliferation [84,85,86,87]. Commonly used in preclinical research 1-MT is a racemic mixture of the two isoforms l-methyl-d-tryptophan and 1-methyl-l-tryptophan (D-1-MT). 1-MT as a racemic mixture mainly inhibits the IDO1 activity. Interestingly, the individual stereoisomers of 1-MT differ in their affinity for the particular isoforms of this enzyme. Authors discovered that the L stereoisomer of 1-MT is more potent in the inhibition of the IDO1 isoform. In turn, D-1-MT is less active in inhibiting IDO1 while it has a much greater affinity for IDO2 [44,88]. Preclinical studies also demonstrated that D-1-MT has superior antitumor activity and is more effective in inhibiting the activity of IDO-expressing tolerogenic DCs, than its L-stereoisomer [88,89]. In effect, d-1-MT, under the name indoximod, is developed clinically as the IDO2 inhibitor for the treatment of several cancers with the general aim of reversing cancer-associated immunosuppression [90,91,92]. Similar results were also observed when CAI was combined with 3′,4′-dimethoxyflavone (DMF), an AhR antagonist. The anticancer properties of CAI combined with DMF or 1-MT are higher than CAI in monotherapy and comparable to an anti-PD-1 antibody. These combinations seem to be an effective strategy for the treatment of various tumors [83]. Preclinical research showed that the combination of melatonin and 1-MT has a synergistic effect combined with tumor-specific immunotherapy and seems to be a new and promising method of growth control of HPV-associated tumors [93]. Also, novel IDO inhibitors, LW106 and NTRC 3883-0 inhibit tumor outgrowth in-vivo, limiting IDO-dependent stroma immune resistance and proliferation of cancer stem cells within the tumor microenvironment (Table 1) [94,95].
3.1.1. Lung Cancer
Preclinical studies showed that anti-PD1 resistance is associated with IDO1 overactivity. PD-L1 expression correlates with IDO1. Therefore, it may be a novel target for lung cancer immunotherapy [110,111]. All current immunotherapies are accompanied by an activation of IDO, which is a negative feedback mechanism. This suggested to the researchers, that IDO inhibitors could potentially be used in combination with immune checkpoint inhibitors in the treatment of the anti-PD1-resistant tumors [112]. So far, these compounds seem to be the most promising solution of tumor resistance to immune checkpoint inhibitors. Li et al. discovered that the treatment with an IDO1 inhibitor navoximod restored the antitumor immune responses in anti-PD1 resistance conditions (Table 1). Therefore, the combination therapy with the inhibitor of IDO1 and the agent targeting PD-1/PD ligand 1 (PD-L1) was expected to be an effective option for overcoming the immunosuppressive tumor microenvironment in anti-PD1-resistant tumors, such as melanoma, glioblastoma multiforme (GBM), or non-small cell lung cancer (Figure 3) [113,146]. However, a phase I clinical trial of navoximod with a PD-L1 inhibitor (atezolizumab) in advanced solid tumors showed that although a pharmacologic effect of navoximod was observed, there was no clear evidence of clinical benefit from concomitant administration of navoximod with atezolizumab [147,148].
Cisplatin resistance (CR) is a significant problem in lung cancer therapy. CR lung cancer cells consume amino acids, such as glutamine and TRP for survival, rather than glucose. Additionally, CR cells can activate KP to reduce the excess of reactive oxygen species (ROS) and maintain conditions suitable for growth and proliferation. Knocking down IDO1 expression using short hairpin RNA or its pharmacological inhibition by epacadostat significantly increases ROS level and inhibits the growth of CR cells (Table 1). Exposing CR cells to antioxidants also leads to the suppression of IDO1 activity, indicating an interrelationship between ROS and IDO1. Moreover, the expression and activation of T regs in the tissues of CR tumors were higher, compared with cisplatin-sensitive tumors. Therefore, the use of KP enzyme inhibitors may be an effective method against CR in tumors [114].
3.1.2. Breast Cancer
The IDO inhibitors ethyl pyruvate and amidoxime slightly suppress the growth of the Ehrlich tumor in mice and potentiates the therapeutic effect of cyclophosphamide [97]. In turn, EOS200271, the novel, selective, non-competitive, orally bioavailable IDO1 inhibitor, demonstrated a potent antitumor effect in the breast cancer animal model. It potently reduces intracellular KYN levels and IDO1-induced T cell anergy, and slows tumor cell growth, both in-vitro and in-vivo, in monotherapy and combined with antibodies directed against PD-L1 [131]. Furthermore, in an in-vitro model of triple-negative breast cancer, TDO2 inhibition by 680C91 significantly reduced intracellular KYN levels, leading to their increased sensitivity to the process of programmed cell death, as well as reduced their ability to proliferate, migrate and invade (Table 1) [71].
Cyclooxygenase-2 (COX-2), the enzyme responsible for prostaglandin synthesis, is upregulated in several tumors. Chen et al. demonstrated the anticancer effect of COX-2 inhibitor nimesulide in COX-2-overexpressing breast cancer in-vivo. They also proved its ability to inhibit IDO activity [123]. Simultaneous inhibition of IDO and COX-2 potently suppresses tumor growth (Table 1) [138,139]. Nimesulide significantly reduced systemic KYN synthesis and therefore could potentially target leukemia-induced IDO immune dysfunction as well [138]. Of course, it cannot be ruled out that, as with celecoxib, the reduction in IDO activity and inhibition of tumor growth may result from COX-2 inhibition, but this points to the potential utility of other COX-2 inhibitors to control IDO activation in cancer treatment [138,139,149].
3.1.3. Leukemias and Lymphomas
Another novel IDO inhibitor, Toho-1, has been investigated for its potential use in cancer immunotherapy. In addition to solid tumors, the increase in the IDO expression induced by IFN-γ or lipopolysaccharide (LPS) is also observed in the PMDC05 leukemic plasmacytoid dendritic cell line. Treatment by Toho-1 enhanced the antigen-presenting and cytotoxic T cells’ activity against PMDC05 cells. It suggests its potential applicability in leukemia immunotherapy (Figure 3) [134]. As mentioned before, indoximod is a D-1-methylated TRP molecule with immune checkpoint inhibitory activity. It is a selective IDO2 inhibitor developed for antitumor therapies (Table 1). In combination with cyclophosphamide, it is effective for IDO-positive non-Hodgkin lymphoma treatment in the mouse model [137].
3.1.4. Head and Neck Neoplasms
In addition to the above, the co-treatment with 1-MT and temozolomide or cytoxan prolonged survival rate in the intracranial GL261 glioblastoma multiforme (GBM) mouse model. It also increases the effectiveness of anticancer drugs against the glioma cell lines without additional side effects [104,150] Furthermore, IDO1 inhibition by 1-MT synergizes with radiotherapy in animal models of glioblastoma and Lewis lung cancer. However, 1-MT poorly penetrates the blood-brain barrier (BBB) [34,82,151]. Therefore, its effectiveness against glioma after systemic administration appears to be limited. In turn, tryptamine (TRY) and N-dimethyltryptamine (DMT) are novel non-competitive inhibitors effective against both constitutively expressed and IFN-γ-induced IDO activity. This suggests that they have the potential to be developed for IDO1- and TDO-related cancer treatment. They effectively inhibit the A172 human glioma cell line growth. Besides, TRY and DMT increased the cytotoxic activity of peripheral blood mononuclear cells in co-culture assays [108]. The commonly used antiviral agent acyclovir also inhibits IDO, leading to a decrease in T regs function in the GBM model [105]. In turn, IDO inhibition by 1-MT or dinaciclib, identified as an indirect inhibitor of this enzyme, in GBM and head and neck squamous cell carcinomas revealed a synergistic effect co-administered with temozolomide (Figure 3) [85,88,150]. Conventional chemotherapeutics, similarly to immune checkpoint blockers commonly activate KP. Numerous reports proved that it reduces their effectiveness [106]. In turn, the administration of IDO inhibitors in combination therapy may increase their efficacy. Therefore, these agents are considered as a promising treatment strategy for lymphoma, melanoma, glioma, glioblastoma and head and neck squamous cell carcinomas (Table 1) [83,105,106,107,108,109,118,119,134,137,151]. Indoximod is currently in clinical trials in combination with either temozolomide or bevacizumab for patients with GBM resistant to initial therapy and few objective responses have been observed [90,91]. This is another evidence that impairing tumor immune resistance seems to be a useful approach in antitumor therapy and IDO inhibition is effective in restoring immune function in malignancies dependent on T regs stimulation, such as GBM. In turn, a phase I clinical trial of EOS200271 showed promising results. It has a tolerable safety profile and significantly inhibits endogenous KYN synthesis, indicating effectiveness and durable clinical benefit in patients with recurrent malignant glioma [120].
3.1.5. Melanoma
Melanoma patients treated with IFN α-2b have increased IDO activity, which leads to an increase in T regs and PD-L1 cytotoxic T cells amount. Therefore, inhibition of IDO1 activity may be an effective solution to this problem [121,152]. In addition, Jia et al. demonstrated that 1-MT co-administered with pimozide revealed combined effectiveness against melanoma in the animal model. 1-MT intensifies the antitumor immunity of pimozide against melanoma via the regulation of tumor proliferation, migration, apoptosis and immune response. In effect, co-treatment with 1-MT significantly prolonged the survival rate of tumor-bearing mice compared to those receiving pimozide in monotherapy. It suggests that the use of these compounds has great potential as a novel treatment strategy for this type of tumor [122]. Similarly, high hopes are attached to navoximod. In the B16F10 mouse model, co-administered with pmel-1 T cells and gp100 peptide vaccination almost completely reduced relative tumor size. Also, DX-03-12, an imidazole-based compound with a phenylimidazole scaffold, seems to be a promising candidate for the treatment of melanoma. It is a potent, orally bioavailable IDO1 inhibitor with low cellular toxicity and high in-vivo antitumor efficacy. Its administration inhibits tumor growth in a B16-F10 melanoma mouse model [153]. Likewise, LW-106 demonstrated a potent antitumor activity based on a similar mechanism in the IDO-overexpressing melanoma mouse model, while NTRC 3883-0 in ovarian cancer cell culture and melanoma mouse model (Table 1) [94,95].
Epacadostat is another IDO inhibitor entered into clinical trials. It appeared to be a promising candidate for the treatment of melanoma, showing decent efficacy in inhibiting its growth in-vivo (Table 1) [94]. However, a phase I clinical trial with epacadostat and PD-L1 blockade in patients with advanced solid tumors did not bring positive results. This combination was not effective to be pursued in PD-L1 negative patients [154]. Despite these unsatisfactory results, this compound is still undergoing further clinical evaluations in combination with ipilimumab and pembrolizumab in melanoma and as monotherapy in advanced ovarian cancer. It recently underwent the phase I/II clinical trials and is currently during phase III trials in combination with checkpoint blockade. However, the latest results from one of the phase III clinical studies indicate that it does not improve the effectiveness of pembrolizumab in patients with unresectable or metastatic melanoma [74,155,156,157].
3.1.6. Ovarian and Cervical Cancer
Preclinical research showed that IDO1 inhibition by 1-MT also counteracts the invasion of carboplatin-resistant ovarian cancer cells via the down-regulation of IDO expression and re-activation of immune cell function. Thus, inhibition of the KP activity seems to be a potentially effective solution in the case of carboplatin-resistant cancers (Table 1) [46,129].
β-Lapachone is a naturally occurring 1,2-naphthoquinone-based compound, that advanced into clinical trials, due to its tumor-selective cytotoxic properties. It turned out to be an uncompetitive IDO1 inhibitor, which potently decreases the proliferation rate of cervical cancer cells in-vitro (Table 1). Its antitumor effect seems to be also related to its impact on TDO activity. Flick et al. showed that the effect of β-lapachone on cancer cells is related to the synergism of its cytotoxic and immunoregulatory properties. This discovery expands its potential use as an anticancer agent [140].
The limited clinical activity of epacadostat prompted the researchers to develop new compounds able to decrease IDO-dependent immune dysregulation in cancer. LVS-019 is a recently discovered potent IDO1 inhibitor. However, the impact of this compound on tumor growth is not known yet. In turn, its derivative, LBJ-10, effectively inhibits the proliferation of cervical cancer cells in-vitro via IDO1 inhibition. Therefore it has a high potential for further development as the agent for anticancer therapy [141]. In turn, BMS-986242 is another novel IDO1 inhibitor that was originally designed to treat various cancers, including melanoma and renal cell carcinoma (RCC). Interestingly, recent in-vitro and animal model studies indicate its high efficacy and safety in the treatment of ovarian cancer (Table 1). Promising results from preclinical studies have led the compound to be included in Phase I/II clinical trials in combination with nivolumab in patients with advanced cancer [130].
3.1.7. Digestive System Cancers
Both systemic and local administration of the IDO inhibitor, ethyl pyruvate accelerated tumor regression in the H-29 liver cancer-bearing mice [142]. In turn, 3-(2-(pyridyl)ethenyl) indole derivatives are new, potent and promising inhibitors of TDO. Their conjugates with irinotecan, designed to reverse tumor immune suppression, significantly improved the efficacy of irinotecan in HepG2 cancer cells. In-vitro studies showed that their use significantly increases the rate of apoptosis of cancer cells by inducing a mitochondria-dependent mechanism. They also potently downregulate the expression level of TDO. This, in turn, intensifies the inhibition of KYN synthesis, leading to intensified proliferation and activation of T cells (Figure 3). In-vivo studies proved that these conjugates can improve the tumor immune microenvironment in a murine model of hepatocellular carcinoma (Table 1). This combination of chemotherapy and immunotherapy may be a promising strategy for treating this type of cancer [143].
Pancreatic ductal adenocarcinoma (PDAC) belongs to tumors resistant to immune checkpoint inhibitors. Blaie et al. showed that a granulocyte-macrophage colony-stimulating factor-secreting allogeneic pancreatic tumor cell vaccine (GVAX) induces intratumoral T cell infiltration, leading to the activation of the tumor microenvironment. They also proved that GVAX therapy induces IDO1 expression within tumor epithelia and vaccine-induced tertiary lymphoid aggregates [132]. In addition, in PDAC, IDO1 expression plays an important role in regulating the activity of TH1, TH17 and probably T regs [131,132] (Figure 3). Epacadostat and EOS200271, a novel IDO1 inhibitor, enhance the antitumor efficacy of GVAX in the PDACs murine model, intensifying intratumoral T cell infiltration and activity caused by the vaccine. Furthermore, an administration of EOS200271 during the vaccine therapy diminished the suppressive effect of intratumoral myeloid-derived suppressor cells (MDSCs) on the proliferation of CD8+ T cells (Table 1). However, the addition of the anti–PD-L1 antibody to this combination did not cause further synergy and in fact may have a negative impact, decreasing the number of intratumoral effector T cells [131,132,133]. It suggests that EOS200271 does not increase the effectiveness of anti–PD-1/PD-L1 antibody for T cell–inflamed tumors such as PDACs treated with GVAX therapy.
Since IDO1-dependent KYN synthesis has been shown to directly promote colorectal cancer (CRC) growth through β-catenin activation, IDO inhibition has emerged as a promising strategy for CRC prevention. IDO overactivity is observed even in the early stages of colon carcinogenesis. Its inhibition suppresses the growth and stimulates the apoptosis of cancer cells [63,64,66,98,158,159,160,161,162,163]. Therefore, the use of IDO inhibitors, such as 1-MT, or epigallocatechin gallate in the animal models suppressed the occurrence of colonic preneoplastic lesions and reduced tumor size and mortality in animals with cancer (Figure 3) [63,98,99,163]. Similar effects were also observed with newer IDO1 inhibitors, such as epacadostat, navoximod and amidoxime. It confirms the potential utility of IDO1 inhibitors as agents for the chemoprevention of IDO-expressing colonic neoplasms. Furthermore, the regulation of IDO1 activity may be considered as a target for the prevention of other malignancies [98,99,100,101,155,163,164].
Also, 4,6-disubstituted indazole derivatives demonstrated the potential utility as IDO1 inhibitors in cancer treatment. In turn, 4,6-substituted-1H-indazole derivatives and 4-bromophenylhydrazinyl benzenesulfonylphenylureas showed high activity against both IDO and TDO and inhibited cancer growth and reduce the tumor weight in-vivo in the CT26 xenograft model (Table 1) [96,100,102]. This suggests that they have the potential to be developed for IDO1- and TDO-related cancer treatment.
3.1.8. Renal Cancer
Changes in TRP metabolism are also observed in the course RCC and lead to the increased synthesis of immunosuppressive metabolites. The inhibition of KP activity in the presence of IFN-α decreases RCC growth in the murine model. The IDO1 inhibitor methyl-thiohydantoin-DL-TRP alters the functionality of intratumoral leukocytes, improving the therapeutic effects of IFN-α (Table 1). Therefore, the combination of IFN-α with IDO inhibitors should be assessed for its utility in the treatment of RCC [135].
3.1.9. Myelodysplastic Syndromes
Recent studies also indicate the role of IDO-dependent immune dysregulation in the pathogenesis of myelodysplastic syndromes (MDS). The expansion of inflammatory hematopoietic suppressor cells, called MDSCs, impairs hematopoiesis and leads to the development of this disease. In turn, increased proliferation and activity of T regs are associated with worse disease outcomes [165,166,167,168,169]. Epacadostat is an oral, potent and selective IDO1 inhibitor. It does not affect IDO2 or TDO activity. Preclinical data suggest that its administration increases proliferation and activity of cytotoxic T cells and decreases T regs and MDSCs. It also promotes proliferation, survival and functions of various immune and immunoregulatory cells. These pleiotropic effects significantly intensify host antitumor immunity in-vivo. Simultaneous treatment with epacadostat, IFN-γ and LPS did not change the phenotype of matured human DCs and decreased KYN production. Peptide-specific T cell lines stimulated with DCs treated with epacadostat produced significantly more proinflammatory cytokines and show higher tumor cell lysis ability (Figure 3). These results indicate that epacadostat could increase the effectiveness of immunotherapeutics, including cancer vaccines, to increase inflammatory responses (Table 1) [124,170].
3.1.10. Sarcomas
Sarcomas revealed limited vulnerability to immunotherapy. In the preclinical studies, PD-L1 blockade led to the increase in the activity of KP enzymes, including both IDO1 and IDO2 [170,171]. IDO1 inhibition by navoximod resulted in a significant decrease in KYN level and KYN/TRP ratio in tumor cells and plasma. However, this compound did not show direct antitumoral activity. Regardless, it induced down-regulation of poliovirus receptor and granzyme expression, as well as up-regulation of inhibin and Dtx4 ubiquitin ligase, suggesting a potential effect of KP metabolites on NK cell function. It indicates that IDO1 inhibition stimulates NK cells against tumor cells (Figure 3). Besides, the co-administration of navoximod with paclitaxel displayed synergistic antitumor effects and did not induce additional side effects. It demonstrates that the combination of IDO inhibitor-based immunotherapy with chemotherapy has great potential as the new strategy for sarcomas treatment (Table 1) [136].
3.2. The Role of Intracellular Mechanisms of Kynurenine Pathway Modulation in Tumor Therapy
Suberoylanilide hydroxamic acid and bortezomib (BTZ) downregulate the IFN-γ-induced IDO expression via inhibition of the phosphorylation and nuclear translocation of STAT1, which causes a suppression of the IDO transcription [126,127]. Similarly, sodium butyrate (NaB) decreases IDO expression, through a mechanism involving increased STAT-1 acetylation. All these compounds reduce IDO-mediated immune tolerance [128]. This suggests that the Janus kinase/signal transducers and activators of transcription (JAK/STAT1) signaling pathway plays an important role in regulating IDO expression. Furthermore, BTZ demonstrated antitumor activity against nasopharyngeal carcinoma (NPC) cells, while in-vitro and in combination with the suberoylanilide hydroxamic potently induces apoptosis and suppresses the growth of NPC in mouse model [127,128]. Therefore, BTZ in combination with NPC-targeted tumor vaccines seems to be useful in the prevention and treatment of this type of cancer (Table 1). High activity of the JAK/STAT1 pathway is also observed, among others, in gallbladder cancer cells. Therefore, its pharmacological modulation may also be a promising method to break tumor immune tolerance and improve immunotherapies of other cancers [123,126,128].
Fludarabine, another inhibitor of STAT1 phosphorylation, also revealed effectiveness in the suppression of IDO activity in breast cancer and melanoma cell lines. However, its mechanism different is from other inhibitors. It is associated with the modulation of STAT1 activation. It does not affect directly the level of IDO transcription but participates in IDO post-transcriptional control. Fludarabine enhances IDO degradation through a proteasome-dependent pathway. Currently used in the treatment of certain malignancies, it has also demonstrated the ability to promote the T cell-mediated antitumor response by inducing degradation of the IDO protein. As it impairs T cell-dependent IDO upregulation, its use in the pre-treatment before immunotherapies, involving the response of T cells, may potentially prevent upregulation of IDO-dependent immunosuppressive mechanisms (Table 1). Also, combining fludarabine with other IDO inhibitors or immune-checkpoint modulators cal significantly reduces immunosuppression accompanying immunotherapies [123].
The therapeutic effect of imatinib, a tyrosine kinase inhibitor, in gastrointestinal stromal tumors (GIST) is also associated with downregulation of IDO1 expression, which, in turn, is driven by the oncogenic tyrosine-protein kinase KIT signaling. Imatinib in experimental tumor models potently reversed IDO1-mediated immunosuppression. This finding suggests that targeting IDO1 may provide a benefit in the context of imatinib treatment of GIST. The associations between cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and IDO expression indicate, that the combination of the KIT blockade with an anti-CTLA-4 therapy seems to be an effective approach in the management GIST (Table 1) [103].
Luo et al. examined the impact of a polysaccharide derived from the Lepista sordid (LSP) on IDO activity in HepG2 cells. LSP treatment potently inhibits IDO-dependent KYN synthesis in IFN-γ-treated HepG2 cells [144]. Furthermore, resveratrol, a stilbenoid, a type of natural phenol, also inhibits the expression and activity of IFN-γ-induced IDO in bone marrow-derived dendritic cells (BMDCs). The mechanism of these compounds seems to be associated with the inhibition of the JAK/STAT1 pathway and protein kinase Cd. Furthermore, in the case of resveratrol, IDO suppression in IFN-γ-stimulated BMDCs plays a key role in the antitumor response, because it modulates the activity of cytotoxic T cells. Systemic administration of resveratrol also suppressed tumor growth in EG7 thymoma-bearing mice in the IDO-dependent manner [117]. It demonstrates that resveratrol modulates the immune response and the IDO-mediated immune tolerance. However, resveratrol not only exerts the antitumor effect by the modulation of immune cells’ activation but also may enhance IDO-mediated tolerance within the tumor environment and tumor cells. Also, Feiji Recipe, a Chinese herbal medicine compound, inhibits tumor growth and prolongs survival in patients with lung cancer. In the mouse model of this cancer, it effectively decreases T cell apoptosis and proliferation of T regs by reducing IDO expression (Table 1). However, its exact mechanism remains unknown [115].
Interferon-induced guanylate-binding protein 1 (GBP1)-mediated extracellular secretion of IDO1 stimulates the progression of lung cancer cells. The interaction between GBP1 and IDO1 proteins additionally enhances the extracellular secretion of this enzyme. Meng et al. demonstrated that inhibition of this interaction by stragaloside IV or astragaloside IV causes a decrease in the extracellular secretion of IDO1 and increases the antitumor effect of PD-1 inhibitors in lung cancer cells both in-vivo and in-vitro (Table 1) [116].
Carbidopa is commonly used to treat Parkinson’s disease (PD) as a peripheral levodopa decarboxylase inhibitor. Patients administering this drug have a significantly lower incidence of various tumors. Recently, it has been shown that it also inhibits the activity of TDO and KYNU within the tumor tissue and its growth. However, it was not effective in inhibiting the proliferation of breast cancer and melanoma cells. It appears to be associated with the increase in an indole-3-acetonitrile level during carbidopa treatment, which enhances the viability of these types of cancer cells. It suggests that treatment with carbidopa may alter TRP metabolism in breast cancer and melanoma in a way that promotes the formation of pro-proliferative TRP metabolites, which may contribute to their development [172].
3.3. Angiogenesis Inhibition
Tumor angiogenesis is a key process necessary for the growth, metastasis and transfer of essential nutrients for solid tumors. Its inhibition is recognized as an effective anticancer strategy for non-small-cell lung carcinoma [173]. IDO revealed non-immune function, in the regulation of this process [174]. Erianin, a natural product isolated from Dendrobium chrysotoxum Lindl, downregulates the IDO-induced tumor cells angiogenesis and endothelial cell-dependent angiogenesis by targeting the JAK2/STAT3 pathway and its downstream genes, such as matrix metalloproteinases-2 and -9 (Table 1). Also, administration of other IDO inhibitors may be effective in inhibiting the IDO-dependent angiogenesis in lung cancer, as well as in other malignancies, providing a potential solution in preventing the processes of tumor growth and metastasis [175,176].
Astragalus membranaceus, commonly used in traditional Chinese medicine, possesses anti-inflammatory and antitumor properties. Several studies indicate that its extract (PG2) significantly inhibits the growth of tumor cells. Connexin 43 (Cx43) is a component of gap junctions, which allow for gap junction intercellular communication between cells. It is omnipresent in cells and involved in the passage of chemotherapeutics into the tumor and its environment cells. The treatment of PG2 upregulates the expression of Cx43, enhancing the distribution of chemotherapeutics, but also downregulates IDO expression. The PG2 and cisplatin co-treatment in animal models of melanoma and lung cancer significantly slowed tumor growth and prolonged survival. This indicates the potential value of the combination therapy of PG2 with chemotherapeutics [177].
3.4. Pharmacological Modulation Activity of KMO Activity
Chauhan et al. discovered that dysfunctional plasmacytoid DCs contribute to the pathogenesis of multiple myeloma (MM). Interaction between plasmacytoid DCs and MM cells triggers upregulation of KMO expression in MM cells [178]. Pharmacological inhibition of this enzyme using Ro-61–8048 activates plasmacytoid DCs and enhances their ability to trigger autologous proliferation of T cells. In effect, the administration of Ro-61–8048 leads to an increase in T cells cytotoxic and NK cells cytolytic activity against tumor cells (Table 1). Furthermore, the combination of Ro-61–8048 and immune checkpoint PD-L1 blockade potently enhances antitumor immunity and cytotoxicity against MM cells, more potent than either agent alone. These preclinical results provide the basis for a novel immune-based therapeutic strategy, targeting KMO, alone or in combination with PD-L1 blockade, to restore anti-MM immune responses in patients with this malignancy [145].
3.5. Gene Silencing
Genetic manipulation is another promising option for targeting TRP metabolism in cancer. Silencing of the IDO1 gene, using RNA interference, significantly reduced melanoma growth in-vivo. Moreover, it also significantly increased the efficacy of DC-based vaccines. Importantly, this approach can target specific cell types and can also inhibit other KP enzymes, even in parallel. Furthermore, gene silencing also eliminates any non-enzymatic tolerance promoting the IDO1 protein activity [179,180]. Another highly effective strategy to block KP activity is to inhibit IDO1 and TDO2 expression using locked nucleic acid (LNA)-modified antisense oligonucleotide. They efficiently downregulate the expression of these enzymes in cancer cells in-vitro without using transfection reagents. Treatment of cancer cells in-vitro with IDO1-specific and/or TDO2 ASOs and small molecule inhibitors can reduce the production of KYN by cancer cells in a synergistic manner and increase proliferation of activated T cells in the co-culture [181]. Therefore, a combination of gene silencing using RNA interference or LNA-modified ASOs and small molecule inhibitors should be considered as a strategy for managing KYN-dependent immunosuppression in cancer immunotherapy.
3.6. Blockade of the AhR
As mentioned before, KYN activates the AhR, inducing target genes of this receptor, including the AhR–IL-6–STAT3 pathway. Due to increased production of KYN, the AhR and, in effect, all signaling pathways dependent on this receptor are overactivated in IDO- and TDO-overexpressing tumors. This, in turn, enhances mechanisms responsible for the development of resistance against immune checkpoint inhibitors and tumor immune escape [18,19,20,71,83,182,183]. AhR-mediated immunosuppression can be reversed by direct blockade of this receptor [18,19,20,71,83,182]. The administration of selective AhR antagonists, such as KYN-101, or the previously mentioned CH-223191 and DMF significantly slows the progression of IDO- and TDO-overexpressing tumors in-vivo [71,83,114,182,183]. Besides, these agents synergize with PD-1 blockade during co-treatment. Campesato et al. also suggested that direct blocking the AhR activation, using its antagonists, in IDO- and TDO- overexpressing tumors could overcome the limitation of single IDO or TDO targeting agents, such as enzyme inhibitors and has the potential as a novel approach to immunotherapy, especially combined with the immune checkpoint inhibitors [182].
4. Summary and Future Directions
IDO inhibitors, especially targeting IDO1 isoform, are an important class of pharmaceuticals because KYN and its metabolites directly participate in many physiological and pathological processes, including tumor immune escape, development of infections, autoimmune, or metabolic diseases and neurodegeneration [2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,37,38,39,40,41,42,43,44,45,46,47,72,77,81,100,184]. They are mostly considered as therapeutics useful in cancer therapy [72,77,82,100,101,125,184,185,186]. Although IDO1 targeting in monotherapy has been shown to have disappointing efficacy in preclinical cancer models, their combinations with conventional treatments, immunogenic chemotherapy, or immunosuppressive drugs have shown satisfactory results. Moreover, the mechanism and safety profile of IDO1 inhibitors are well suited to their use in combination therapies. This indicates that drugs targeting KP should be combined with other anticancer therapies, to improve their efficacy [74,77,81,101,164,185,186,187]. To date, only a few oral IDO inhibitors have entered clinical trials, including epacadostat, indoximod, navoximod, BMS-986242 and EOS200271. In contrast, newer compounds capable of inducing an effective reduction in intracellular KYN levels remain in preclinical studies [76,82,90,91,121,125,130,147,148,152,184,186,187,188,189].
It is worth mentioning a few controversies regarding the use of IDO inhibitors. Competitive inhibitors, such as 1-MT, widely used in preclinical studies, or norharmane, hardly induce therapeutic effect in-vivo due to their low potency. They reach effectiveness only at a high, comparable with TRP, plasma level [77,81,88,164,189,190,191]. Besides, they fulfill many of the structural and functional criteria as the potential AhR agonists with a binding affinity similar to KYN. Therefore, their low systemic immunomodulatory effect seems to be also to a certain degree related to their off-target effects, such as the activation of AhR [146,192]. This should be taken into account when evaluating their clinical outcomes and potential utility. In contrast, newer, more potent inhibitors such as epacadostat, navoximod, or BMS-986242 effectively inhibit IDO activity even on nanomolar concentrations, efficiently decreasing KYN production in-vivo. However, their direct antitumor effect in monotherapy remains low [88,154,157,164,193,194,195,196]. This all indicates that a better understanding of the basic biology of the KP enzymes is still needed to develop new, more effective and safer drugs and identify patients who are best suited to benefit from them. Despite these limitations and the failures observed in clinical trials of epacadostat, combination therapies with IDO1 inhibitors and other immuno- or chemotherapeutics for the treatment of cancer still appear to be clinically justified because IDO1 is commonly overactivated in tumors and their microenvironment, both constitutively and induced by the antitumor immune response. [43,44,45,46,47,181]. Additionally, recent preclinical evidence indicates that the IDO1 inhibitors synergize the effects of tumor immuno- and chemotherapies and help to solve the problem of tumor resistance against them [83,93,105,106,107,108,112,113,119,120,129,132,133,136,137,143,146,151,154,155,156,157,171]. In turn, the discoveries concerning the role of IDO1 in tumor angiogenesis suggest that inhibiting its activity may effectively suppress this process, providing a potentially effective solution in the prevention of tumor development and metastasis. However, the impact of IDO inhibitors on tumor vasculature requires better examination [174,175,176].
Beyond the direct inhibition of IDO1, the authors suggest other approaches, which may serve as potential therapeutic targets associated with impaired TRP catabolism. Inhibition of TDO and IDO2 may limit KYN synthesis when inhibition of IDO1 is not enough to achieve satisfactory clinical effects [78,89,100,136,143]. The direct pharmacological targeting of TDO is considered for cancer immunotherapy [73,74,75,96,100,140,143]. Also, the development of dual IDO and TDO inhibitors should extend the therapeutic possibilities and improve the effectiveness of cancer treatment. Targeting more than one enzyme at once may induce broad and synergistic responses of the immune system. It appears to be an effective solution in the case of inherent or acquired resistance to currently available IDO1 inhibitors [76,77,100]. Because tumor cells can upregulate multiple immune checkpoint pathways to evade the immune response, therefore, selective inhibition of IDO1 may not be sufficient to promote tumor regression in most patients. The idea of dual IDO1 and TDO inhibitors is also desirable, because inhibition of IDO may induce the activation of TDO [96,100,164]. Moreover, targeting IDO2 together with IDO1 may open new options for cancer immunotherapy [78,81,101,136]. However, they are upstream enzymes of KP and their inhibition may cause more adverse effects than selective more selective approaches [78,100,164].
Pharmacological inhibition of KMO also seems to have great potential as the therapeutic target for the treatment of certain neoplasms, such as MM [145]. Furthermore, some authors observed that the expression levels of KMO and KYNU are potently upregulated in the course of HER2-enriched and triple-negative breast cancer subtypes, as well as cutaneous squamous cell carcinoma. It leads to excessive production of 3-HAA and AA, other potent immunosuppressive metabolites of KP. Their accumulation intensifies the progression of these cancers. KMO and KYNU inhibitors may therefore be considered as potential options for the treatment of breast cancer and cutaneous squamous cell carcinoma [197,198]. However, the efficacy of these compounds in cancer treatment has not yet been evaluated.
In addition to direct inhibition of the enzyme, the authors outlined other targets that may be considered as potential therapeutic solutions for KP overactivation accompanying cancer progression. They include downregulation of IDO1, IDO2, TDO and KMO expression, upregulation of their proteasomal degradation rate, targeting upstream regulators of IDO1 expression and activation, such as JAK/STAT3 pathway and KIT tyrosine kinase or downstream effectors of the KP metabolites, such as general control nonderepressible-2 kinase and mammalian target of rapamycin kinase [40,103,115,116,123,126,127,128,144]. Furthermore, inhibition of extracellular IDO secretion or the direct targeting of particular KP metabolites using monoclonal antibodies as well as blocking the KP receptors such as AhR and G protein-coupled receptor 35, or TRP cellular transport mechanisms also should be taken into consideration for further therapeutic strategies [40,116]. These mechanisms seem to be the most promising, because they may reduce the immunosuppressive tumor microenvironment conditions, regardless of the source and intensity of KYN synthesis. Especially AhR antagonists, such as DMF, CH-223191 and KYN-101 showed promising results in antitumor therapy in animal models [71,83,114,182].
5. Conclusions
Numerous studies indicate that activation of the KP is observed during the course of various malignancies. In turn, the accumulation of metabolites belonging to this pathway may enhance neoplastic progression, because they stimulate tumor growth and inhibit the immune system’s response against cancer cells. They also appear to be at least partially responsible for the development of the resistance to antitumor therapy. It indicates that the modulation of the KP activity can be an effective solution as an adjunctive treatment in the management of numerous neoplasms. Among the enzymes belonging to this pathway, IDO has attracted the greatest interest among researchers Inhibition of its activity has so far provided the most promising results, both in-vitro and in-vivo. Also, TDO and KMO appear to have potential as possible targets for antitumor therapy. Despite the unpromising results of previous clinical trials with IDO inhibitors, numerous preclinical studies suggest that the inhibition of the KP enzymes may still prove to be an effective solution in cases remaining a challenge for modern oncology, especially the tumor immune escape and resistance to chemotherapy and immunotherapy. This demonstrates the rationale for further research in this area, especially for their clinical applications.
Author Contributions
Conceptualization, A.M. and A.T.-K.; writing—original draft preparation, A.M.; writing—review and editing, A.T.-K. and D.P.; figures preparation, A.T.-K.; supervision, D.P.; funding acquisition, D.P. All authors have read and agreed to the published version of the manuscript.
Funding
The APC was funded by Medical University of Bialystok.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data sharing not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Perez-Gracia, E.; Blanco, R.; Carmona, M.; Carro, E.; Ferrer, I. Oxidative Stress Damage and Oxidative Stress Responses in the Choroid Plexus in Alzheimer’s Disease. Acta Neuropathol. 2009, 118, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Ocampo, J.R.; Huitrón, R.L.; González-Esquivel, D.; Ugalde-Muñiz, P.; Jiménez-Anguiano, A.; Pineda, B.; Pedraza-Chaverri, J.; Ríos, C.; de la Cruz, V.P. Kynurenines with Neuroactive and Redox Properties: Relevance to Aging and Brain Diseases. Oxid. Med. Cell. Longev. 2014, 2014, 646909. [Google Scholar] [CrossRef]
- Bender, D.A. Biochemistry of Tryptophan in Health and Disease. Mol. Asp. Med. 1983, 6, 101–197. [Google Scholar] [CrossRef]
- Badawy, A.A. Tryptophan Metabolism in Alcoholism. Adv. Exp. Med. Biol. 1999, 467, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Badawy, A.A.-B. Tryptophan Metabolism, Disposition and Utilization in Pregnancy. Biosci. Rep. 2015, 35, e00261. [Google Scholar] [CrossRef] [PubMed]
- Badawy, A.A.-B. Pellagra and Alcoholism: A Biochemical Perspective. Alcohol Alcohol. 2014, 49, 238–250. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Liu, D.; Song, P.; Zou, M.-H. Deregulated Tryptophan-Kynurenine Pathway Is Linked to Inflammation, Oxidative Stress, and Immune Activation Pathway in Cardiovascular Diseases. Front. Biosci. Landmark Ed. 2015, 20, 1116–1143. [Google Scholar] [PubMed] [Green Version]
- Heyes, M.P.; Saito, K.; Crowley, J.S.; Davis, L.E.; Demitrack, M.A.; Der, M.; Dilling, L.A.; Elia, J.; Kruesi, M.J.; Lackner, A. Quinolinic Acid and Kynurenine Pathway Metabolism in Inflammatory and Non-Inflammatory Neurological Disease. Brain J. Neurol. 1992, 115 (Pt 5), 1249–1273. [Google Scholar] [CrossRef]
- Fallarino, F.; Grohmann, U.; Vacca, C.; Bianchi, R.; Orabona, C.; Spreca, A.; Fioretti, M.C.; Puccetti, P. T Cell Apoptosis by Tryptophan Catabolism. Cell Death Differ. 2002, 9, 1069–1077. [Google Scholar] [CrossRef]
- Topczewska-Bruns, J.; Pawlak, D.; Chabielska, E.; Tankiewicz, A.; Buczko, W. Increased Levels of 3-Hydroxykynurenine in Different Brain Regions of Rats with Chronic Renal Insufficiency. Brain Res. Bull. 2002, 58, 423–428. [Google Scholar] [CrossRef]
- Topczewska-Bruns, J.; Tankiewicz, A.; Pawlak, D.; Buczko, W. Behavioral Changes in the Course of Chronic Renal Insufficiency in Rats. Pol. J. Pharmacol. 2001, 53, 263–269. [Google Scholar]
- Forrest, C.M.; Mackay, G.M.; Oxford, L.; Stoy, N.; Stone, T.W.; Darlington, L.G. Kynurenine Pathway Metabolism in Patients with Osteoporosis after 2 Years of Drug Treatment. Clin. Exp. Pharmacol. Physiol. 2006, 33, 1078–1087. [Google Scholar] [CrossRef]
- Dayer, M.R.; Safari, I.; Dayer, M.S. New Evidence on Hypoglycemic Effect of Quinolinic Acid in Diabetic Rats. Pak. J. Biol. Sci. PJBS 2009, 12, 1025–1030. [Google Scholar] [CrossRef] [Green Version]
- Munipally, P.K.; Agraharm, S.G.; Valavala, V.K.; Gundae, S.; Turlapati, N.R. Evaluation of Indoleamine 2,3-Dioxygenase Expression and Kynurenine Pathway Metabolites Levels in Serum Samples of Diabetic Retinopathy Patients. Arch. Physiol. Biochem. 2011, 117, 254–258. [Google Scholar] [CrossRef]
- Prendergast, G.C. Cancer: Why Tumours Eat Tryptophan. Nature 2011, 478, 192–194. [Google Scholar] [CrossRef]
- Apalset, E.M.; Gjesdal, C.G.; Ueland, P.M.; Midttun, Ø.; Ulvik, A.; Eide, G.E.; Meyer, K.; Tell, G.S. Interferon (IFN)-γ-Mediated Inflammation and the Kynurenine Pathway in Relation to Bone Mineral Density: The Hordaland Health Study. Clin. Exp. Immunol. 2014, 176, 452–460. [Google Scholar] [CrossRef] [Green Version]
- Esquivel, D.G.; Ramírez-Ortega, D.; Pineda, B.; Castro, N.; Ríos, C.; de la Cruz, V.P. Kynurenine Pathway Metabolites and Enzymes Involved in Redox Reactions. Neuropharmacology 2017, 112, 331–345. [Google Scholar] [CrossRef]
- Kawajiri, K.; Fujii-Kuriyama, Y. The Aryl Hydrocarbon Receptor: A Multifunctional Chemical Sensor for Host Defense and Homeostatic Maintenance. Exp. Anim. 2017, 66, 75–89. [Google Scholar] [CrossRef] [Green Version]
- Gutiérrez-Vázquez, C.; Quintana, F.J. Regulation of the Immune Response by the Aryl Hydrocarbon Receptor. Immunity 2018, 48, 19–33. [Google Scholar] [CrossRef] [Green Version]
- Bessede, A.; Gargaro, M.; Pallotta, M.T.; Matino, D.; Servillo, G.; Brunacci, C.; Bicciato, S.; Mazza, E.M.C.; Macchiarulo, A.; Vacca, C.; et al. Aryl Hydrocarbon Receptor Control of a Disease Tolerance Defence Pathway. Nature 2014, 511, 184–190. [Google Scholar] [CrossRef] [Green Version]
- Kolachalama, V.B.; Shashar, M.; Alousi, F.; Shivanna, S.; Rijal, K.; Belghasem, M.E.; Walker, J.; Matsuura, S.; Chang, G.H.; Gibson, C.M.; et al. Uremic Solute-Aryl Hydrocarbon Receptor-Tissue Factor Axis Associates with Thrombosis after Vascular Injury in Humans. J. Am. Soc. Nephrol. JASN 2018, 29, 1063–1072. [Google Scholar] [CrossRef]
- Nguyen, N.T.; Nakahama, T.; Le, D.H.; Van Son, L.; Chu, H.H.; Kishimoto, T. Aryl Hydrocarbon Receptor and Kynurenine: Recent Advances in Autoimmune Disease Research. Front. Immunol. 2014, 5, 551. [Google Scholar] [CrossRef] [Green Version]
- King, N.J.C.; Thomas, S.R. Molecules in Focus: Indoleamine 2,3-Dioxygenase. Int. J. Biochem. Cell Biol. 2007, 39, 2167–2172. [Google Scholar] [CrossRef]
- Capece, L.; Lewis-Ballester, A.; Marti, M.A.; Estrin, D.A.; Yeh, S.-R. Molecular Basis for the Substrate Stereoselectivity in Tryptophan Dioxygenase. Biochemistry 2011, 50, 10910–10918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knox, W.E.; Greengard, O. The Regulation of Some Enzymes of Nitrogen Metabolism—an Introduction to Enzyme Physiology. Adv. Enzyme Regul. 1965, 3, 247–313. [Google Scholar] [CrossRef]
- Feigelson, M.; Feigelson, P. Metabolic Effects of Glucocorticoids as Related to Enzyme Induction. Adv. Enzyme Regul. 1965, 3, 11–27. [Google Scholar] [CrossRef]
- Schimke, R.T.; Sweeney, E.W.; Berlin, C.M. The Roles of Synthesis And Degradation in the Control of Rat Liver Tryptophan Pyrrolase. J. Biol. Chem. 1965, 240, 322–331. [Google Scholar] [CrossRef]
- Badawy, A.A.-B. Kynurenine Pathway of Tryptophan Metabolism: Regulatory and Functional Aspects. Int. J. Tryptophan Res. IJTR 2017, 10. [Google Scholar] [CrossRef] [Green Version]
- Badawy, A.A.-B.; Bano, S. Tryptophan Metabolism in Rat Liver After Administration of Tryptophan, Kynurenine Metabolites, and Kynureninase Inhibitors. Int. J. Tryptophan Res. IJTR 2016, 9, 51–65. [Google Scholar] [CrossRef] [Green Version]
- Brouns, R.; Verkerk, R.; Aerts, T.; De Surgeloose, D.; Wauters, A.; Scharpé, S.; De Deyn, P.P. The Role of Tryptophan Catabolism along the Kynurenine Pathway in Acute Ischemic Stroke. Neurochem. Res. 2010, 35, 1315–1322. [Google Scholar] [CrossRef]
- Amori, L.; Guidetti, P.; Pellicciari, R.; Kajii, Y.; Schwarcz, R. On the Relationship between the Two Branches of the Kynurenine Pathway in the Rat Brain in Vivo. J. Neurochem. 2009, 109, 316–325. [Google Scholar] [CrossRef] [Green Version]
- Barone, P. The “Yin” and the “Yang” of the Kynurenine Pathway: Excitotoxicity and Neuroprotection Imbalance in Stress-Induced Disorders. Behav. Pharmacol. 2019, 30, 163–186. [Google Scholar] [CrossRef]
- Walsh, H.A.; Botting, N.P. Purification and Biochemical Characterization of Some of the Properties of Recombinant Human Kynureninase. Eur. J. Biochem. 2002, 269, 2069–2074. [Google Scholar] [CrossRef]
- Okuda, S.; Nishiyama, N.; Saito, H.; Katsuki, H. 3-Hydroxykynurenine, an Endogenous Oxidative Stress Generator, Causes Neuronal Cell Death with Apoptotic Features and Region Selectivity. J. Neurochem. 1998, 70, 299–307. [Google Scholar] [CrossRef]
- Stone, T.W. Neuropharmacology of Quinolinic and Kynurenic Acids. Pharmacol. Rev. 1993, 45, 309–379. [Google Scholar]
- Schwarcz, R.; Pellicciari, R. Manipulation of Brain Kynurenines: Glial Targets, Neuronal Effects, and Clinical Opportunities. J. Pharmacol. Exp. Ther. 2002, 303, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Muller, A.J.; DuHadaway, J.B.; Donover, P.S.; Sutanto-Ward, E.; Prendergast, G.C. Inhibition of Indoleamine 2,3-Dioxygenase, an Immunoregulatory Target of the Cancer Suppression Gene Bin1, Potentiates Cancer Chemotherapy. Nat. Med. 2005, 11, 312–319. [Google Scholar] [CrossRef]
- Prendergast, G.C.; Malachowski, W.P.; DuHadaway, J.B.; Muller, A.J. Discovery of IDO1 Inhibitors: From Bench to Bedside. Cancer Res. 2017, 77, 6795–6811. [Google Scholar] [CrossRef] [Green Version]
- Schramme, F.; Crosignani, S.; Frederix, K.; Hoffmann, D.; Pilotte, L.; Stroobant, V.; Preillon, J.; Driessens, G.; Van den Eynde, B.J. Inhibition of Tryptophan-Dioxygenase Activity Increases the Antitumor Efficacy of Immune Checkpoint Inhibitors. Cancer Immunol. Res. 2020, 8, 32–45. [Google Scholar] [CrossRef]
- Platten, M.; von Knebel Doeberitz, N.; Oezen, I.; Wick, W.; Ochs, K. Cancer Immunotherapy by Targeting IDO1/TDO and Their Downstream Effectors. Front. Immunol. 2014, 5, 673. [Google Scholar] [CrossRef]
- Lee, G.K.; Park, H.J.; Macleod, M.; Chandler, P.; Munn, D.H.; Mellor, A.L. Tryptophan Deprivation Sensitizes Activated T Cells to Apoptosis Prior to Cell Division. Immunology 2002, 107, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Apetoh, L.; Quintana, F.J.; Pot, C.; Joller, N.; Xiao, S.; Kumar, D.; Burns, E.J.; Sherr, D.H.; Weiner, H.L.; Kuchroo, V.K. The Aryl Hydrocarbon Receptor Interacts with C-Maf to Promote the Differentiation of Type 1 Regulatory T Cells Induced by IL-27. Nat. Immunol. 2010, 11, 854–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmgaard, R.B.; Zamarin, D.; Munn, D.H.; Wolchok, J.D.; Allison, J.P. Indoleamine 2,3-Dioxygenase Is a Critical Resistance Mechanism in Antitumor T Cell Immunotherapy Targeting CTLA-4. J. Exp. Med. 2013, 210, 1389–1402. [Google Scholar] [CrossRef] [PubMed]
- Munn, D.H.; Mellor, A.L. IDO in the Tumor Microenvironment: Inflammation, Counter-Regulation, and Tolerance. Trends Immunol. 2016, 37, 193–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Creelan, B.C.; Antonia, S.; Bepler, G.; Garrett, T.J.; Simon, G.R.; Soliman, H.H. Indoleamine 2,3-Dioxygenase Activity and Clinical Outcome Following Induction Chemotherapy and Concurrent Chemoradiation in Stage III Non-Small Cell Lung Cancer. Oncoimmunology 2013, 2, e23428. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, A.; Nikaido, T.; Ochiai, K.; Takakura, S.; Saito, M.; Aoki, Y.; Ishii, N.; Yanaihara, N.; Yamada, K.; Takikawa, O.; et al. Indoleamine 2,3-Dioxygenase Serves as a Marker of Poor Prognosis in Gene Expression Profiles of Serous Ovarian Cancer Cells. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2005, 11, 6030–6039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sim, S.H.; Ahn, Y.-O.; Yoon, J.; Kim, T.M.; Lee, S.-H.; Kim, D.-W.; Heo, D.S. Influence of Chemotherapy on Nitric Oxide Synthase, Indole-Amine-2,3-Dioxygenase and CD124 Expression in Granulocytes and Monocytes of Non-Small Cell Lung Cancer. Cancer Sci. 2012, 103, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, R.; Urade, Y.; Tokuda, M.; Hayaishi, O. Induction of Indoleamine 2,3-Dioxygenase in Mouse Lung during Virus Infection. Proc. Natl. Acad. Sci. USA 1979, 76, 4084–4086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.; Li, L.; Klonowski, K.D.; Tompkins, S.M.; Tripp, R.A.; Mellor, A.L. Induction and Role of Indoleamine 2,3 Dioxygenase in Mouse Models of Influenza a Virus Infection. PLoS ONE 2013, 8, e66546. [Google Scholar] [CrossRef] [Green Version]
- Robinson, C.M.; Hale, P.T.; Carlin, J.M. The Role of IFN-Gamma and TNF-Alpha-Responsive Regulatory Elements in the Synergistic Induction of Indoleamine Dioxygenase. J. Interferon Cytokine Res. 2005, 25, 20–30. [Google Scholar] [CrossRef]
- Uyttenhove, C.; Pilotte, L.; Théate, I.; Stroobant, V.; Colau, D.; Parmentier, N.; Boon, T.; Van den Eynde, B.J. Evidence for a Tumoral Immune Resistance Mechanism Based on Tryptophan Degradation by Indoleamine 2,3-Dioxygenase. Nat. Med. 2003, 9, 1269–1274. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zhang, R.; Li, S.; Liu, J. IDO1: An Important Immunotherapy Target in Cancer Treatment. Int. Immunopharmacol. 2017, 47, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Le Naour, S.; Knol, A.-C.; Pandolfino, M.-C.; Khammari, A.; Dréno, B. Effect of Indoleamine 2,3 Dioxygenase Inhibitor on the Cytotoxic Activity of Tumour-Infiltrating Lymphocytes. Acta Derm. Venereol. 2019, 99, 1186–1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munn, D.H.; Sharma, M.D.; Baban, B.; Harding, H.P.; Zhang, Y.; Ron, D.; Mellor, A.L. GCN2 Kinase in T Cells Mediates Proliferative Arrest and Anergy Induction in Response to Indoleamine 2,3-Dioxygenase. Immunity 2005, 22, 633–642. [Google Scholar] [CrossRef] [Green Version]
- McGaha, T.L. IDO-GCN2 and Autophagy in Inflammation. Oncotarget 2015, 6, 21771–21772. [Google Scholar] [CrossRef]
- Metz, R.; Rust, S.; Duhadaway, J.B.; Mautino, M.R.; Munn, D.H.; Vahanian, N.N.; Link, C.J.; Prendergast, G.C. IDO Inhibits a Tryptophan Sufficiency Signal That Stimulates MTOR: A Novel IDO Effector Pathway Targeted by D-1-Methyl-Tryptophan. Oncoimmunology 2012, 1, 1460–1468. [Google Scholar] [CrossRef] [Green Version]
- Zaher, S.S.; Germain, C.; Fu, H.; Larkin, D.F.P.; George, A.J.T. 3-Hydroxykynurenine Suppresses CD4+ T-Cell Proliferation, Induces T-Regulatory-Cell Development, and Prolongs Corneal Allograft Survival. Investig. Ophthalmol. Vis. Sci. 2011, 52, 2640–2648. [Google Scholar] [CrossRef] [Green Version]
- Turner, M.S.; Kane, L.P.; Morel, P.A. Dominant Role of Antigen Dose in CD4+Foxp3+ Regulatory T Cell Induction and Expansion. J. Immunol. 2009, 183, 4895–4903. [Google Scholar] [CrossRef] [Green Version]
- Kohlhaas, S.; Garden, O.A.; Scudamore, C.; Turner, M.; Okkenhaug, K.; Vigorito, E. Cutting Edge: The Foxp3 Target MiR-155 Contributes to the Development of Regulatory T Cells. J. Immunol. 2009, 182, 2578–2582. [Google Scholar] [CrossRef]
- Chen, W.; Liang, X.; Peterson, A.J.; Munn, D.H.; Blazar, B.R. The Indoleamine 2,3-Dioxygenase Pathway Is Essential for Human Plasmacytoid Dendritic Cell-Induced Adaptive T Regulatory Cell Generation. J. Immunol. 2008, 181, 5396–5404. [Google Scholar] [CrossRef]
- Pallotta, M.T.; Orabona, C.; Volpi, C.; Vacca, C.; Belladonna, M.L.; Bianchi, R.; Servillo, G.; Brunacci, C.; Calvitti, M.; Bicciato, S.; et al. Indoleamine 2,3-Dioxygenase Is a Signaling Protein in Long-Term Tolerance by Dendritic Cells. Nat. Immunol. 2011, 12, 870–878. [Google Scholar] [CrossRef] [Green Version]
- Cribbs, A.P.; Kennedy, A.; Penn, H.; Read, J.E.; Amjadi, P.; Green, P.; Syed, K.; Manka, S.W.; Brennan, F.M.; Gregory, B.; et al. Treg Cell Function in Rheumatoid Arthritis Is Compromised by Ctla-4 Promoter Methylation Resulting in a Failure to Activate the Indoleamine 2,3-Dioxygenase Pathway. Arthritis Rheumatol. 2014, 66, 2344–2354. [Google Scholar] [CrossRef]
- Bishnupuri, K.S.; Alvarado, D.M.; Khouri, A.N.; Shabsovich, M.; Chen, B.; Dieckgraefe, B.K.; Ciorba, M.A. IDO1 and Kynurenine Pathway Metabolites Activate PI3K-Akt Signaling in the Neoplastic Colon Epithelium to Promote Cancer Cell Proliferation and Inhibit Apoptosis. Cancer Res. 2019, 79, 1138–1150. [Google Scholar] [CrossRef] [Green Version]
- Thaker, A.I.; Rao, M.S.; Bishnupuri, K.S.; Kerr, T.A.; Foster, L.; Marinshaw, J.M.; Newberry, R.D.; Stenson, W.F.; Ciorba, M.A. IDO1 Metabolites Activate β-Catenin Signaling to Promote Cancer Cell Proliferation and Colon Tumorigenesis in Mice. Gastroenterology 2013, 145, 416–425.e4. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Gong, J.; Liu, Y. Indoleamine 2, 3-Dioxygenase Regulation of Immune Response (Review). Mol. Med. Rep. 2018, 17, 4867–4873. [Google Scholar] [CrossRef] [Green Version]
- Brandacher, G.; Perathoner, A.; Ladurner, R.; Schneeberger, S.; Obrist, P.; Winkler, C.; Werner, E.R.; Werner-Felmayer, G.; Weiss, H.G.; Göbel, G.; et al. Prognostic Value of Indoleamine 2,3-Dioxygenase Expression in Colorectal Cancer: Effect on Tumor-Infiltrating T Cells. Clin. Cancer Res. 2006, 12, 1144–1151. [Google Scholar] [CrossRef] [Green Version]
- Ninomiya, S.; Hara, T.; Tsurumi, H.; Hoshi, M.; Kanemura, N.; Goto, N.; Kasahara, S.; Shimizu, M.; Ito, H.; Saito, K.; et al. Indoleamine 2,3-Dioxygenase in Tumor Tissue Indicates Prognosis in Patients with Diffuse Large B-Cell Lymphoma Treated with R-CHOP. Ann. Hematol. 2011, 90, 409–416. [Google Scholar] [CrossRef]
- Sansone, P.; Bromberg, J. Targeting the Interleukin-6/Jak/Stat Pathway in Human Malignancies. J. Clin. Oncol. 2012, 30, 1005–1014. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Pardoll, D.; Jove, R. STATs in Cancer Inflammation and Immunity: A Leading Role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef]
- Litzenburger, U.M.; Opitz, C.A.; Sahm, F.; Rauschenbach, K.J.; Trump, S.; Winter, M.; Ott, M.; Ochs, K.; Lutz, C.; Liu, X.; et al. Constitutive IDO Expression in Human Cancer Is Sustained by an Autocrine Signaling Loop Involving IL-6, STAT3 and the AHR. Oncotarget 2014, 5, 1038–1051. [Google Scholar] [CrossRef] [Green Version]
- D’Amato, N.C.; Rogers, T.J.; Gordon, M.A.; Greene, L.I.; Cochrane, D.R.; Spoelstra, N.S.; Nemkov, T.G.; D’Alessandro, A.; Hansen, K.C.; Richer, J.K. A TDO2-AhR Signaling Axis Facilitates Anoikis Resistance and Metastasis in Triple-Negative Breast Cancer. Cancer Res. 2015, 75, 4651–4664. [Google Scholar] [CrossRef] [Green Version]
- Labadie, B.W.; Bao, R.; Luke, J.J. Reimagining IDO Pathway Inhibition in Cancer Immunotherapy via Downstream Focus on the Tryptophan-Kynurenine-Aryl Hydrocarbon Axis. Clin. Cancer Res. 2019, 25, 1462–1471. [Google Scholar] [CrossRef] [Green Version]
- Pantouris, G.; Mowat, C.G. Antitumour Agents as Inhibitors of Tryptophan 2,3-Dioxygenase. Biochem. Biophys. Res. Commun. 2014, 443, 28–31. [Google Scholar] [CrossRef]
- Santhanam, S.; Alvarado, D.M.; Ciorba, M.A. Therapeutic Targeting of Inflammation and Tryptophan Metabolism in Colon and Gastrointestinal Cancer. Transl. Res. 2016, 167, 67–79. [Google Scholar] [CrossRef] [Green Version]
- Pilotte, L.; Larrieu, P.; Stroobant, V.; Colau, D.; Dolusic, E.; Frédérick, R.; De Plaen, E.; Uyttenhove, C.; Wouters, J.; Masereel, B.; et al. Reversal of Tumoral Immune Resistance by Inhibition of Tryptophan 2,3-Dioxygenase. Proc. Natl. Acad. Sci. USA 2012, 109, 2497–2502. [Google Scholar] [CrossRef] [Green Version]
- Pei, Z.; Mendonca, R.; Gazzard, L.; Pastor, R.; Goon, L.; Gustafson, A.; VanderPorten, E.; Hatzivassiliou, G.; Dement, K.; Cass, R.; et al. Aminoisoxazoles as Potent Inhibitors of Tryptophan 2,3-Dioxygenase 2 (TDO2). ACS Med. Chem. Lett. 2018, 9, 417–421. [Google Scholar] [CrossRef]
- Prendergast, G.C.; Malachowski, W.J.; Mondal, A.; Scherle, P.; Muller, A.J. Indoleamine 2,3-Dioxygenase and Its Therapeutic Inhibition in Cancer. Int. Rev. Cell Mol. Biol. 2018, 336, 175–203. [Google Scholar] [CrossRef]
- Röhrig, U.F.; Majjigapu, S.R.; Caldelari, D.; Dilek, N.; Reichenbach, P.; Ascencao, K.; Irving, M.; Coukos, G.; Vogel, P.; Zoete, V.; et al. 1,2,3-Triazoles as Inhibitors of Indoleamine 2,3-Dioxygenase 2 (IDO2). Bioorg. Med. Chem. Lett. 2016, 26, 4330–4333. [Google Scholar] [CrossRef]
- Ball, H.J.; Yuasa, H.J.; Austin, C.J.D.; Weiser, S.; Hunt, N.H. Indoleamine 2,3-Dioxygenase-2; a New Enzyme in the Kynurenine Pathway. Int. J. Biochem. Cell Biol. 2009, 41, 467–471. [Google Scholar] [CrossRef]
- Pantouris, G.; Serys, M.; Yuasa, H.J.; Ball, H.J.; Mowat, C.G. Human Indoleamine 2,3-Dioxygenase-2 Has Substrate Specificity and Inhibition Characteristics Distinct from Those of Indoleamine 2,3-Dioxygenase-1. Amino Acids 2014, 46, 2155–2163. [Google Scholar] [CrossRef]
- Liu, M.; Wang, X.; Wang, L.; Ma, X.; Gong, Z.; Zhang, S.; Li, Y. Targeting the IDO1 Pathway in Cancer: From Bench to Bedside. J. Hematol. Oncol. 2018, 11, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyazaki, T.; Moritake, K.; Yamada, K.; Hara, N.; Osago, H.; Shibata, T.; Akiyama, Y.; Tsuchiya, M. Indoleamine 2,3-Dioxygenase as a New Target for Malignant Glioma Therapy. J. Neurosurg. 2009, 111, 230–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, J.; Chen, C.; Ju, R.; Wang, Q.; Li, J.; Guo, L.; Ye, C.; Zhang, D. Carboxyamidotriazole Combined with IDO1-Kyn-AhR Pathway Inhibitors Profoundly Enhances Cancer Immunotherapy. J. Immunother. Cancer 2019, 7, 246. [Google Scholar] [CrossRef] [PubMed]
- Munn, D.H.; Shafizadeh, E.; Attwood, J.T.; Bondarev, I.; Pashine, A.; Mellor, A.L. Inhibition of T Cell Proliferation by Macrophage Tryptophan Catabolism. J. Exp. Med. 1999, 189, 1363–1372. [Google Scholar] [CrossRef] [PubMed]
- Hwu, P.; Du, M.X.; Lapointe, R.; Do, M.; Taylor, M.W.; Young, H.A. Indoleamine 2,3-Dioxygenase Production by Human Dendritic Cells Results in the Inhibition of T Cell Proliferation. J. Immunol. 2000, 164, 3596–3599. [Google Scholar] [CrossRef]
- Munn, D.H.; Sharma, M.D.; Lee, J.R.; Jhaver, K.G.; Johnson, T.S.; Keskin, D.B.; Marshall, B.; Chandler, P.; Antonia, S.J.; Burgess, R.; et al. Potential Regulatory Function of Human Dendritic Cells Expressing Indoleamine 2,3-Dioxygenase. Science 2002, 297, 1867–1870. [Google Scholar] [CrossRef]
- Mellor, A.L.; Baban, B.; Chandler, P.; Marshall, B.; Jhaver, K.; Hansen, A.; Koni, P.A.; Iwashima, M.; Munn, D.H. Cutting Edge: Induced Indoleamine 2,3 Dioxygenase Expression in Dendritic Cell Subsets Suppresses T Cell Clonal Expansion. J. Immunol. 2003, 171. [Google Scholar] [CrossRef] [Green Version]
- Lob, S.; Konigsrainer, A.; Schafer, R.; Rammensee, H.-G.; Opelz, G.; Terness, P. Levo- but Not Dextro-1-Methyl Tryptophan Abrogates the IDO Activity of Human Dendritic Cells. Blood 2008, 111, 2152–2154. [Google Scholar] [CrossRef]
- Li, J.; Li, Y.; Yang, D.; Hu, N.; Guo, Z.; Kuang, C.; Yang, Q. Establishment of a human indoleamine 2, 3-dioxygenase 2 (hIDO2) bioassay system and discovery of tryptanthrin derivatives as potent hIDO2 inhibitors. Eur. J. Med. Chem. 2016, 123, 171–179. [Google Scholar] [CrossRef]
- Zakharia, Y.; Johnson, T.S.; Colman, H.; Vahanian, N.N.; Link, C.J.; Kennedy, E.; Sadek, R.F.; Kong, F.M.; Vender, J.; Munn, D.; et al. A Phase I/II Study of the Combination of Indoximod and Temozolomide for Adult Patients with Temozolomide-Refractory Primary Malignant Brain Tumors. J. Clin. Oncol. 2014, 32, TPS2107. [Google Scholar] [CrossRef]
- Colman, H.; Mott, F.; Spira, A.I.; Johnson, T.S.; Zakharia, Y.; Vahanian, N.N.; Link, C.J.; Kennedy, E.P.; Sadek, R.F.; Munn, D.; et al. A Phase 1b/2 Study of the Combination of the IDO Pathway Inhibitor Indoximod and Temozolomide for Adult Patients with Temozolomide-Refractory Primary Malignant Brain Tumors: Safety Analysis and Preliminary Efficacy of the Phase 1b Component. J. Clin. Oncol. 2015, 33, 2070. [Google Scholar] [CrossRef]
- Bakmiwewa, S.M.; Fatokun, A.A.; Tran, A.; Payne, R.J.; Hunt, N.H.; Ball, H.J. Identification of Selective Inhibitors of Indoleamine 2,3-Dioxygenase 2. Bioorg. Med. Chem. Lett. 2012, 22, 7641–7646. [Google Scholar] [CrossRef]
- Moreno, A.C.R.; Porchia, B.F.M.M.; Pagni, R.L.; Souza, P.d.C.; Pegoraro, R.; Rodrigues, K.B.; Barros, T.B.; Aps, L.R.d.M.M.; de Araújo, E.F.; Calich, V.L.G.; et al. The Combined Use of Melatonin and an Indoleamine 2,3-Dioxygenase-1 Inhibitor Enhances Vaccine-Induced Protective Cellular Immunity to HPV16-Associated Tumors. Front. Immunol. 2018, 9, 1914. [Google Scholar] [CrossRef]
- Fu, R.; Zhang, Y.-W.; Li, H.-M.; Lv, W.-C.; Zhao, L.; Guo, Q.-L.; Lu, T.; Weiss, S.J.; Li, Z.-Y.; Wu, Z.-Q. LW106, a Novel Indoleamine 2,3-Dioxygenase 1 Inhibitor, Suppresses Tumour Progression by Limiting Stroma-Immune Crosstalk and Cancer Stem Cell Enrichment in Tumour Micro-Environment. Br. J. Pharmacol. 2018, 175, 3034–3049. [Google Scholar] [CrossRef]
- Grobben, Y.; de Man, J.; van Doornmalen, A.M.; Muller, M.; Willemsen-Seegers, N.; Vu-Pham, D.; Mulder, W.R.; Prinsen, M.B.W.; de Wit, J.; Sterrenburg, J.G.; et al. Targeting Indoleamine 2,3-Dioxygenase in Cancer Models Using the Novel Small Molecule Inhibitor NTRC 3883-0. Front. Immunol. 2020, 11, 609490. [Google Scholar] [CrossRef]
- Yang, L.; Chen, Y.; He, J.; Njoya, E.M.; Chen, J.; Liu, S.; Xie, C.; Huang, W.; Wang, F.; Wang, Z.; et al. 4,6-Substituted-1H-Indazoles as Potent IDO1/TDO Dual Inhibitors. Bioorg. Med. Chem. 2019, 27, 1087–1098. [Google Scholar] [CrossRef]
- Bogdanova, L.A.; Morozkova, T.S.; Amitina, S.A.; Mazhukin, D.G.; Nikolin, V.P.; Popova, N.A.; Kaledin, V.I. Dual Effects of Indoleamine 2,3-Dioxygenase Inhibitors on the Therapeutic Effects of Cyclophosphamide and Cycloplatam on Ehrlich Ascites Tumor in Mice. Bull. Exp. Biol. Med. 2014, 157, 506–509. [Google Scholar] [CrossRef]
- Ogawa, K.; Hara, T.; Shimizu, M.; Nagano, J.; Ohno, T.; Hoshi, M.; Ito, H.; Tsurumi, H.; Saito, K.; Seishima, M.; et al. (-)-Epigallocatechin Gallate Inhibits the Expression of Indoleamine 2,3-Dioxygenase in Human Colorectal Cancer Cells. Oncol. Lett. 2012, 4, 546–550. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, K.; Hara, T.; Shimizu, M.; Ninomiya, S.; Nagano, J.; Sakai, H.; Hoshi, M.; Ito, H.; Tsurumi, H.; Saito, K.; et al. Suppression of Azoxymethane-Induced Colonic Preneoplastic Lesions in Rats by 1-Methyltryptophan, an Inhibitor of Indoleamine 2,3-Dioxygenase. Cancer Sci. 2012, 103, 951–958. [Google Scholar] [CrossRef]
- Ye, Z.; Yue, L.; Shi, J.; Shao, M.; Wu, T. Role of IDO and TDO in Cancers and Related Diseases and the Therapeutic Implications. J. Cancer 2019, 10, 2771–2782. [Google Scholar] [CrossRef] [Green Version]
- Cheong, J.E.; Sun, L. Targeting the IDO1/TDO2-KYN-AhR Pathway for Cancer Immunotherapy—Challenges and Opportunities. Trends Pharmacol. Sci. 2018, 39, 307–325. [Google Scholar] [CrossRef]
- Poncelet, L.; Ait-Belkacem, R.; Marillier, R.; Gomes, B.; Stauber, J. Target Exposure and Pharmacodynamics Study of the Indoleamine 2,3-Dioxygenase-1 (IDO-1) Inhibitor Epacadostat in the CT26 Mouse Tumor Model. J. Pharm. Biomed. Anal. 2019, 170, 220–227. [Google Scholar] [CrossRef]
- Balachandran, V.P.; Cavnar, M.J.; Zeng, S.; Bamboat, Z.M.; Ocuin, L.M.; Obaid, H.; Sorenson, E.C.; Popow, R.; Ariyan, C.; Rossi, F.; et al. Imatinib Potentiates Antitumor T Cell Responses in Gastrointestinal Stromal Tumor through the Inhibition of Ido. Nat. Med. 2011, 17, 1094–1100. [Google Scholar] [CrossRef]
- Li, M.; Bolduc, A.R.; Hoda, M.N.; Gamble, D.N.; Dolisca, S.-B.; Bolduc, A.K.; Hoang, K.; Ashley, C.; McCall, D.; Rojiani, A.M.; et al. The Indoleamine 2,3-Dioxygenase Pathway Controls Complement-Dependent Enhancement of Chemo-Radiation Therapy against Murine Glioblastoma. J. Immunother. Cancer 2014, 2, 21. [Google Scholar] [CrossRef] [Green Version]
- Söderlund, J.; Erhardt, S.; Kast, R.E. Acyclovir Inhibition of IDO to Decrease Tregs as a Glioblastoma Treatment Adjunct. J. Neuroinflamm. 2010, 7, 44. [Google Scholar] [CrossRef] [Green Version]
- Riess, C.; Schneider, B.; Kehnscherper, H.; Gesche, J.; Irmscher, N.; Shokraie, F.; Classen, C.F.; Wirthgen, E.; Domanska, G.; Zimpfer, A.; et al. Activation of the Kynurenine Pathway in Human Malignancies Can Be Suppressed by the Cyclin-Dependent Kinase Inhibitor Dinaciclib. Front. Immunol. 2020, 11, 55. [Google Scholar] [CrossRef] [Green Version]
- Adams, S.; Braidy, N.; Bessede, A.; Bessesde, A.; Brew, B.J.; Grant, R.; Teo, C.; Guillemin, G.J. The Kynurenine Pathway in Brain Tumor Pathogenesis. Cancer Res. 2012, 72, 5649–5657. [Google Scholar] [CrossRef] [Green Version]
- Tourino, M.C.; de Oliveira, E.M.; Bellé, L.P.; Knebel, F.H.; Albuquerque, R.C.; Dörr, F.A.; Okada, S.S.; Migliorini, S.; Soares, I.S.; Campa, A. Tryptamine and Dimethyltryptamine Inhibit Indoleamine 2,3 Dioxygenase and Increase the Tumor-Reactive Effect of Peripheral Blood Mononuclear Cells. Cell Biochem. Funct. 2013, 31, 361–364. [Google Scholar] [CrossRef]
- Du, L.; Xing, Z.; Tao, B.; Li, T.; Yang, D.; Li, W.; Zheng, Y.; Kuang, C.; Yang, Q. Both IDO1 and TDO Contribute to the Malignancy of Gliomas via the Kyn-AhR-AQP4 Signaling Pathway. Signal Transduct. Target. Ther. 2020, 5, 10. [Google Scholar] [CrossRef] [Green Version]
- Takada, K.; Kohashi, K.; Shimokawa, M.; Haro, A.; Osoegawa, A.; Tagawa, T.; Seto, T.; Oda, Y.; Maehara, Y. Co-Expression of IDO1 and PD-L1 in Lung Squamous Cell Carcinoma: Potential Targets of Novel Combination Therapy. Lung Cancer 2019, 128, 26–32. [Google Scholar] [CrossRef]
- Volaric, A.; Gentzler, R.; Hall, R.; Mehaffey, J.H.; Stelow, E.B.; Bullock, T.N.; Martin, L.W.; Mills, A.M. Indoleamine-2,3-Dioxygenase in Non-Small Cell Lung Cancer: A Targetable Mechanism of Immune Resistance Frequently Coexpressed with PD-L1. Am. J. Surg. Pathol. 2018, 42, 1216–1223. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Barsoumian, H.B.; Schoenhals, J.E.; Cushman, T.R.; Caetano, M.S.; Wang, X.; Valdecanas, D.R.; Niknam, S.; Younes, A.I.; Li, G.; et al. Indoleamine 2,3-Dioxygenase 1 Inhibition Targets Anti-PD1-Resistant Lung Tumors by Blocking Myeloid-Derived Suppressor Cells. Cancer Lett. 2018, 431, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Stone, E.; Marshall, N.; Donkor, M.; Triplett, K.; Blazek, J.; Triplett, T.; Ehrlich, L.; Georgiou, G. Abstract LB-226: Depletion of Kynurenine Using an Engineered Therapeutic Enzyme Potently Inhibits Cancer Immune Checkpoints Both as a Monotherapy and in Combination with Anti-PD-1. Cancer Res. 2015, 75, 226. [Google Scholar] [CrossRef]
- Nguyen, D.J.M.; Theodoropoulos, G.; Li, Y.-Y.; Wu, C.; Sha, W.; Feun, L.G.; Lampidis, T.J.; Savaraj, N.; Wangpaichitr, M. Targeting the Kynurenine Pathway for the Treatment of Cisplatin-Resistant Lung Cancer. Mol. Cancer Res. 2020, 18, 105–117. [Google Scholar] [CrossRef] [Green Version]
- Luo, B.; Que, Z.-J.; Zhou, Z.-Y.; Wang, Q.; Dong, C.-S.; Jiang, Y.; Hu, B.; Shi, H.; Jin, Y.; Liu, J.-W.; et al. Feiji Recipe Inhibits the Growth of Lung Cancer by Modulating T-Cell Immunity through Indoleamine-2,3-Dioxygenase Pathway in an Orthotopic Implantation Model. J. Integr. Med. 2018, 16, 283–289. [Google Scholar] [CrossRef]
- Meng, Y.; Wang, W.; Chen, M.; Chen, K.; Xia, X.; Zhou, S.; Yang, H. GBP1 Facilitates Indoleamine 2,3-Dioxygenase Extracellular Secretion to Promote the Malignant Progression of Lung Cancer. Front. Immunol. 2020, 11, 622467. [Google Scholar] [CrossRef]
- Noh, K.T.; Chae, S.H.; Chun, S.H.; Jung, I.D.; Kang, H.K.; Park, Y.-M. Resveratrol Suppresses Tumor Progression via the Regulation of Indoleamine 2,3-Dioxygenase. Biochem. Biophys. Res. Commun. 2013, 431, 348–353. [Google Scholar] [CrossRef]
- Meng, X.; Du, G.; Ye, L.; Sun, S.; Liu, Q.; Wang, H.; Wang, W.; Wu, Z.; Tian, J. Combinatorial Antitumor Effects of Indoleamine 2,3-Dioxygenase Inhibitor NLG919 and Paclitaxel in a Murine B16-F10 Melanoma Model. Int. J. Immunopathol. Pharmacol. 2017, 30, 215–226. [Google Scholar] [CrossRef]
- Sandri, S.; Watanabe, L.R.M.; Oliveira, E.A.d.; Faião-Flores, F.; Migliorini, S.; Tiago, M.; Felipe-Silva, A.; Vazquez, V.d.L.; da Costa Souza, P.; Consolaro, M.E.L.; et al. Indoleamine 2,3-Dioxygenase in Melanoma Progression and BRAF Inhibitor Resistance. Pharmacol. Res. 2020, 159, 104998. [Google Scholar] [CrossRef]
- Pour, S.R.; Morikawa, H.; Kiani, N.A.; Gomez-Cabrero, D.; Hayes, A.; Zheng, X.; Pernemalm, M.; Lehtiö, J.; Mole, D.J.; Hansson, J.; et al. Immunometabolic Network Interactions of the Kynurenine Pathway in Cutaneous Malignant Melanoma. Front. Oncol. 2020, 10, 51. [Google Scholar] [CrossRef]
- Chevolet, I.; Schreuer, M.; Speeckaert, R.; Neyns, B.; Hoorens, I.; van Geel, N.; Krüse, V.; Hennart, B.; Allorge, D.; Van Gele, M.; et al. Systemic Immune Changes Associated with Adjuvant Interferon-A2b-Therapy in Stage III Melanoma Patients: Failure at the Effector Phase? Melanoma Res. 2015, 25, 357–361. [Google Scholar] [CrossRef]
- Jia, H.; Ren, W.; Feng, Y.; Wei, T.; Guo, M.; Guo, J.; Zhao, J.; Song, X.; Wang, M.; Zhao, T.; et al. The Enhanced Antitumour Response of Pimozide Combined with the IDO Inhibitor L-MT in Melanoma. Int. J. Oncol. 2018, 53, 949–960. [Google Scholar] [CrossRef]
- Hanafi, L.-A.; Gauchat, D.; Godin-Ethier, J.; Possamaï, D.; Duvignaud, J.-B.; Leclerc, D.; Grandvaux, N.; Lapointe, R. Fludarabine Downregulates Indoleamine 2,3-Dioxygenase in Tumors via a Proteasome-Mediated Degradation Mechanism. PLoS ONE 2014, 9, e99211. [Google Scholar] [CrossRef] [Green Version]
- Komrokji, R.S.; Wei, S.; Mailloux, A.W.; Zhang, L.; Padron, E.; Sallman, D.; Lancet, J.E.; Tinsley, S.; Nardelli, L.A.; Pinilla-Ibarz, J.; et al. A Phase II Study to Determine the Safety and Efficacy of the Oral Inhibitor of Indoleamine 2,3-Dioxygenase (IDO) Enzyme INCB024360 in Patients with Myelodysplastic Syndromes. Clin. Lymphoma Myeloma Leuk. 2019, 19, 157–161. [Google Scholar] [CrossRef]
- Liu, X.; Shin, N.; Koblish, H.K.; Yang, G.; Wang, Q.; Wang, K.; Leffet, L.; Hansbury, M.J.; Thomas, B.; Rupar, M.; et al. Selective Inhibition of IDO1 Effectively Regulates Mediators of Antitumor Immunity. Blood 2010, 115, 3520–3530. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Jiang, G.; Gao, J.; Li, L.; Du, J.; Jiao, X. SAHA Down-Regulates the Expression of Indoleamine 2,3-Dioxygenase via Inhibition of the JAK/STAT1 Signaling Pathway in Gallbladder Carcinoma Cells. Oncol. Rep. 2013, 29, 269–275. [Google Scholar] [CrossRef] [Green Version]
- Jiang, G.-M.; Wang, H.-S.; Du, J.; Ma, W.-F.; Wang, H.; Qiu, Y.; Zhang, Q.-G.; Xu, W.; Liu, H.-F.; Liang, J.-P. Bortezomib Relieves Immune Tolerance in Nasopharyngeal Carcinoma via STAT1 Suppression and Indoleamine 2,3-Dioxygenase Downregulation. Cancer Immunol. Res. 2017, 5, 42–51. [Google Scholar] [CrossRef] [Green Version]
- Hui, K.F.; Lam, B.H.; Ho, D.N.; Tsao, S.W.; Chiang, A.K. Bortezomib and SAHA synergistically induce ROS-driven caspase-dependent apoptosis of nasopharyngeal carcinoma and block replication of Epstein-Barr virus. Mol. Cancer Ther. 2013, 12, 747–758. [Google Scholar] [CrossRef] [Green Version]
- Ma, H.; Qin, Q.; Mi, J.; Feng, Q. 1-MT Inhibits the Invasion of CBP-Resistant Ovarian Cancer Cells via down-Regulating IDO Expression and Re-Activating Immune Cells Function. BMC Pharmacol. Toxicol. 2020, 21, 67. [Google Scholar] [CrossRef]
- Cherney, E.C.; Zhang, L.; Nara, S.; Zhu, X.; Gullo-Brown, J.; Maley, D.; Lin, T.-A.; Hunt, J.T.; Huang, C.; Yang, Z.; et al. Discovery and Preclinical Evaluation of BMS-986242, a Potent, Selective Inhibitor of Indoleamine-2,3-Dioxygenase 1. ACS Med. Chem. Lett. 2021, 12, 288–294. [Google Scholar] [CrossRef]
- Gomes, B.; Driessens, G.; Bartlett, D.; Cai, D.; Cauwenberghs, S.; Crosignani, S.; Dalvie, D.; Denies, S.; Dillon, C.P.; Fantin, V.R.; et al. Characterization of the Selective Indoleamine 2,3-Dioxygenase-1 (IDO1) Catalytic Inhibitor EOS200271/PF-06840003 Supports IDO1 as a Critical Resistance Mechanism to PD-(L)1 Blockade Therapy. Mol. Cancer Ther. 2018, 17, 2530–2542. [Google Scholar] [CrossRef] [Green Version]
- Blair, A.B.; Kleponis, J.; Thomas, D.L.; Muth, S.T.; Murphy, A.G.; Kim, V.; Zheng, L. IDO1 Inhibition Potentiates Vaccine-Induced Immunity against Pancreatic Adenocarcinoma. J. Clin. Investig. 2019, 129, 1742–1755. [Google Scholar] [CrossRef]
- Crosignani, S.; Bingham, P.; Bottemanne, P.; Cannelle, H.; Cauwenberghs, S.; Cordonnier, M.; Dalvie, D.; Deroose, F.; Feng, J.L.; Gomes, B.; et al. Discovery of a Novel and Selective Indoleamine 2,3-Dioxygenase (IDO-1) Inhibitor 3-(5-Fluoro-1H-Indol-3-Yl)Pyrrolidine-2,5-Dione (EOS200271/PF-06840003) and Its Characterization as a Potential Clinical Candidate. J. Med. Chem. 2017, 60, 9617–9629. [Google Scholar] [CrossRef]
- Yamahira, A.; Narita, M.; Iwabuchi, M.; Uchiyama, T.; Iwaya, S.; Ohiwa, R.; Nishizawa, Y.; Suzuki, T.; Yokoyama, Y.; Hashimoto, S.; et al. Activation of the Leukemia Plasmacytoid Dendritic Cell Line PMDC05 by Toho-1, a Novel IDO Inhibitor. Anticancer Res. 2014, 34, 4021–4028. [Google Scholar]
- Trott, J.F.; Kim, J.; Abu Aboud, O.; Wettersten, H.; Stewart, B.; Berryhill, G.; Uzal, F.; Hovey, R.C.; Chen, C.-H.; Anderson, K.; et al. Inhibiting Tryptophan Metabolism Enhances Interferon Therapy in Kidney Cancer. Oncotarget 2016, 7, 66540–66557. [Google Scholar] [CrossRef] [Green Version]
- Nafia, I.; Toulmonde, M.; Bortolotto, D.; Chaibi, A.; Bodet, D.; Rey, C.; Velasco, V.; Larmonier, C.B.; Cerf, L.; Adam, J.; et al. IDO Targeting in Sarcoma: Biological and Clinical Implications. Front. Immunol. 2020, 11, 274. [Google Scholar] [CrossRef]
- Nakamura, N.; Hara, T.; Shimizu, M.; Mabuchi, R.; Nagano, J.; Ohno, T.; Kochi, T.; Kubota, M.; Shirakami, Y.; Goto, N.; et al. Effects of Indoleamine 2,3-Dioxygenase Inhibitor in Non-Hodgkin Lymphoma Model Mice. Int. J. Hematol. 2015, 102, 327–334. [Google Scholar] [CrossRef]
- Chen, J.-Y.; Li, C.-F.; Kuo, C.-C.; Tsai, K.K.; Hou, M.-F.; Hung, W.-C. Cancer/Stroma Interplay via Cyclooxygenase-2 and Indoleamine 2,3-Dioxygenase Promotes Breast Cancer Progression. Breast Cancer Res. 2014, 16, 410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basu, G.D.; Tinder, T.L.; Bradley, J.M.; Tu, T.; Hattrup, C.L.; Pockaj, B.A.; Mukherjee, P. Cyclooxygenase-2 Inhibitor Enhances the Efficacy of a Breast Cancer Vaccine: Role of IDO. J. Immunol. 2006, 177, 2391–2402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flick, H.E.; Lalonde, J.M.; Malachowski, W.P.; Muller, A.J. The Tumor-Selective Cytotoxic Agent β-Lapachone Is a Potent Inhibitor of IDO1. Int. J. Tryptophan Res. 2013, 6, 35–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Ren, J.; Ma, Y.; Liu, H.; Rong, Q.; Feng, Y.; Wang, Y.; Cheng, Y.; Ge, R.; Li, Z.; et al. Discovery of Cyanopyridine Scaffold as Novel Indoleamine-2,3-Dioxygenase 1 (IDO1) Inhibitors through Virtual Screening and Preliminary Hit Optimisation. J. Enzyme Inhib. Med. Chem. 2019, 34, 250–263. [Google Scholar] [CrossRef]
- Vasilyeva, E.D.; Kaledin, V.; Nikolin, V.P.; Popova, N.A.; Kirilyuk, I.A.; Grigor’ev, I.A. Accelerated Rejection of the Second Transplants of Immunogenic Tumor in Mice under Inhibition of Indoleamine 2,3-Dioxygenase Activity by Ethyl Pyruvate. Exp. Oncol. 2012, 34, 66–68. [Google Scholar]
- Liu, Q.; Hua, S.; Wang, X.; Chen, F.; Gou, S. The Introduction of Immunosuppressor (TDO Inhibitor) Significantly Improved the Efficacy of Irinotecan in Treating Hepatocellular Carcinoma. Cancer Immunol. Immunother. 2021, 70, 497–508. [Google Scholar] [CrossRef]
- Luo, Q.; Yan, L.; Xu, P.; Xiong, C.; Yang, Z.; Hu, P.; Hu, H.; Hong, R. Discovery of a Polysaccharide from the Fruiting Bodies of Lepista Sordida as Potent Inhibitors of Indoleamine 2, 3-Dioxygenase (IDO) in HepG2 Cells via Blocking of STAT1-Mediated JAK-PKC-δ Signaling Pathways. Carbohydr. Polym. 2018, 197, 540–547. [Google Scholar] [CrossRef]
- Ray, A.; Song, Y.; Du, T.; Tai, Y.-T.; Chauhan, D.; Anderson, K.C. Targeting Tryptophan Catabolic Kynurenine Pathway Enhances Antitumor Immunity and Cytotoxicity in Multiple Myeloma. Leukemia 2020, 34, 567–577. [Google Scholar] [CrossRef]
- Moyer, B.J.; Rojas, I.Y.; Murray, I.A.; Lee, S.; Hazlett, H.F.; Perdew, G.H.; Tomlinson, C.R. Indoleamine 2,3-Dioxygenase 1 (IDO1) Inhibitors Activate the Aryl Hydrocarbon Receptor. Toxicol. Appl. Pharmacol. 2017, 323, 74–80. [Google Scholar] [CrossRef] [Green Version]
- Jung, K.H.; LoRusso, P.; Burris, H.; Gordon, M.; Bang, Y.-J.; Hellmann, M.D.; Cervantes, A.; de Olza, M.O.; Marabelle, A.; Hodi, F.S.; et al. Phase I Study of the Indoleamine 2,3-Dioxygenase 1 (IDO1) Inhibitor Navoximod (GDC-0919) Administered with PD-L1 Inhibitor (Atezolizumab) in Advanced Solid Tumors. Clin. Cancer Res. 2019, 25, 3220–3228. [Google Scholar] [CrossRef] [Green Version]
- Ebata, T.; Shimizu, T.; Fujiwara, Y.; Tamura, K.; Kondo, S.; Iwasa, S.; Yonemori, K.; Shimomura, A.; Kitano, S.; Koyama, T.; et al. Phase I Study of the Indoleamine 2,3-Dioxygenase 1 Inhibitor Navoximod (GDC-0919) as Monotherapy and in Combination with the PD-L1 Inhibitor Atezolizumab in Japanese Patients with Advanced Solid Tumours. Investig. New Drugs 2020, 38, 468–477. [Google Scholar] [CrossRef] [Green Version]
- Iachininoto, M.G.; Nuzzolo, E.R.; Bonanno, G.; Mariotti, A.; Procoli, A.; Locatelli, F.; De Cristofaro, R.; Rutella, S. Cyclooxygenase-2 (COX-2) Inhibition Constrains Indoleamine 2,3-Dioxygenase 1 (IDO1) Activity in Acute Myeloid Leukaemia Cells. Molecules 2013, 18, 10132–10145. [Google Scholar] [CrossRef] [Green Version]
- Hanihara, M.; Kawataki, T.; Oh-Oka, K.; Mitsuka, K.; Nakao, A.; Kinouchi, H. Synergistic Antitumor Effect with Indoleamine 2,3-Dioxygenase Inhibition and Temozolomide in a Murine Glioma Model. J. Neurosurg. 2016, 124, 1594–1601. [Google Scholar] [CrossRef] [Green Version]
- Li, A.; Barsoumian, H.B.; Schoenhals, J.E.; Caetano, M.S.; Wang, X.; Menon, H.; Valdecanas, D.R.; Niknam, S.; Younes, A.I.; Cortez, M.A.; et al. IDO1 Inhibition Overcomes Radiation-Induced “Rebound Immune Suppression” by Reducing Numbers of IDO1-Expressing Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Int. J. Radiat. Oncol. Biol. Phys. 2019, 104, 903–912. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Desjardins, A.; Rixe, O.; Cloughesy, T.; Alekar, S.; Williams, J.H.; Li, R.; Taylor, C.T.; Lassman, A.B. A Phase 1 Study of PF-06840003, an Oral Indoleamine 2,3-Dioxygenase 1 (IDO1) Inhibitor in Patients with Recurrent Malignant Glioma. Investig. New Drugs 2020, 38, 1784–1795. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.; Liu, Y.; Wang, S.; Wang, T.; Zhang, G.; Chen, X.; Li, Y.; Cui, H.; Lai, F.; Sheng, L. Design and Synthesis of Indoleamine 2,3-Dioxygenase 1 Inhibitors and Evaluation of Their Use as Anti-Tumor Agents. Molecules 2019, 24, 2124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, G.V.; Dummer, R.; Hamid, O.; Gajewski, T.; Caglevic, C.; Dalle, S.; Arance, A.; Carlino, M.S.; Grob, J.-J.; Kim, T.M.; et al. Epacadostat (E) plus Pembrolizumab (P) versus Pembrolizumab Alone in Patients (Pts) with Unresectable or Metastatic Melanoma: Results of the Phase 3 ECHO-301/KEYNOTE-252 Study. J. Clin. Oncol. 2018, 36, 108. [Google Scholar] [CrossRef]
- Yentz, S.; Smith, D. Indoleamine 2,3-Dioxygenase (IDO) Inhibition as a Strategy to Augment Cancer Immunotherapy. BioDrugs 2018, 32, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-X.; Sun, S.-Y.; Dong, Q.-Q.; Wu, X.-X.; Tang, W.; Xing, Y.-Q. Recent Advances in the Discovery of Indoleamine 2,3-Dioxygenase 1 (IDO1) Inhibitors. MedChemComm 2019, 10, 1740–1754. [Google Scholar] [CrossRef]
- Long, G.V.; Dummer, R.; Hamid, O.; Gajewski, T.F.; Caglevic, C.; Dalle, S.; Arance, A.; Carlino, M.S.; Grob, J.-J.; Kim, T.M.; et al. Epacadostat plus Pembrolizumab versus Placebo plus Pembrolizumab in Patients with Unresectable or Metastatic Melanoma (ECHO-301/KEYNOTE-252): A Phase 3, Randomised, Double-Blind Study. Lancet Oncol. 2019, 20, 1083–1097. [Google Scholar] [CrossRef]
- Ferdinande, L.; Decaestecker, C.; Verset, L.; Mathieu, A.; Lopez, X.M.; Negulescu, A.-M.; Van Maerken, T.; Salmon, I.; Cuvelier, C.A.; Demetter, P. Clinicopathological Significance of Indoleamine 2,3-Dioxygenase 1 Expression in Colorectal Cancer. Br. J. Cancer 2012, 106, 141–147. [Google Scholar] [CrossRef] [Green Version]
- Park, J.-H.; Lee, J.-M.; Lee, E.-J.; Kim, D.-J.; Hwang, W.-B. Kynurenine Promotes the Goblet Cell Differentiation of HT-29 Colon Carcinoma Cells by Modulating Wnt, Notch and AhR Signals. Oncol. Rep. 2018, 39, 1930–1938. [Google Scholar] [CrossRef] [Green Version]
- Hacking, S.; Vitkovski, T.; Jain, S.; Jin, C.; Chavarria, H.; Wu, D.; Nasim, M. Clinical Significance of Program Death Ligand-1 and Indoleamine-2,3-Dioxygenase Expression in Colorectal Carcinoma. Appl. Immunohistochem. Mol. Morphol. AIMM 2021, 29, 201–208. [Google Scholar] [CrossRef]
- Gao, Y.-F.; Peng, R.-Q.; Li, J.; Ding, Y.; Zhang, X.; Wu, X.-J.; Pan, Z.-Z.; Wan, D.-S.; Zeng, Y.-X.; Zhang, X.-S. The Paradoxical Patterns of Expression of Indoleamine 2,3-Dioxygenase in Colon Cancer. J. Transl. Med. 2009, 7, 71. [Google Scholar] [CrossRef] [Green Version]
- Takamatsu, M.; Hirata, A.; Ohtaki, H.; Hoshi, M.; Ando, T.; Ito, H.; Hatano, Y.; Tomita, H.; Kuno, T.; Saito, K.; et al. Inhibition of Indoleamine 2,3-Dioxygenase 1 Expression Alters Immune Response in Colon Tumor Microenvironment in Mice. Cancer Sci. 2015, 106, 1008–1015. [Google Scholar] [CrossRef]
- Liu, X.; Zhou, W.; Zhang, X.; Ding, Y.; Du, Q.; Hu, R. 1-L-MT, an IDO Inhibitor, Prevented Colitis-Associated Cancer by Inducing CDC20 Inhibition-Mediated Mitotic Death of Colon Cancer Cells. Int. J. Cancer 2018, 143, 1516–1529. [Google Scholar] [CrossRef] [Green Version]
- Günther, J.; Däbritz, J.; Wirthgen, E. Limitations and Off-Target Effects of Tryptophan-Related IDO Inhibitors in Cancer Treatment. Front. Immunol. 2019, 10, 1801. [Google Scholar] [CrossRef]
- Zambetti, N.A.; Ping, Z.; Chen, S.; Kenswil, K.J.G.; Mylona, M.A.; Sanders, M.A.; Hoogenboezem, R.M.; Bindels, E.M.J.; Adisty, M.N.; Van Strien, P.M.H.; et al. Mesenchymal Inflammation Drives Genotoxic Stress in Hematopoietic Stem Cells and Predicts Disease Evolution in Human Pre-Leukemia. Cell Stem Cell 2016, 19, 613–627. [Google Scholar] [CrossRef] [Green Version]
- Mailloux, A.W.; Sugimori, C.; Komrokji, R.S.; Yang, L.; Maciejewski, J.P.; Sekeres, M.A.; Paquette, R.; Loughran, T.P.; List, A.F.; Epling-Burnette, P.K. Expansion of Effector Memory Regulatory T Cells Represents a Novel Prognostic Factor in Lower Risk Myelodysplastic Syndrome. J. Immunol. 2012, 189, 3198–3208. [Google Scholar] [CrossRef] [Green Version]
- Epling-Burnette, P.K.; List, A.F. Advancements in the Molecular Pathogenesis of Myelodysplastic Syndrome. Curr. Opin. Hematol. 2009, 16, 70–76. [Google Scholar] [CrossRef]
- Yang, L.; Qian, Y.; Eksioglu, E.; Epling-Burnette, P.K.; Wei, S. The Inflammatory Microenvironment in MDS. Cell. Mol. Life Sci. 2015, 72, 1959–1966. [Google Scholar] [CrossRef]
- Berthon, C.; Fontenay, M.; Corm, S.; Briche, I.; Allorge, D.; Hennart, B.; Lhermitte, M.; Quesnel, B. Metabolites of Tryptophan Catabolism Are Elevated in Sera of Patients with Myelodysplastic Syndromes and Inhibit Hematopoietic Progenitor Amplification. Leuk. Res. 2013, 37, 573–579. [Google Scholar] [CrossRef]
- Toulmonde, M.; Italiano, A. PD-1 Inhibition in Sarcoma Still Needs Investigation. Lancet Oncol. 2018, 19, e6. [Google Scholar] [CrossRef] [Green Version]
- Toulmonde, M.; Penel, N.; Adam, J.; Chevreau, C.; Blay, J.-Y.; Le Cesne, A.; Bompas, E.; Piperno-Neumann, S.; Cousin, S.; Grellety, T.; et al. Use of PD-1 Targeting, Macrophage Infiltration, and IDO Pathway Activation in Sarcomas: A Phase 2 Clinical Trial. JAMA Oncol. 2018, 4, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Duarte, D.; Amaro, F.; Silva, I.; Silva, D.; Fresco, P.; Oliveira, J.C.; Reguengo, H.; Gonçalves, J.; Vale, N. Carbidopa Alters Tryptophan Metabolism in Breast Cancer and Melanoma Cells Leading to the Formation of Indole-3-Acetonitrile, a Pro-Proliferative Metabolite. Biomolecules 2019, 9, 409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, A.; Yang, P.C.; Yu, C.J.; Lee, Y.C.; Yao, Y.T.; Chen, C.L.; Lee, L.N.; Kuo, S.H.; Luh, K.T. Tumor Angiogenesis Correlates with Histologic Type and Metastasis in Non-Small-Cell Lung Cancer. Am. J. Respir. Crit. Care Med. 1995, 152, 2157–2162. [Google Scholar] [CrossRef] [PubMed]
- Nonaka, H.; Saga, Y.; Fujiwara, H.; Akimoto, H.; Yamada, A.; Kagawa, S.; Takei, Y.; Machida, S.; Takikawa, O.; Suzuki, M. Indoleamine 2,3-Dioxygenase Promotes Peritoneal Dissemination of Ovarian Cancer through Inhibition of Natural Killercell Function and Angiogenesis Promotion. Int. J. Oncol. 2011, 38, 113–120. [Google Scholar]
- Su, C.; Zhang, P.; Liu, J.; Cao, Y. Erianin Inhibits Indoleamine 2, 3-Dioxygenase -Induced Tumor Angiogenesis. Biomed. Pharmacother. 2017, 88, 521–528. [Google Scholar] [CrossRef]
- Gong, Y.-Q.; Fan, Y.; Wu, D.-Z.; Yang, H.; Hu, Z.-B.; Wang, Z.-T. In Vivo and in Vitro Evaluation of Erianin, a Novel Anti-Angiogenic Agent. Eur. J. Cancer 2004, 40, 1554–1565. [Google Scholar] [CrossRef]
- Phacharapiyangkul, N.; Wu, L.-H.; Lee, W.-Y.; Kuo, Y.-H.; Wu, Y.-J.; Liou, H.-P.; Tsai, Y.-E.; Lee, C.-H. The Extracts of Astragalus Membranaceus Enhance Chemosensitivity and Reduce Tumor Indoleamine 2, 3-Dioxygenase Expression. Int. J. Med. Sci. 2019, 16, 1107–1115. [Google Scholar] [CrossRef] [Green Version]
- Chauhan, D.; Singh, A.V.; Brahmandam, M.; Carrasco, R.; Bandi, M.; Hideshima, T.; Bianchi, G.; Podar, K.; Tai, Y.-T.; Mitsiades, C.; et al. Functional Interaction of Plasmacytoid Dendritic Cells with Multiple Myeloma Cells: A Therapeutic Target. Cancer Cell 2009, 16, 309–323. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Koropatnick, J.; Li, M.; Zhang, X.; Ling, F.; Ren, X.; Hao, X.; Sun, H.; Vladau, C.; Franek, J.A.; et al. Reinstalling Antitumor Immunity by Inhibiting Tumor-Derived Immunosuppressive Molecule IDO through RNA Interference. J. Immunol. 2006, 177, 5639–5646. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Koropatnick, J.; Chen, D.; Velenosi, T.; Ling, H.; Zhang, X.; Jiang, N.; Navarro, B.; Ichim, T.E.; Urquhart, B.; et al. Silencing IDO in Dendritic Cells: A Novel Approach to Enhance Cancer Immunotherapy in a Murine Breast Cancer Model. Int. J. Cancer 2013, 132, 967–977. [Google Scholar] [CrossRef]
- Klar, R.; Michel, S.; Schell, M.; Hinterwimmer, L.; Zippelius, A.; Jaschinski, F. A Highly Efficient Modality to Block the Degradation of Tryptophan for Cancer Immunotherapy: Locked Nucleic Acid-Modified Antisense Oligonucleotides to Inhibit Human Indoleamine 2,3-Dioxygenase 1/Tryptophan 2,3-Dioxygenase Expression. Cancer Immunol. Immunother. 2020, 69, 57–67. [Google Scholar] [CrossRef]
- Campesato, L.F.; Budhu, S.; Tchaicha, J.; Weng, C.-H.; Gigoux, M.; Cohen, I.J.; Redmond, D.; Mangarin, L.; Pourpe, S.; Liu, C.; et al. Blockade of the AHR Restricts a Treg-Macrophage Suppressive Axis Induced by L-Kynurenine. Nat. Commun. 2020, 11, 4011. [Google Scholar] [CrossRef]
- Liu, Y.; Liang, X.; Yin, X.; Lv, J.; Tang, K.; Ma, J.; Ji, T.; Zhang, H.; Dong, W.; Jin, X.; et al. Blockade of IDO-Kynurenine-AhR Metabolic Circuitry Abrogates IFN-γ-Induced Immunologic Dormancy of Tumor-Repopulating Cells. Nat. Commun. 2017, 8, 15207. [Google Scholar] [CrossRef]
- Amobi, A.; Qian, F.; Lugade, A.A.; Odunsi, K. Tryptophan Catabolism and Cancer Immunotherapy Targeting IDO Mediated Immune Suppression. Adv. Exp. Med. Biol. 2017, 1036, 129–144. [Google Scholar] [CrossRef]
- Brochez, L.; Chevolet, I.; Kruse, V. The Rationale of Indoleamine 2,3-Dioxygenase Inhibition for Cancer Therapy. Eur. J. Cancer 2017, 76, 167–182. [Google Scholar] [CrossRef]
- Ricciuti, B.; Leonardi, G.C.; Puccetti, P.; Fallarino, F.; Bianconi, V.; Sahebkar, A.; Baglivo, S.; Chiari, R.; Pirro, M. Targeting Indoleamine-2,3-Dioxygenase in Cancer: Scientific Rationale and Clinical Evidence. Pharmacol. Ther. 2019, 196, 105–116. [Google Scholar] [CrossRef]
- Davar, D.; Bahary, N. Modulating Tumor Immunology by Inhibiting Indoleamine 2,3-Dioxygenase (IDO): Recent Developments and First Clinical Experiences. Target. Oncol. 2018, 13, 125–140. [Google Scholar] [CrossRef]
- Le Naour, J.; Galluzzi, L.; Zitvogel, L.; Kroemer, G.; Vacchelli, E. Trial Watch: IDO Inhibitors in Cancer Therapy. Oncoimmunology 2020, 9, 1777625. [Google Scholar] [CrossRef]
- Soliman, H.H.; Jackson, E.; Neuger, T.; Dees, E.C.; Harvey, R.D.; Han, H.; Ismail-Khan, R.; Minton, S.; Vahanian, N.N.; Link, C.; et al. A First in Man Phase I Trial of the Oral Immunomodulator, Indoximod, Combined with Docetaxel in Patients with Metastatic Solid Tumors. Oncotarget 2014, 5, 8136–8146. [Google Scholar] [CrossRef] [Green Version]
- Hou, D.-Y.; Muller, A.J.; Sharma, M.D.; DuHadaway, J.; Banerjee, T.; Johnson, M.; Mellor, A.L.; Prendergast, G.C.; Munn, D.H. Inhibition of Indoleamine 2,3-Dioxygenase in Dendritic Cells by Stereoisomers of 1-Methyl-Tryptophan Correlates with Antitumor Responses. Cancer Res. 2007, 67, 792–801. [Google Scholar] [CrossRef] [Green Version]
- Kudo, Y.; Boyd, C.A. Human Placental Indoleamine 2,3-Dioxygenase: Cellular Localization and Characterization of an Enzyme Preventing Fetal Rejection. Biochim. Biophys. Acta 2000, 1500, 119–124. [Google Scholar] [CrossRef] [Green Version]
- Lewis, H.C.; Chinnadurai, R.; Bosinger, S.E.; Galipeau, J. The IDO Inhibitor 1-Methyl Tryptophan Activates the Aryl Hydrocarbon Receptor Response in Mesenchymal Stromal Cells. Oncotarget 2017, 8, 91914–91927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siu, L.L.; Gelmon, K.; Chu, Q.; Pachynski, R.; Alese, O.; Basciano, P.; Walker, J.; Mitra, P.; Zhu, L.; Phillips, P.; et al. Abstract CT116: BMS-986205, an Optimized Indoleamine 2,3-Dioxygenase 1 (IDO1) Inhibitor, Is Well Tolerated with Potent Pharmacodynamic (PD) Activity, Alone and in Combination with Nivolumab (Nivo) in Advanced Cancers in a Phase 1/2a Trial. Cancer Res. 2017, 77, CT116. [Google Scholar] [CrossRef]
- Mautino, M.R.; Jaipuri, F.A.; Waldo, J.; Kumar, S.; Adams, J.; Allen, C.V.; Marcinowicz-Flick, A.; Munn, D.; Vahanian, N.; Link, C.J. Abstract 491: NLG919, a Novel Indoleamine-2,3-Dioxygenase (IDO)-Pathway Inhibitor Drug Candidate for Cancer Therapy. Cancer Res. 2013, 73, 491. [Google Scholar] [CrossRef]
- Ma, S.; Suchomel, J.; Yanez, E.; Yost, E.; Liang, X.; Zhu, R.; Le, H.; Siebers, N.; Joas, L.; Morley, R.; et al. Investigation of the Absolute Bioavailability and Human Mass Balance of Navoximod, a Novel IDO1 Inhibitor. Br. J. Clin. Pharmacol. 2019, 85, 1751–1760. [Google Scholar] [CrossRef]
- Krähenbühl, L.; Goldinger, S.M.; Mangana, J.; Kerl, K.; Chevolet, I.; Brochez, L.; Horak, C.; Levesque, M.; Dummer, R.; Cheng, P.F. A Longitudinal Analysis of IDO and PDL1 Expression during Immune- or Targeted Therapy in Advanced Melanoma. Neoplasia 2018, 20, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Heng, B.; Bilgin, A.A.; Lovejoy, D.B.; Tan, V.X.; Milioli, H.H.; Gluch, L.; Bustamante, S.; Sabaretnam, T.; Moscato, P.; Lim, C.K.; et al. Differential Kynurenine Pathway Metabolism in Highly Metastatic Aggressive Breast Cancer Subtypes: Beyond IDO1-Induced Immunosuppression. Breast Cancer Res. 2020, 22, 113. [Google Scholar] [CrossRef] [PubMed]
- Ci, C.; Wu, C.; Lyu, D.; Chang, X.; He, C.; Liu, W.; Chen, L.; Ding, W. Downregulation of Kynureninase Restrains Cutaneous Squamous Cell Carcinoma Proliferation and Represses the PI3K/AKT Pathway. Clin. Exp. Dermatol. 2020, 45, 194–201. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The course of the kynurenine pathway in neoplastic cells. The kynurenine pathway enzymes are in italics, while those involved in carcinogenesis are marked in red. AMO: aminocarboxymuconatesemialdehyde decarboxylase, IDO: indoleamine 2,3-dioxygenase, KAT: kynurenine aminotransferase, KMO: kynurenine 3-monooxygenase, KYN: kynurenine, KYNU: kynureninase, TDO: tryptophan 2,3-dioxygenase.
Figure 1.
The course of the kynurenine pathway in neoplastic cells. The kynurenine pathway enzymes are in italics, while those involved in carcinogenesis are marked in red. AMO: aminocarboxymuconatesemialdehyde decarboxylase, IDO: indoleamine 2,3-dioxygenase, KAT: kynurenine aminotransferase, KMO: kynurenine 3-monooxygenase, KYN: kynurenine, KYNU: kynureninase, TDO: tryptophan 2,3-dioxygenase.

Figure 2.
Upstream regulators of 2,3-dioxygenases activity and downstream effectors of kynurenine involved in neoplastic progression. IL-1, IL-6, TNF-α and IFN-γ stimulates IDO leading to enhanced synthesis of kynurenine. Its main effectors are presented in the circles. AhR: aryl hydrocarbon receptor, IDO: indoleamine 2,3-dioxygenase, IFN-γ: interferon-γ, IL-1: interleukin 1, IL-6: interleukin 6, KYN: kynurenine, STAT3: signal transducer and activator of transcription 3, TDO: tryptophan 2,3-dioxygenase, TNF-α: tumor necrosis factor-α, TRP: tryptophan.
Figure 2.
Upstream regulators of 2,3-dioxygenases activity and downstream effectors of kynurenine involved in neoplastic progression. IL-1, IL-6, TNF-α and IFN-γ stimulates IDO leading to enhanced synthesis of kynurenine. Its main effectors are presented in the circles. AhR: aryl hydrocarbon receptor, IDO: indoleamine 2,3-dioxygenase, IFN-γ: interferon-γ, IL-1: interleukin 1, IL-6: interleukin 6, KYN: kynurenine, STAT3: signal transducer and activator of transcription 3, TDO: tryptophan 2,3-dioxygenase, TNF-α: tumor necrosis factor-α, TRP: tryptophan.

Figure 3.
Effects of inhibition of 2,3-dioxygenases activity in selected types of cancers. APC: antigen presenting cells, CD8+ T cells: cytotoxic T cells IDO: indoleamine 2,3-dioxygenase, KYN: kynurenine, MDSCs: myeloid-derived suppressor cells, NK: natural killer cells, PBMCs: peripheral blood mononuclear cells, PD-L1: programmed cell death protein-1 ligand, ROS: reactive oxygen species, TDO: tryptophan 2,3-dioxygenase, Th: Helper T cells, TRP: tryptophan, Tregs: T regulatory cell.
Figure 3.
Effects of inhibition of 2,3-dioxygenases activity in selected types of cancers. APC: antigen presenting cells, CD8+ T cells: cytotoxic T cells IDO: indoleamine 2,3-dioxygenase, KYN: kynurenine, MDSCs: myeloid-derived suppressor cells, NK: natural killer cells, PBMCs: peripheral blood mononuclear cells, PD-L1: programmed cell death protein-1 ligand, ROS: reactive oxygen species, TDO: tryptophan 2,3-dioxygenase, Th: Helper T cells, TRP: tryptophan, Tregs: T regulatory cell.


{kind=link}
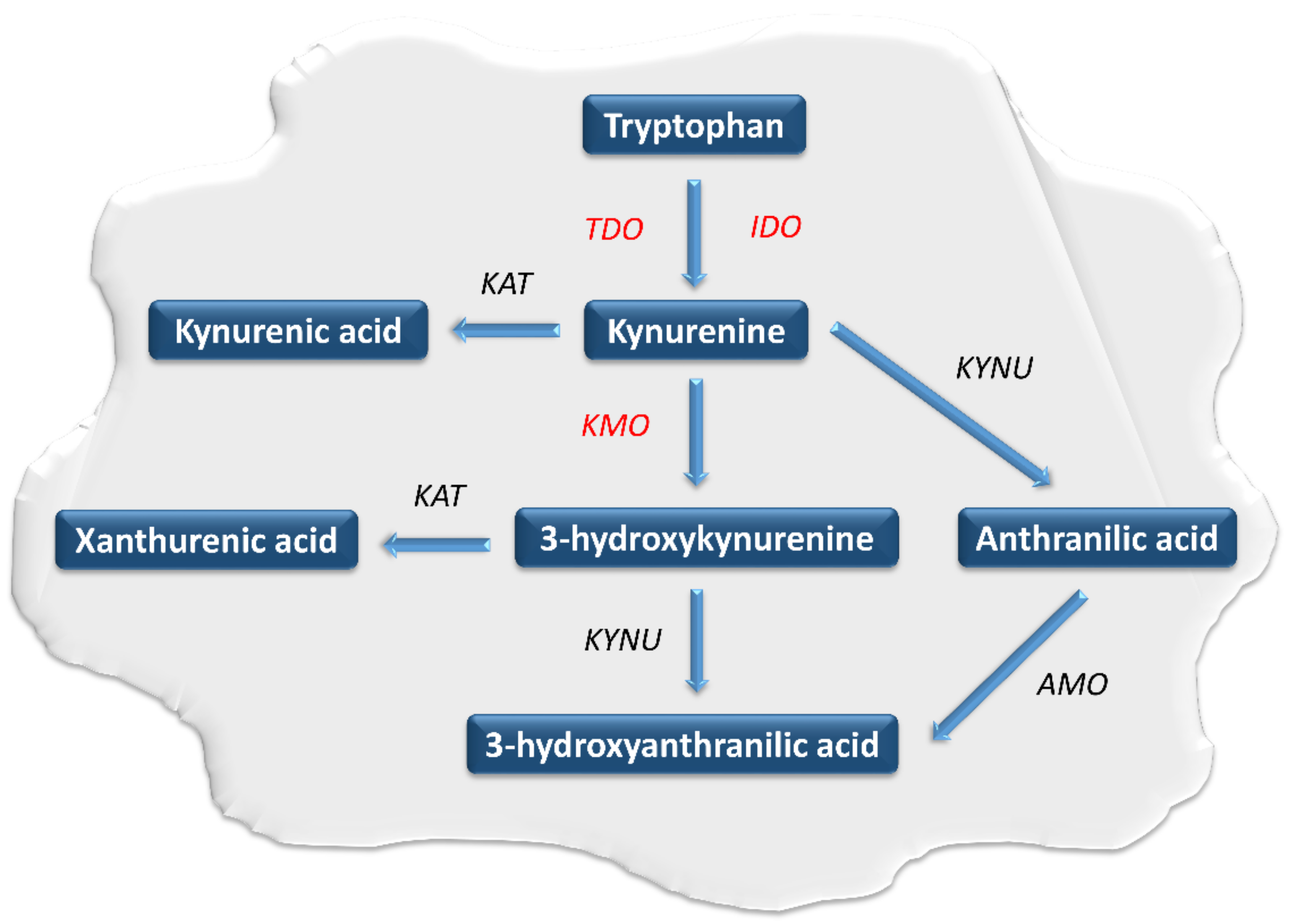{kind=link}
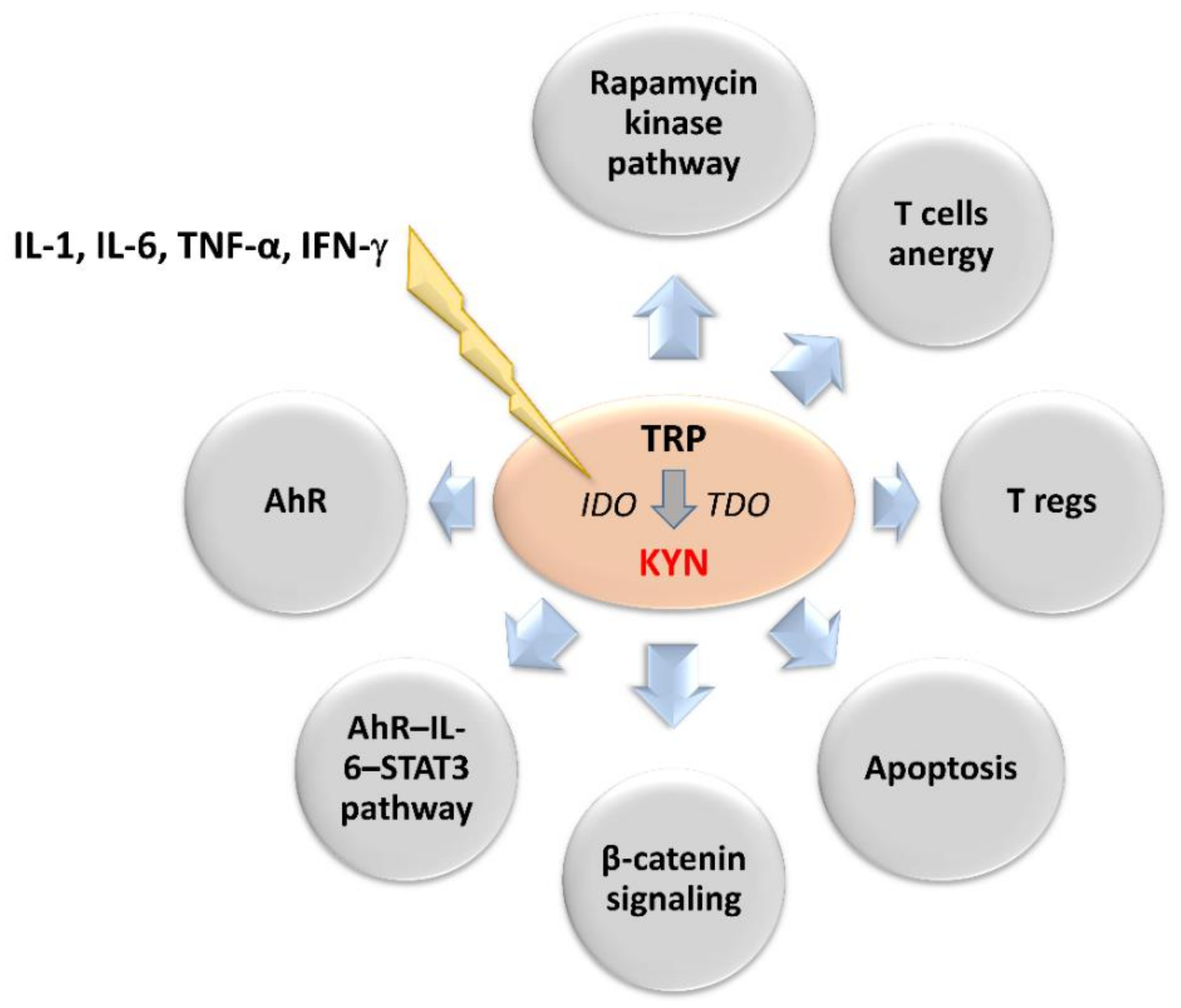{kind=link}
{kind=link}
Table 1.
Targeting KP enzymes in preclinical studies on antineoplastic therapy.
| Tumor | Target | Mechanism of Action and Agent | Suppression of Tumor Growth | Comments | References |
|---|---|---|---|---|---|
| Colon cancer | IDO1 | Direct enzyme inhibition | In-vitro SW837 cell line culture In-vivo Balb/c mice bearing CT26 tumor | [64,96,97,98,99,100,101,102] | |
| 1-MT | 7% | ||||
| 4,6-substituted-1H-indazole derivatives | 38% | ||||
| Amidoxime | N/A | ||||
| Epacadostat | 56% | ||||
| epigallocatechin gallate, Navoximod | N/A 75% (in co-treatment with oxaliplatin) | ||||
| 4-Bromophenylhydrazinyl benzenesulfonylphenylureas | 25% | ||||
| Ehrlich ascites carcinoma | IDO1 | Direct enzyme inhibition | In-vivo ICR mice bearing Erlich tumor | [97] | |
| Ethyl pyruvate | 78% (in co-treatment with cyclophosphamide); 79% (in co-treatment with cycloplatam) | ||||
| Amidoxime | 88% (in co-treatment with cyclophosphamid); 85% (in co-treatment with cycloplatam) | ||||
| Gastrointestinal stromal tumors | IDO1 | Downregulation of oncogenic KIT tyrosine-protein kinase signaling | In-vivo C57Bl/6J mice bearing GIST-T1 tumor | [103] | |
| Imatinib | 70% | ||||
| Glioblastoma | IDO1 | Direct enzyme inhibition | In-vivo C57BL/6 mice bearing GL261 glioblastoma | [83,104,105,106,107] | |
| 1-MT | 63% (in conjunction with two-fraction radiotherapy) | ||||
| Dinaciclib | N/A | ||||
| Acyclovir | N/A | ||||
| Glioma | IDO1 | Direct enzyme inhibition | In-vitro A172 human glioma cell line culture In-vivo C57BL/6 mice bearing LN229 glioma | [107,108,109] | |
| 1-MT | 87% (in co-treatment with temozonlomide) | ||||
| Tryptamine | N/A | ||||
| N-dimethyltryptamine | 64% (in combiantion with carboxyamidotriazole and cytotoxic T lymphocytes therapy) | ||||
| HPV-associated tumors | IDO1 | Direct enzyme inhibition | In-vivo 57BL/6 mice bearing TC-1 tumor | [93] | |
| 1-MT | 60% (in combiantion with gDE7-based immunotherapy) | ||||
| Lung cancer | IDO1 | Direct enzyme inhibition | In-vitro NCI-H460 and A549 cell line cultures In-vivo C57BL6/N mice bearing LLC, LL24 tumor lines, Balb/c nude mice bearing NCI-H1299 tumor | [94,95,110,111,112,113,114,115,116] | |
| 1-MT | 47% | ||||
| Navoximod | 65% | ||||
| EOS200271 | 45% | ||||
| LW106 | 68% | ||||
| Epacadostat | 51% | ||||
| INCB023843 | 22% | ||||
| Downregualtion of enzyme expression | |||||
| Feiji Recipe | 60% | ||||
| Inhibition of the extracellular IDO1 secretion | |||||
| Stragaloside IV | N/A | ||||
| Astragaloside IV | 72% (in combination with anti-PD1 antibody) | ||||
| Thymona | Downregualtion of enzyme expression | In-vivo C57BL/6 mice bearnng EG7 tumor line | [117] | ||
| Resveratrol | 51% | ||||
| Melanoma | IDO1 | Direct enzyme inhibition | In-vitro 624.38mel cell line culture In-vivo BALB/c mice bearing B16F10 melanoma | [94,95,113,118,119,120,121,122,123] | |
| 1-MT | 52% (in co-treatment with cyclophosphamide) 48% (in co-treatment with pimozide) 46% (in monotherapy), | ||||
| Navoximod | 95% (in co-treatment with pmel-1 T cells and gp100 peptide vaccination) | ||||
| Epacadostat | 50% | ||||
| DX-03-12 | 72.2% | ||||
| LW106 | 65% | ||||
| NTRC 3883-0 | 20% | ||||
| Increased proteasomal degradation | |||||
| Fludarabine | N/A | ||||
| Myelodysplastic syndrome | IDO1 | Direct enzyme inhibition | In-vivo C57BL/6N mice bearing MDS92 tumor | [124,125] | |
| Epacadostat | 50% | ||||
| Nasolaryngeal carcinoma | IDO1 | STAT1 acetylation | In-vitro CNE2 and CNE1 cell line cultures In-vivo BALB/C (NU/NU) mice bearing C666-1 tumor | [126,127,128] | |
| Bortezomib | 23% | ||||
| Suberoylanilide hydroxamic acid | 41% | ||||
| Combination of bortezomib with suberoylanilide hydroxamic acid | 76% | ||||
| Sodium butyrate | N/A | ||||
| Ovarian cancer | IDO1 | Direct enzyme inhibition | In-vitro SKOV3 cell line, patient-derived ovarian cancer cell culture In-vivo Balb/c NU/NU mice bearing SKOV3 tumor | [46,94,95,129,130] | |
| 1-MT | 10% | ||||
| NTRC 3883-0 | N/A | ||||
| LW106 | N/A | ||||
| BMS-986242 | N/A | ||||
| Pancreatic Ductal Adenocarcinoma | IDO1 | Direct enzyme inhibition | In-vitro Human PDAC specimens | [124,131,132,133] | |
| Epacadostat | 55% | ||||
| EOS200271 | 32% | ||||
| Plasmacytoid dendritic cell leukemia | IDO1 | Direct enzyme inhibition | In-vitro PMDC05 cell line culture | [134] | |
| Toho-1 | N/A | ||||
| Renal Cancer | IDO1 | Direct enzyme inhibition | In-vivo Balb/cJ mice bearing 786-O tumor | [135] | |
| Methyl-thiohydantoin-DL-TRP | 45% (in co-treatment with IFN-α) | ||||
| Sarcomas | IDO1 | Direct enzyme inhibition | In-vivo C57BL/6N mice bearing MCA205 tumor | [136] | |
| Navoximod | 0% (did not show antitumoral activity, but has a potential impact on the NK cell functions). | ||||
| Non-Hodgkin Lymphoma | IDO2 | Direct enzyme inhibition Indoximod | 77% (in co-treatment with cyclophosphamide) | In-vivo BalB/c NU/NU mice bearing A20 B-cell lymphoma | [137] |
| Breast cancer | IDO 1 | Direct enzyme inhibition | In-vitro MDA-231and BT549 cell lines In-vivo C57BL/6 mice bearing MDA-231 tumor | [71,123,131,138,139] | |
| EOS200271 | 21% | ||||
| Downregulation of enzyme expression | |||||
| Nimesulide | 54% | ||||
| Celecoxib | 52% | ||||
| Increased proteasomal degradation | |||||
| Fludarabine | N/A | ||||
| TDO2 | Direct enzyme inhibition | ||||
| 680C91 | 85% | ||||
| Cervical cancer | IDO1 | Direct enzyme inhibition | In-vitro HeLa cell line | [140,141] | |
| LBJ-10 | N/A | ||||
| IDO1 and TDO | Direct enzyme inhibition | ||||
| β-lapachone | N/A | ||||
| Liver cancer | IDO1 | Direct enzyme inhibition | In-vitro HepG2 cell line In-vivo BALB/c mice bearing H22 tumor C3HA mice bearing H-29 tumor | [142,143,144] | |
| Ethyl pyruvate | 39% | ||||
| JAK/STAT1 pathway inhibition | |||||
| Polysaccharide from Lepista sordid | N/A | ||||
| TDO | Direct enzyme inhibition | ||||
| 3-(2-(Pyridyl)ethenyl) indole derivatives | 65% (in combination with irinotecan) | ||||
| Multiple Myeloma | KMO | Direct enzyme inhibition | In-vitro Patient-derived multiple myeloma cell culture | [145] | |
| Ro-61–8048 | N/A |
IFN-α—interferon-α, N/A—Data not available.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Mor, A.; Tankiewicz-Kwedlo, A.; Pawlak, D. Kynurenines as a Novel Target for the Treatment of Malignancies. Pharmaceuticals 2021, 14, 606. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14070606
AMA Style
Mor A, Tankiewicz-Kwedlo A, Pawlak D. Kynurenines as a Novel Target for the Treatment of Malignancies. Pharmaceuticals. 2021; 14(7):606. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14070606
Chicago/Turabian StyleMor, Adrian, Anna Tankiewicz-Kwedlo, and Dariusz Pawlak. 2021. "Kynurenines as a Novel Target for the Treatment of Malignancies" Pharmaceuticals 14, no. 7: 606. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14070606
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.