
Discovery of Novel Chemical Series of OXA-48 β-Lactamase Inhibitors by High-Throughput Screening

 , , ,
, , ,
Abstract
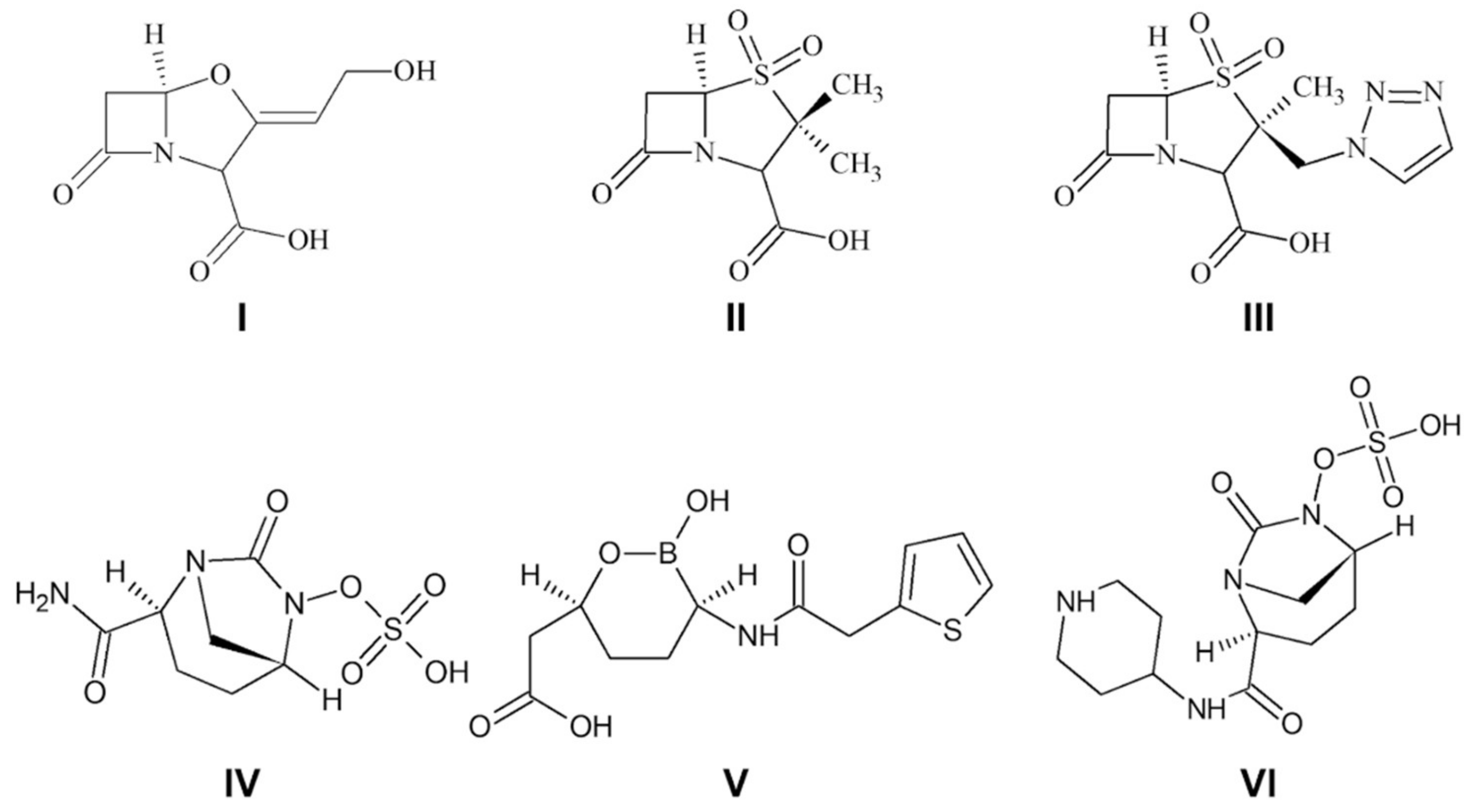
:1. Introduction
2. Results and Discussion
2.1. Compound Collection
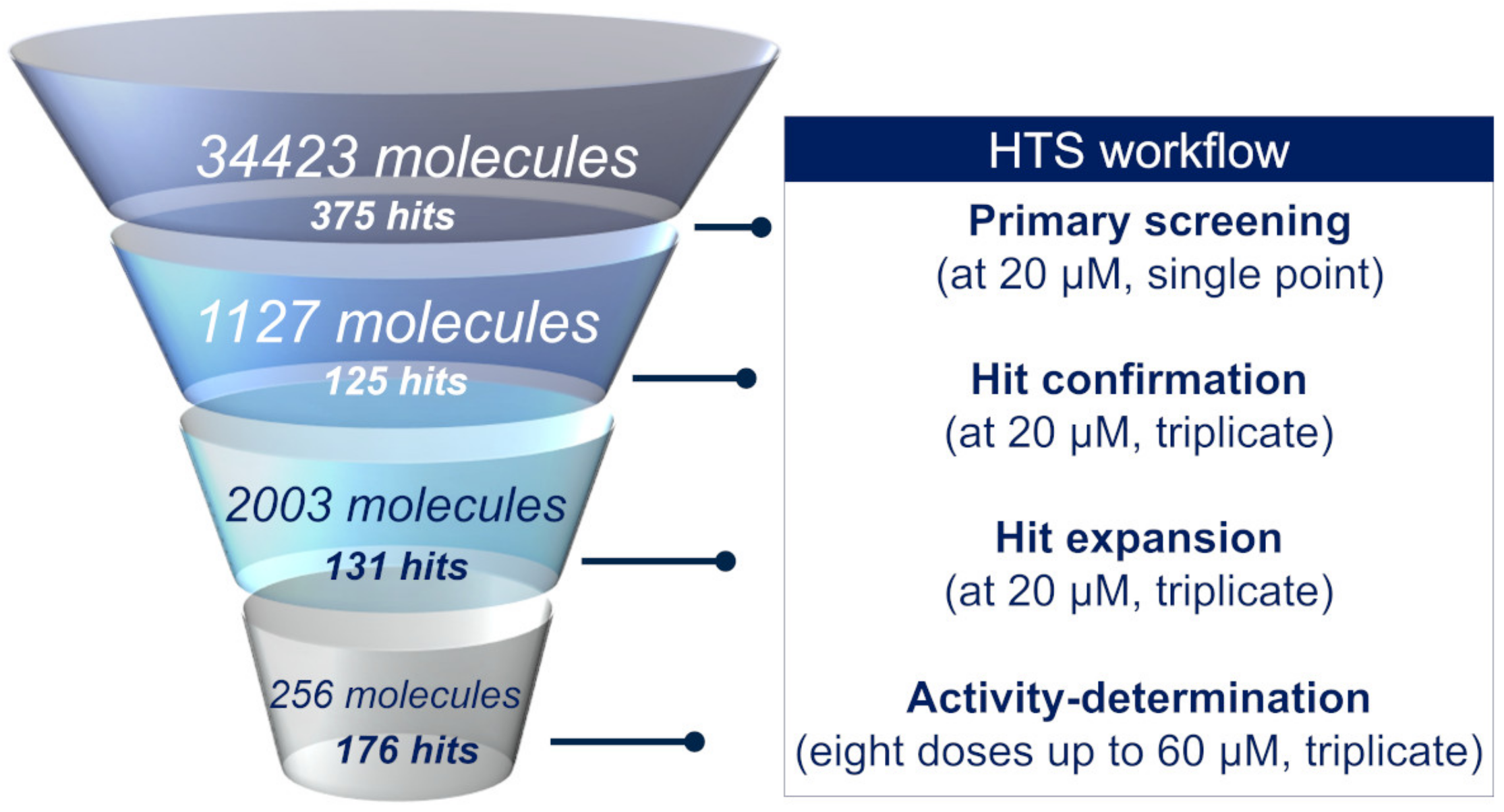
2.2. High-Throughput Screening
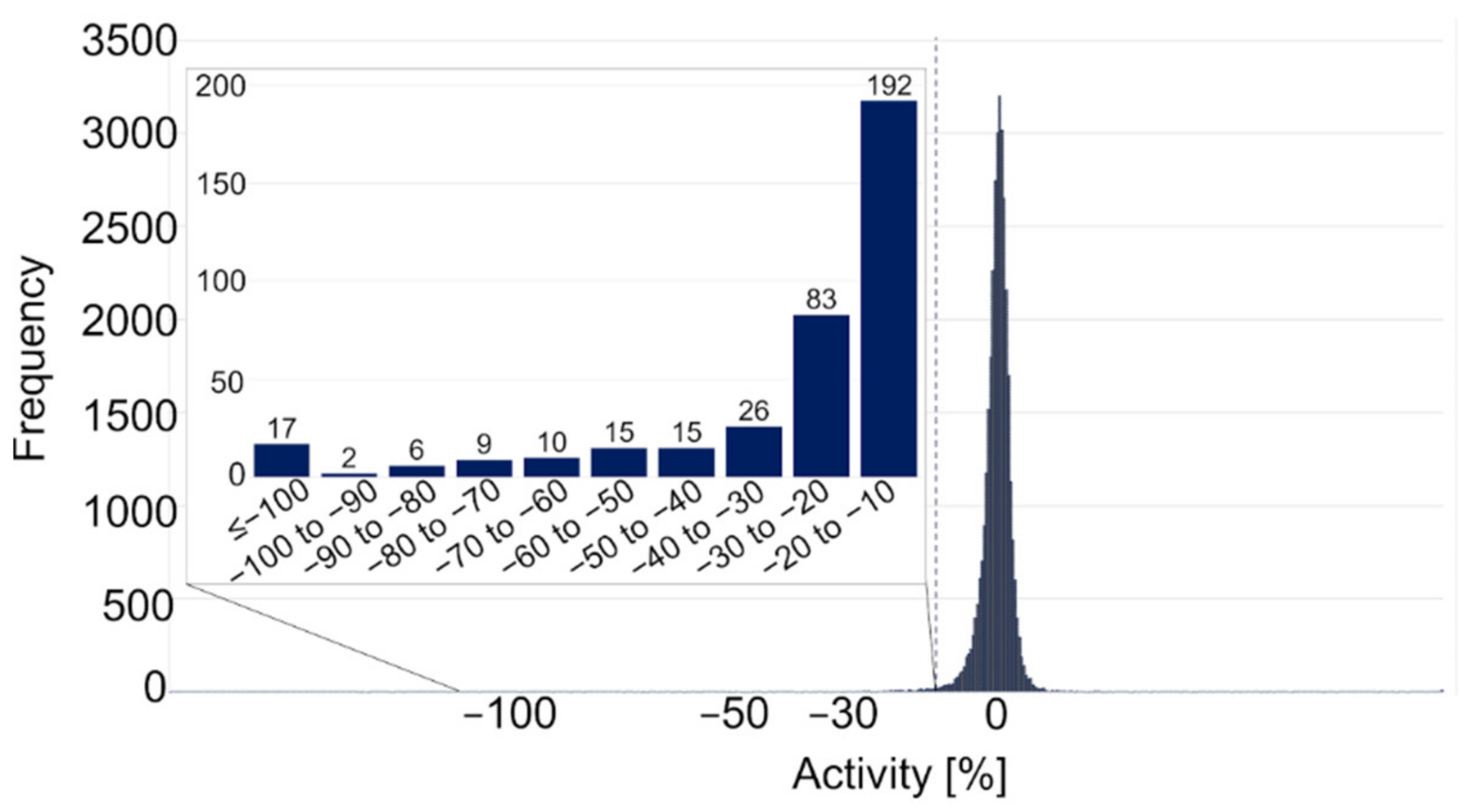
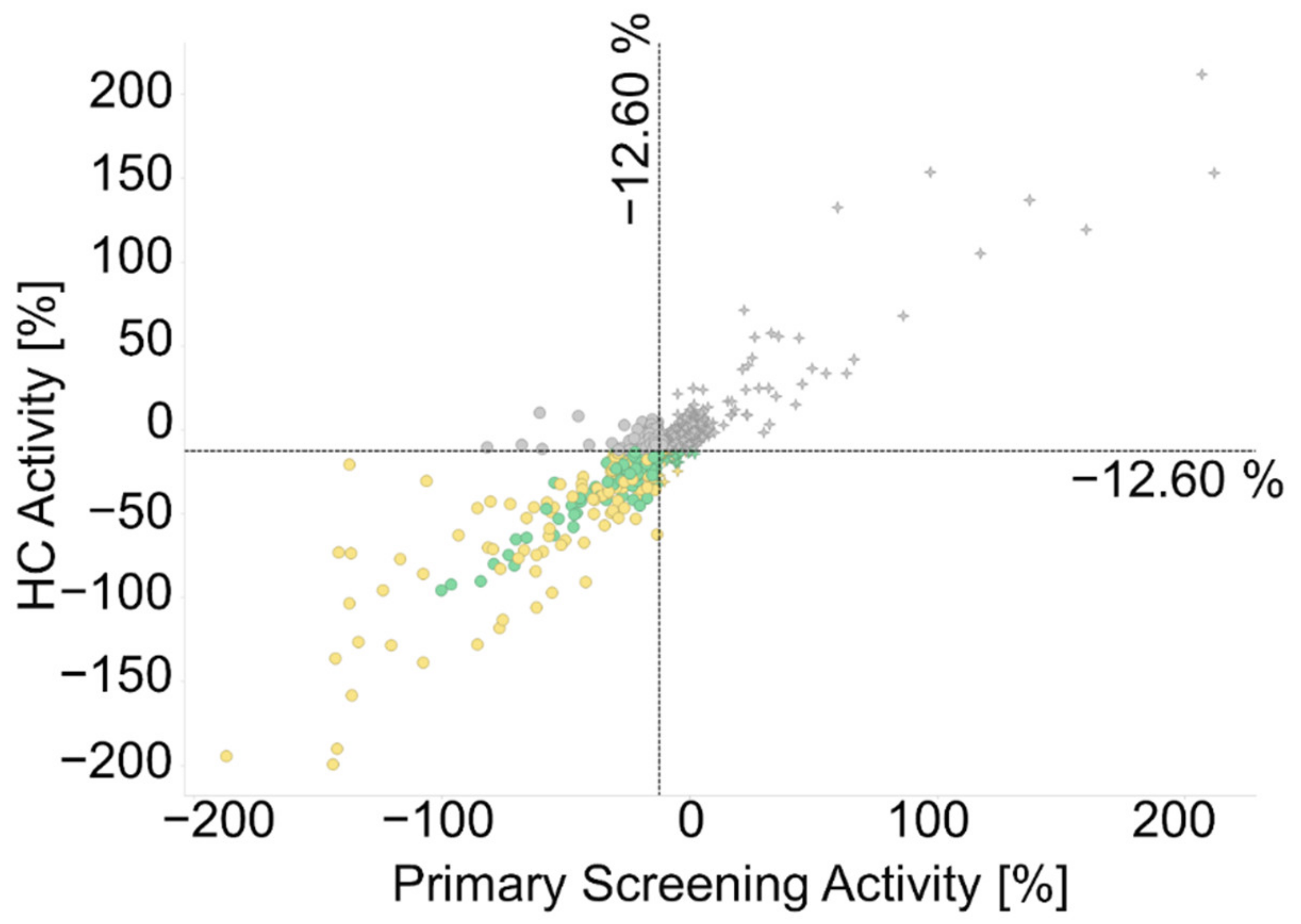
2.2.1. Primary Screening
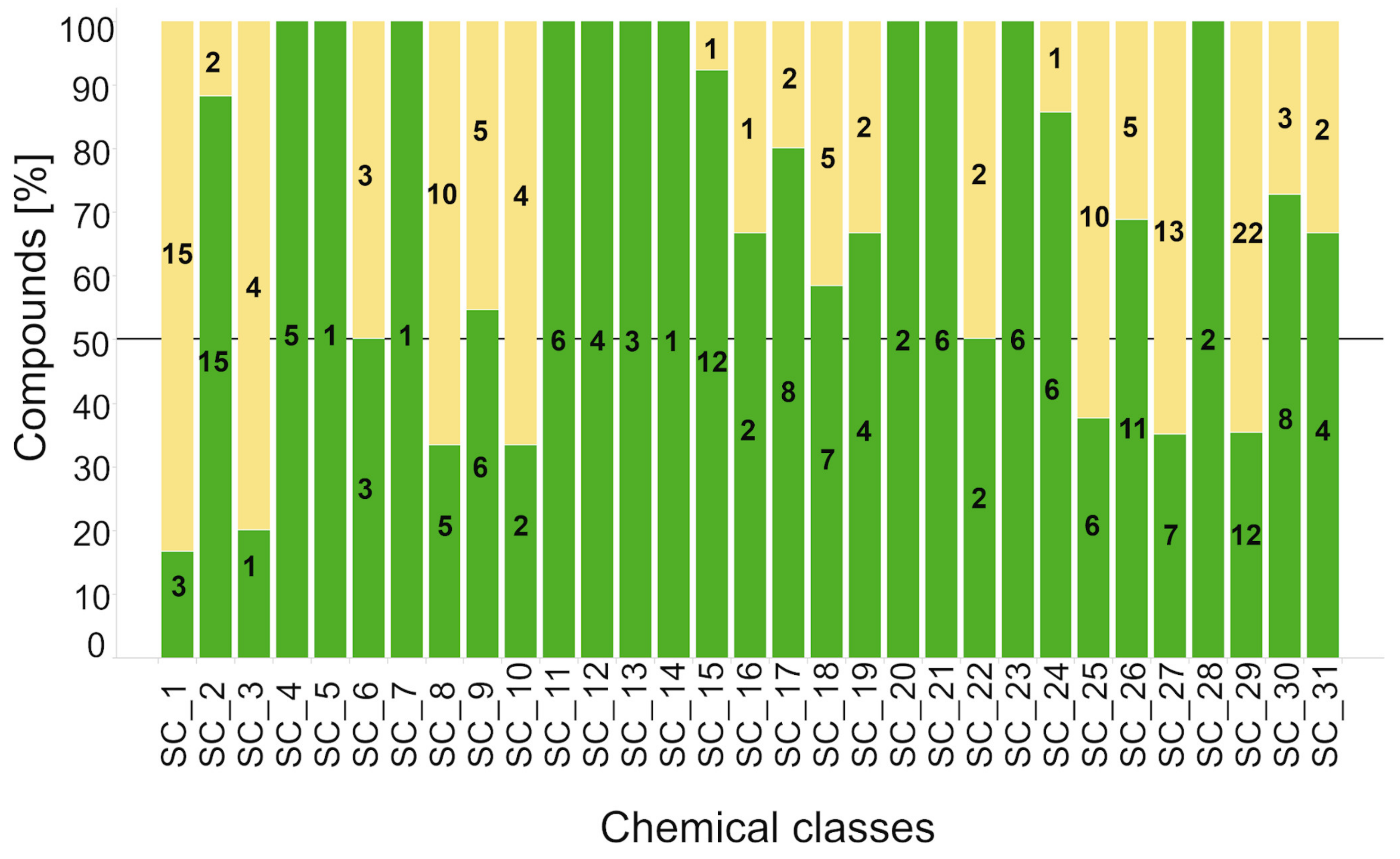
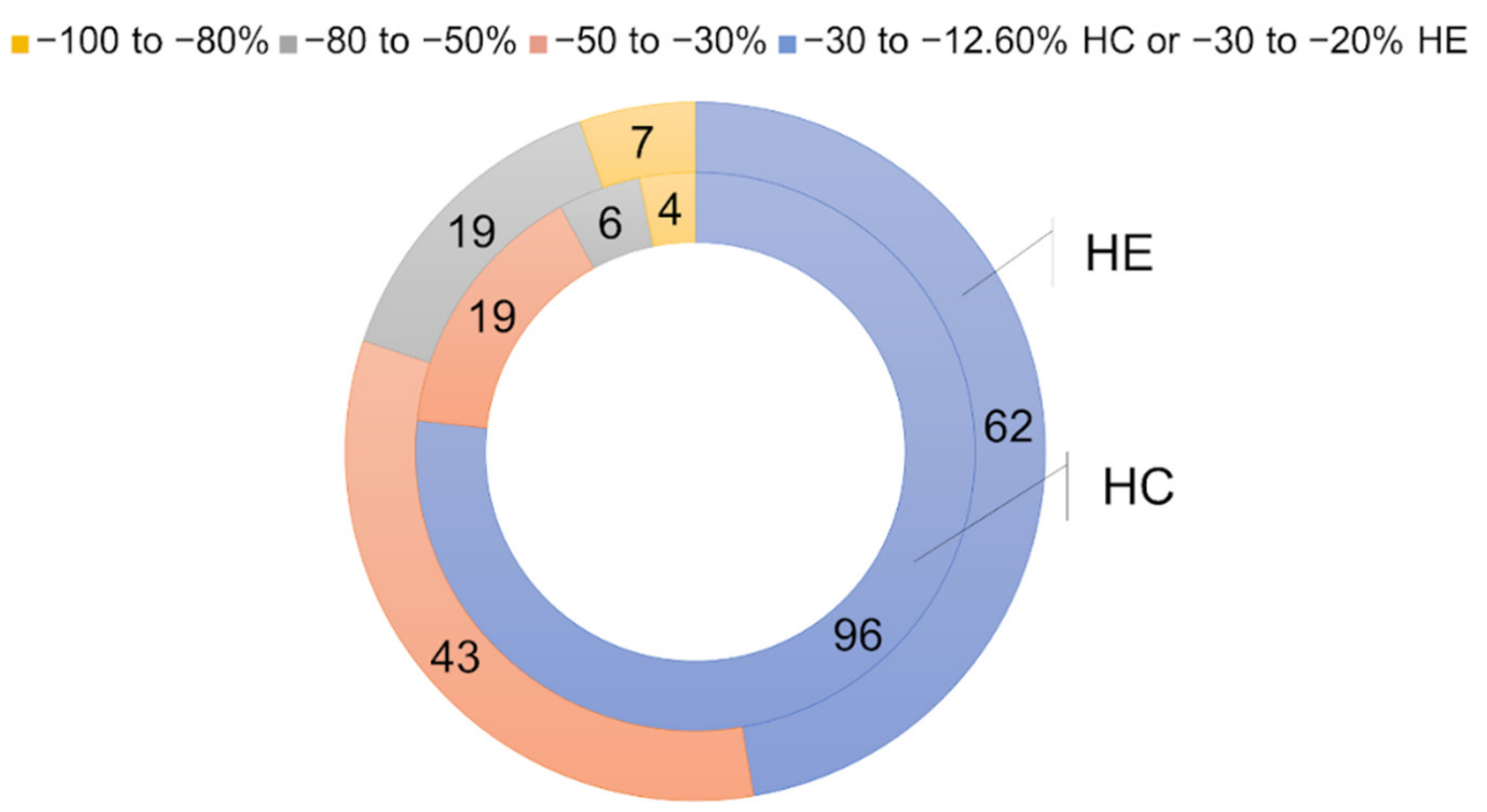
2.2.2. Hit Confirmation
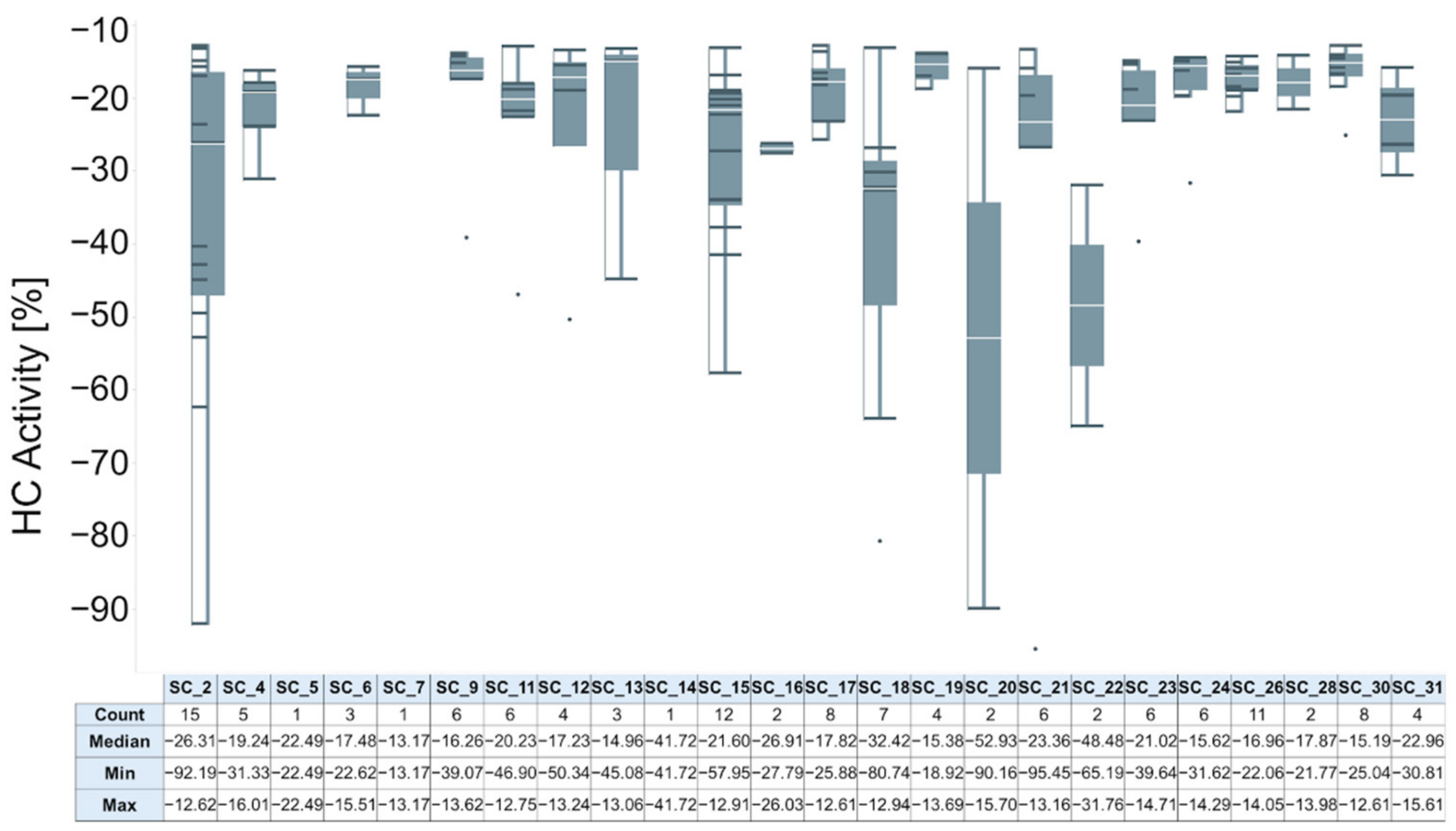
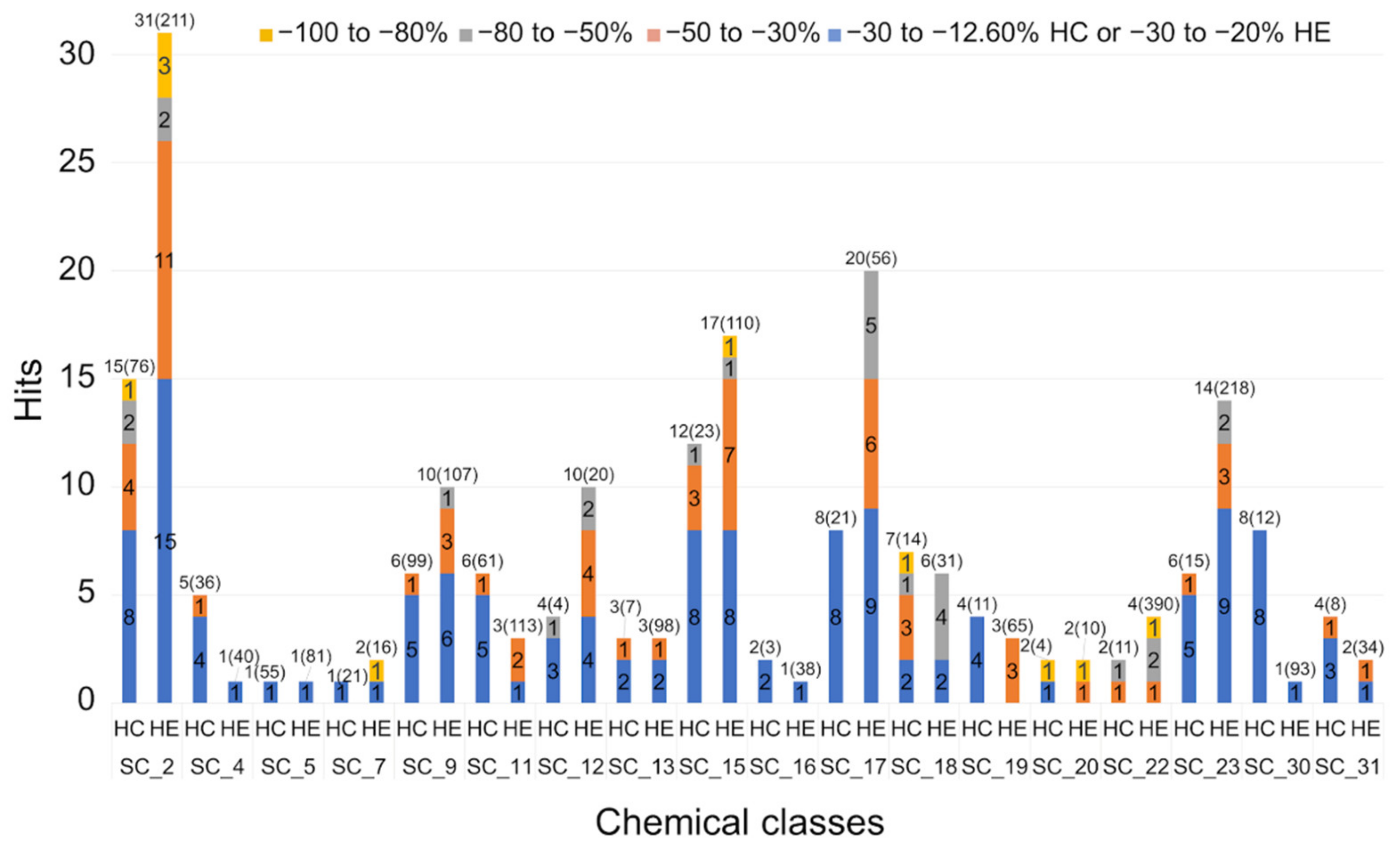
2.2.3. Hit Expansion
2.2.4. Activity Determination
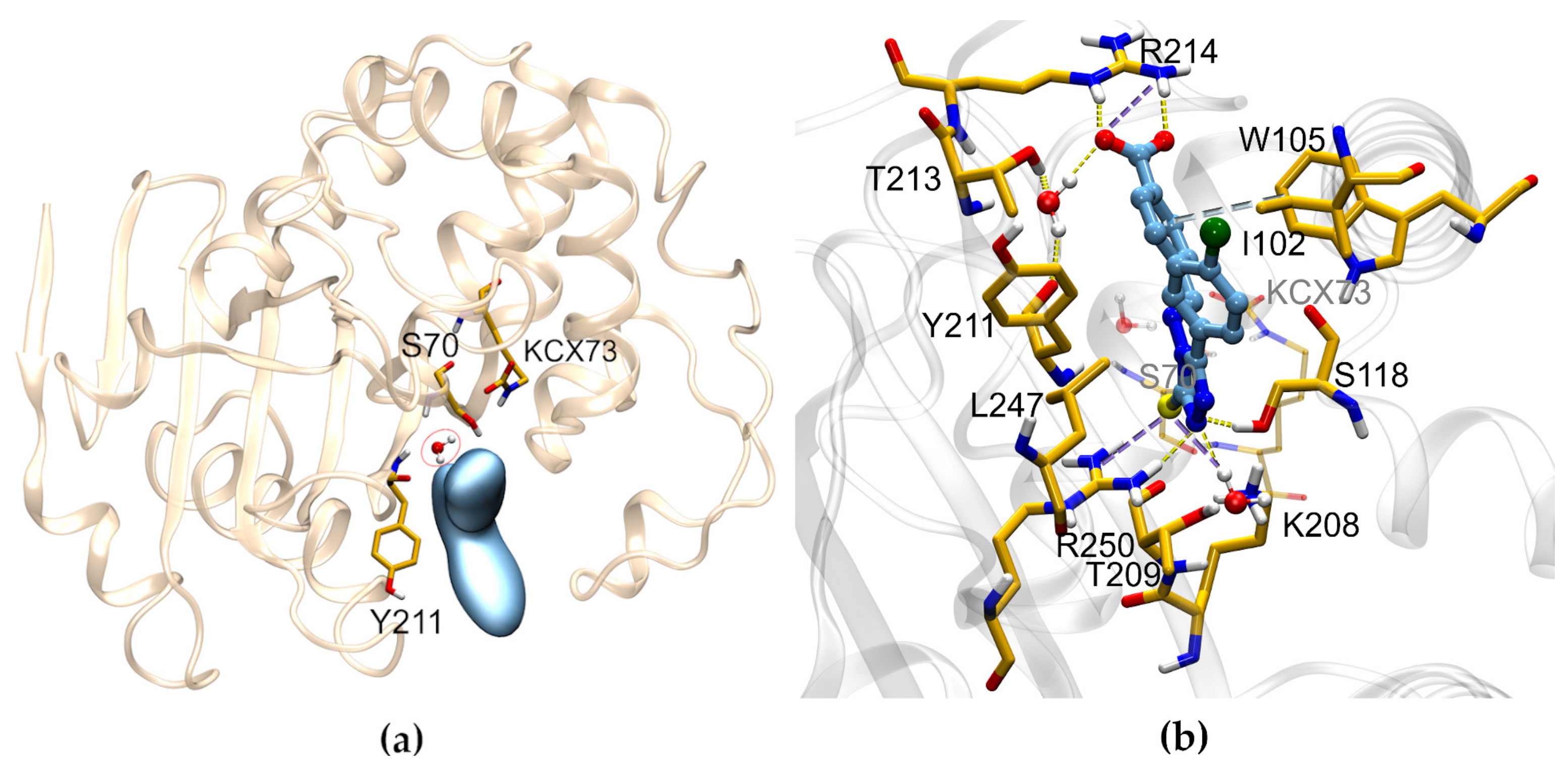
2.3. Mechanism of Action
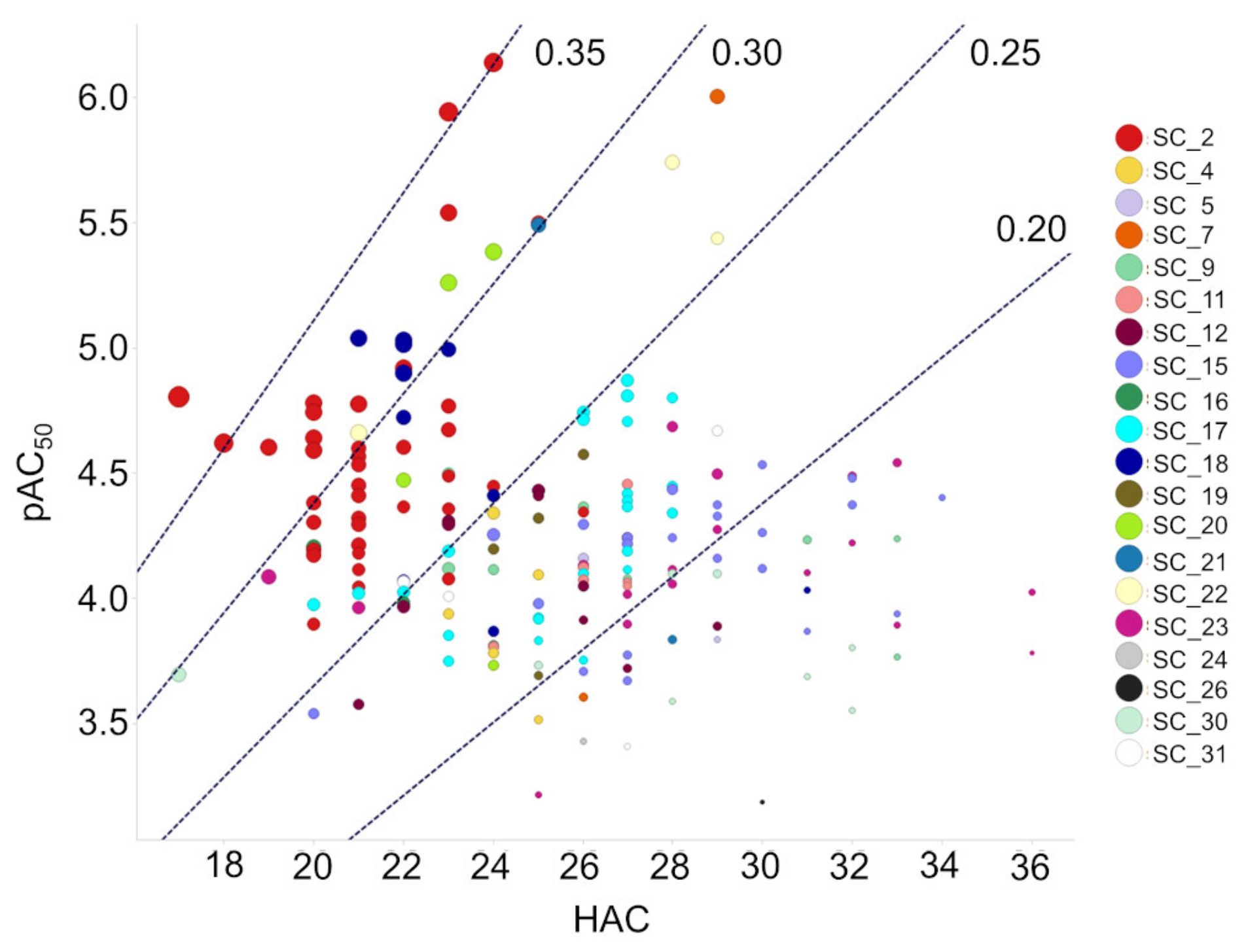
2.4. SC_2: SAR from HTS
2.5. SC_7: SAR from HTS
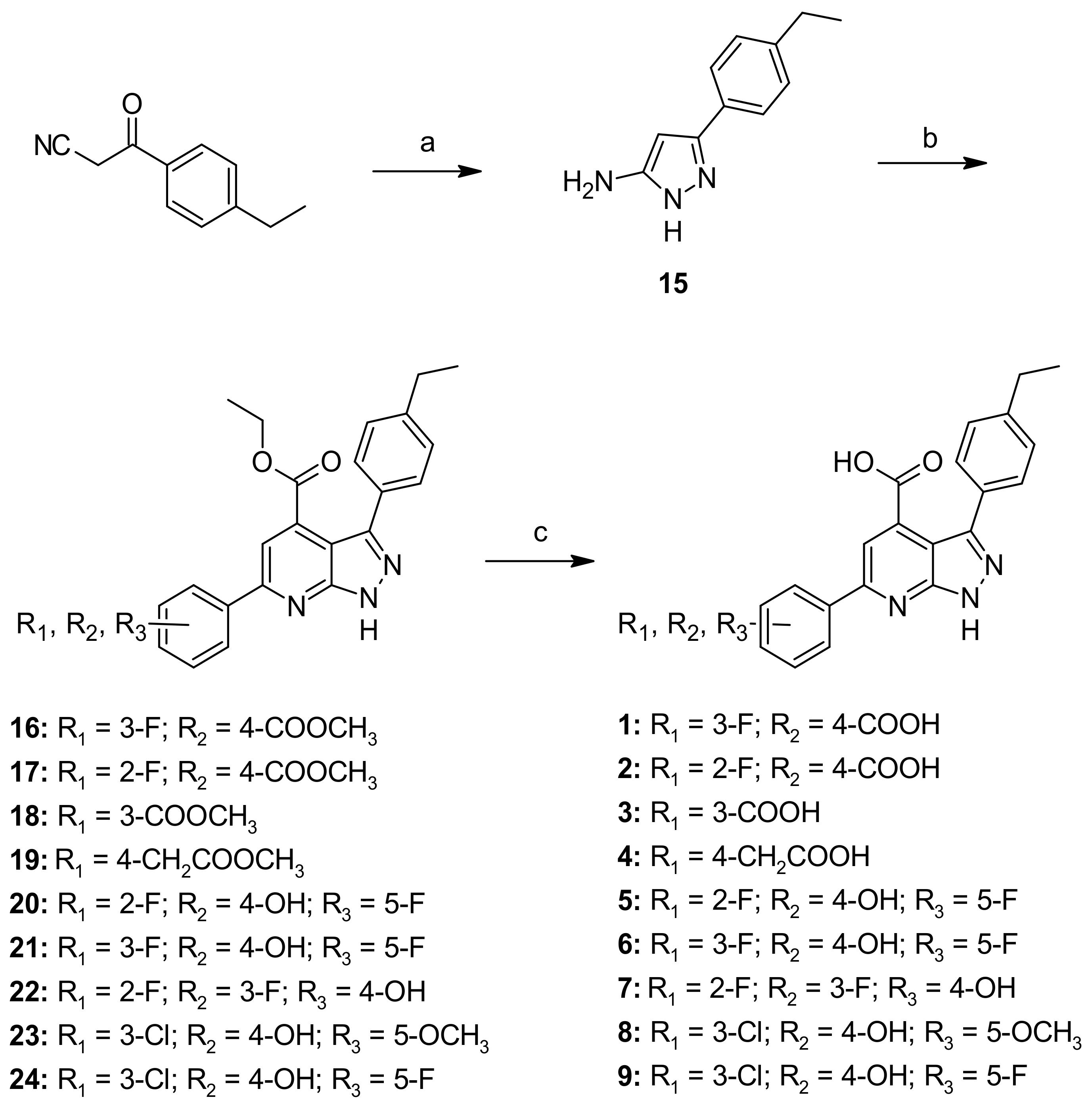
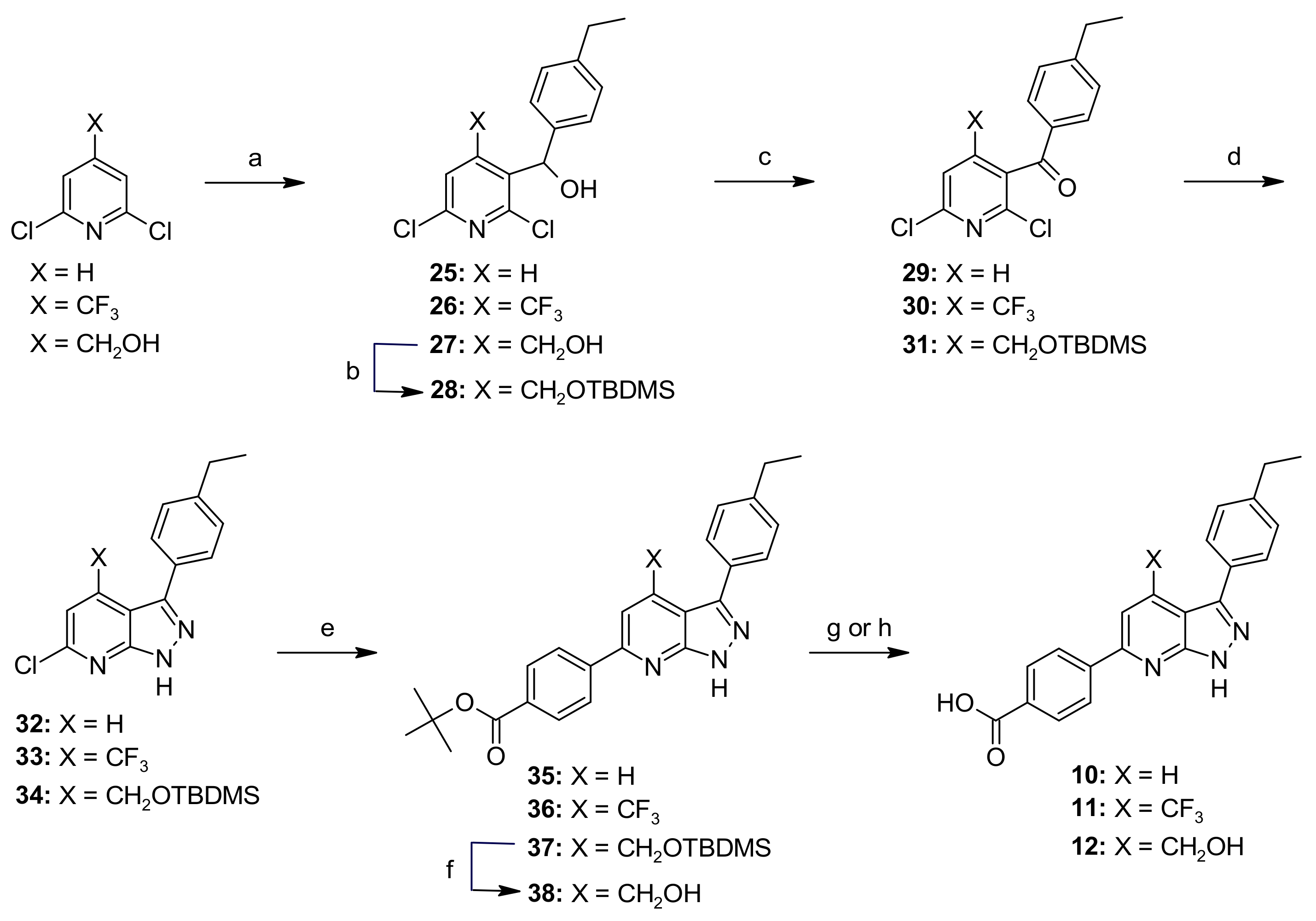
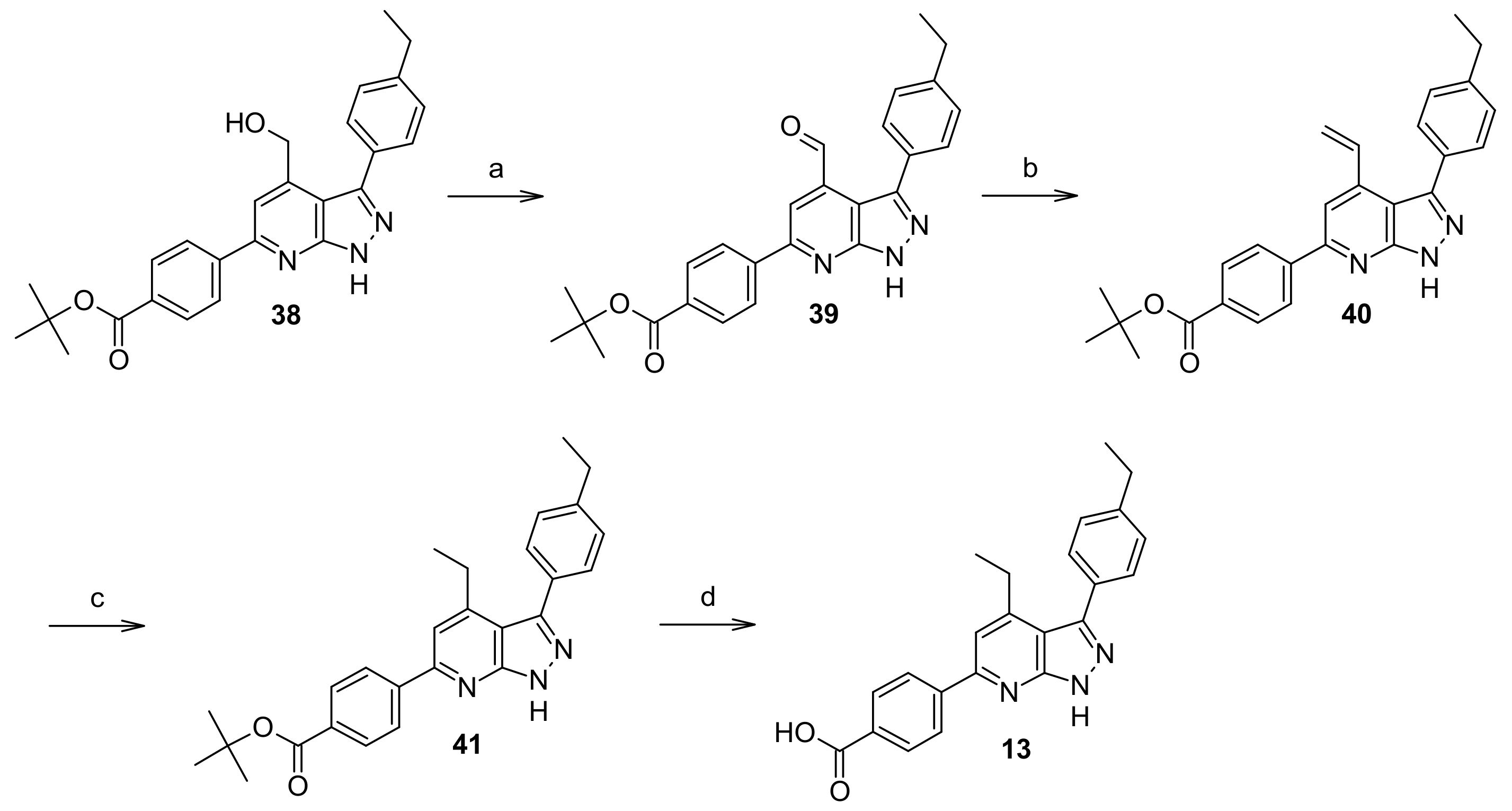
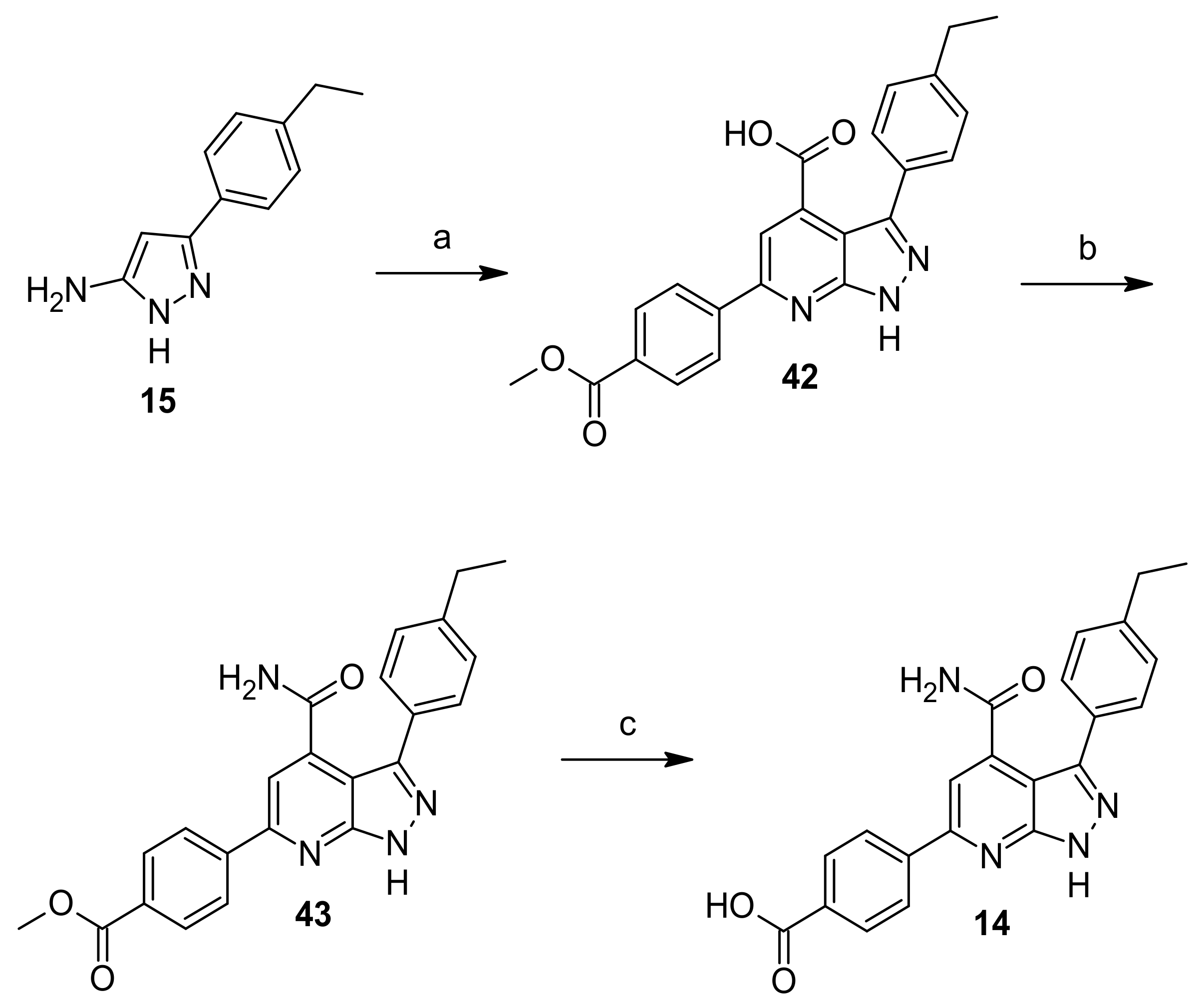
2.6. Chemistry
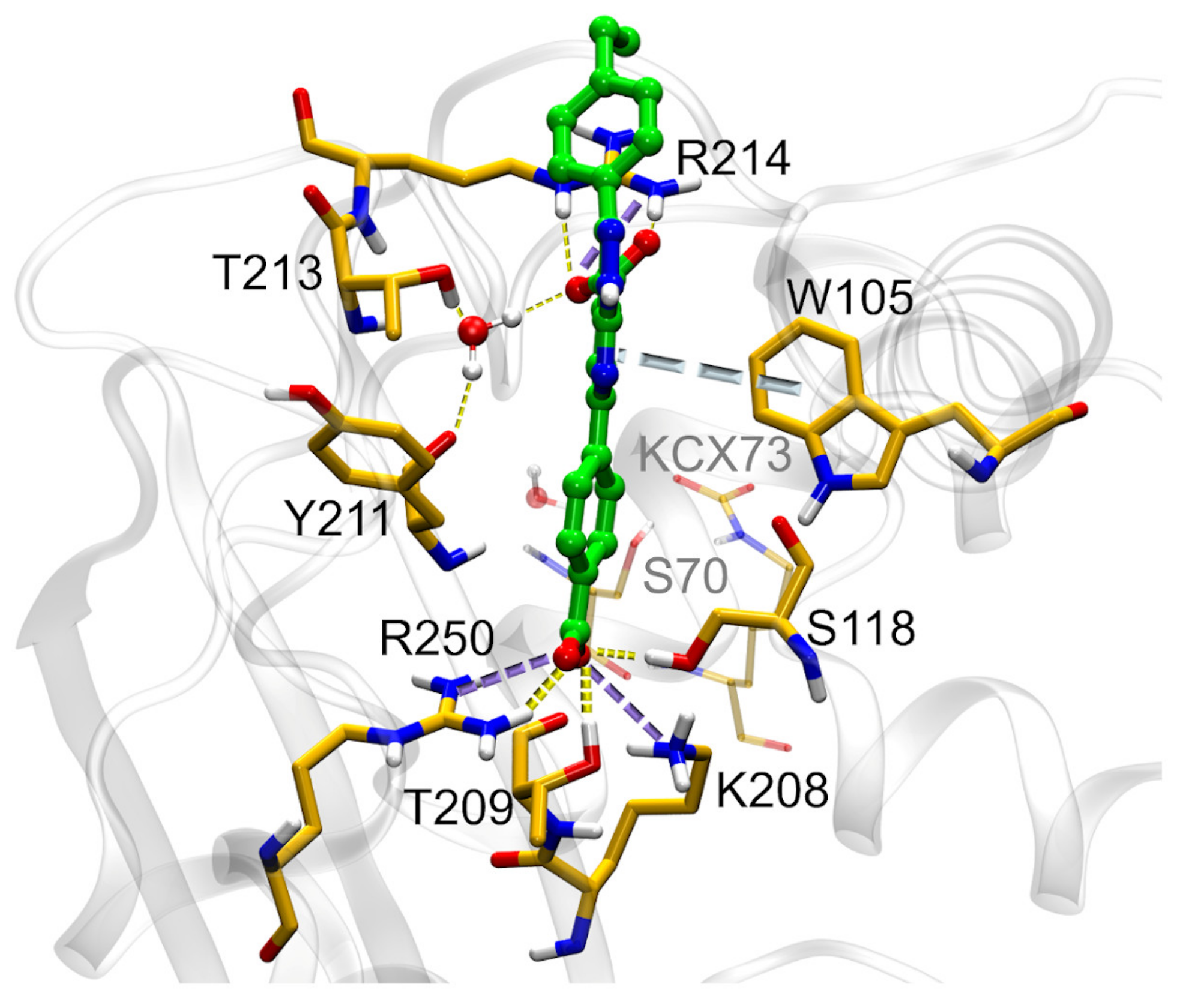
2.7. SC_7: Explorative SAR
2.8. Determination of MIC, Cytotoxicity and Solubility
3. Materials and Methods
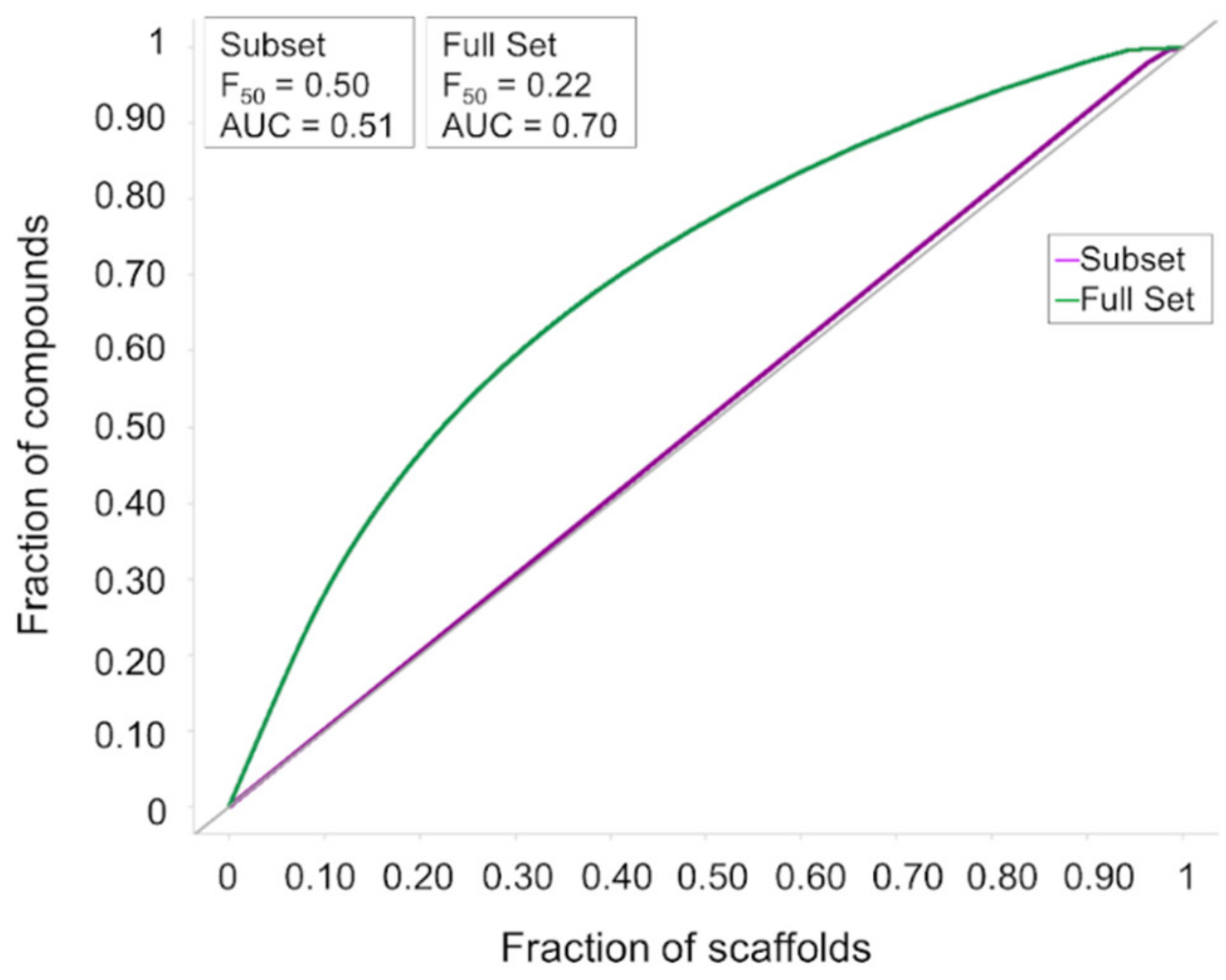
3.1. Cheminformatics
3.2. Chemistry
3.2.1. General Information
3.2.2. General Procedures
General Procedure A—Synthesis of Compounds 16–24, 42
General Procedure B—Synthesis of Compounds 25–27
General Procedure C—Synthesis of Compounds 29–31
General Procedure D—Synthesis of Compounds 32–34
General Procedure E—Synthesis of Compounds 35–37
General Procedure F—Synthesis of Compounds 1–9
General Procedure G—Synthesis of Compounds 10 and 11
3.2.3. Synthesis of Intermediates and Final Compounds
3-(4-Ethylphenyl)-1H-pyrazol-5-amine (15)
Ethyl-3-(4-ethylphenyl)-6-(3-fluoro-4-(methoxycarbonyl)phenyl)-1H-pyrazolo[3,4-b]pyridine-4-carboxylate (16)
Ethyl-3-(4-ethylphenyl)-6-(2-fluoro-4-(methoxycarbonyl)phenyl)-1H-pyrazolo[3,4-b]pyridine-4-carboxylate (17)
Ethyl-3-(4-ethylphenyl)-6-(3-(methoxycarbonyl)phenyl)-1H-pyrazolo[3,4-b]pyridine-4-carboxylate (18)
Ethyl-3-(4-ethylphenyl)-6-(4-(2-methoxy-2-oxoethyl)phenyl)-1H-pyrazolo[3,4-b]pyridine-4-carboxylate (19)
Ethyl-6-(2,5-difluoro-4-hydroxyphenyl)-3-(4-ethylphenyl)-1H-pyrazolo[3,4-b]pyridine-4-carboxylate (20)
Ethyl-6-(3,5-difluoro-4-hydroxyphenyl)-3-(4-ethylphenyl)-1H-pyrazolo[3,4-b]pyridine-4-carboxylate (21)
Ethyl-6-(2,3-difluoro-4-hydroxyphenyl)-3-(4-ethylphenyl)-1H-pyrazolo[3,4-b]pyridine-4-carboxylate (22)
Ethyl-6-(3-chloro-4-hydroxy-5-methoxyphenyl)-3-(4-ethylphenyl)-1H-pyrazolo[3,4-b]pyridine-4-carboxylate (23)
Ethyl-6-(3-chloro-5-fluoro-4-hydroxyphenyl)-3-(4-ethylphenyl)-1H-pyrazolo[3,4-b]pyridine-4-carboxylate (24)
(2,6-Dichloropyridin-3-yl)(4-ethylphenyl)methanol (25)
(2,6-Dichloro-4-(trifluoromethyl)pyridin-3-yl)(4-ethylphenyl)methanol (26)
(2,6-Dichloro-4-(hydroxymethyl)pyridin-3-yl)(4-ethylphenyl)methanol (27)
(4-(((Tert-butyldimethylsilyl)oxy)methyl)-2,6-dichloropyridin-3-yl)(4-ethylphenyl)methanol (28)
(2,6-Dichloropyridin-3-yl)(4-ethylphenyl)methanone (29)
2,6-Dichloro-4-(trifluoromethyl)pyridin-3-yl)(4-ethylphenyl)methanone (30)
(4-(((Tert-butyldimethylsilyl)oxy)methyl)-2,6-dichloropyridin-3-yl)(4-ethylphenyl)methanone (31)
6-Chloro-3-(4-ethylphenyl)-1H-pyrazolo[3,4-b]pyridine (32)
6-Chloro-3-(4-ethylphenyl)-4-(trifluoromethyl)-1H-pyrazolo[3,4-b]pyridine (33)
4-(((Tert-butyldimethylsilyl)oxy)methyl)-6-chloro-3-(4-ethylphenyl)-1H-pyrazolo[3,4-b]pyridine (34)
Tert-butyl 4-(3-(4-ethylphenyl)-1H-pyrazolo[3,4-b]pyridin-6-yl)benzoate (35)
Tert-butyl-4-(3-(4-ethylphenyl)-4-(trifluoromethyl)-1H-pyrazolo[3,4-b]pyridin-6-yl)benzoate (36)
Tert-butyl-4-(4-(((tert-butyldimethylsilyl)oxy)methyl)-3-(4-ethylphenyl)-1H-pyrazolo[3,4-b]pyridin-6-yl)benzoate (37)
Tert-butyl-4-(3-(4-ethylphenyl)-4-(hydroxymethyl)-1H-pyrazolo[3,4-b]pyridin-6-yl)benzoate (38)
Tert-butyl-4-(3-(4-ethylphenyl)-4-formyl-1H-pyrazolo[3,4-b]pyridin-6-yl)benzoate (39)
Tert-butyl-4-(3-(4-ethylphenyl)-4-vinyl-1H-pyrazolo[3,4-b]pyridin-6-yl)benzoate (40)
Tert-butyl-4-(4-ethyl-3-(4-ethylphenyl)-1H-pyrazolo[3,4-b]pyridin-6-yl)benzoate (41)
3-(4-Ethylphenyl)-6-(4-(methoxycarbonyl)phenyl)-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid (42)
Methyl 4-(4-carbamoyl-3-(4-ethylphenyl)-1H-pyrazolo[3,4-b]pyridin-6-yl)benzoate (43)
6-(4-Carboxy-3-fluorophenyl)-3-(4-ethylphenyl)-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid (1)
6-(4-Carboxy-2-fluorophenyl)-3-(4-ethylphenyl)-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid (2)
6-(3-Carboxyphenyl)-3-(4-ethylphenyl)-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid (3)
6-(4-(Carboxymethyl)phenyl)-3-(4-ethylphenyl)-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid (4)
6-(2,5-Difluoro-4-hydroxyphenyl)-3-(4-ethylphenyl)-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid (5)
6-(3,5-Difluoro-4-hydroxyphenyl)-3-(4-ethylphenyl)-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid (6)
6-(2,3-Difluoro-4-hydroxyphenyl)-3-(4-ethylphenyl)-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid (7)
6-(3-Chloro-4-hydroxy-5-methoxyphenyl)-3-(4-ethylphenyl)-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid (8)
6-(3-Chloro-5-fluoro-4-hydroxyphenyl)-3-(4-ethylphenyl)-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid (9)
4-(3-(4-Ethylphenyl)-1H-pyrazolo[3,4-b]pyridin-6-yl)benzoic acid (10)
4-(3-(4-Ethylphenyl)-4-(trifluoromethyl)-1H-pyrazolo[3,4-b]pyridin-6-yl)benzoic acid (11)
4-(3-(4-Ethylphenyl)-4-(hydroxymethyl)-1H-pyrazolo[3,4-b]pyridin-6-yl)benzoic acid (12)
4-(4-Ethyl-3-(4-ethylphenyl)-1H-pyrazolo[3,4-b]pyridin-6-yl)benzoic acid (13)
4-(4-Carbamoyl-3-(4-ethylphenyl)-1H-pyrazolo[3,4-b]pyridin-6-yl)benzoic acid (14)
3.3. Protein Production and Purification
3.4. X-ray Crystallography
3.5. Primary Enzymatic Assay for OXA-48
3.6. Counter-Screening Assay
3.7. HTS Campaign Mathematical Background
3.8. Mechanism of Action of OXA-48 Inhibiotors
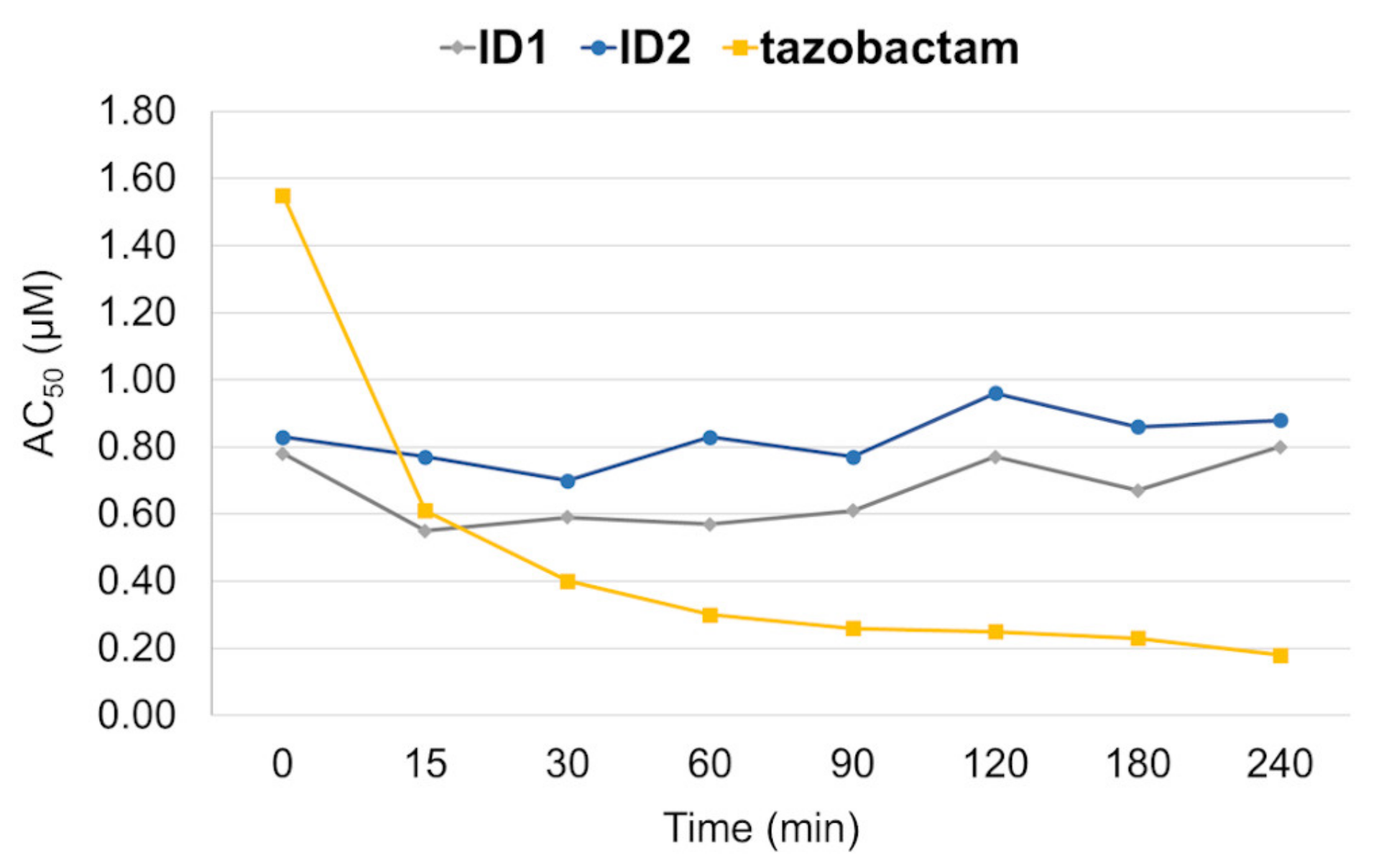
3.8.1. Time-Dependent Inhibition of OXA-48
3.8.2. Dilution after Preincubation of OXA-48 with Selected Inhibitors
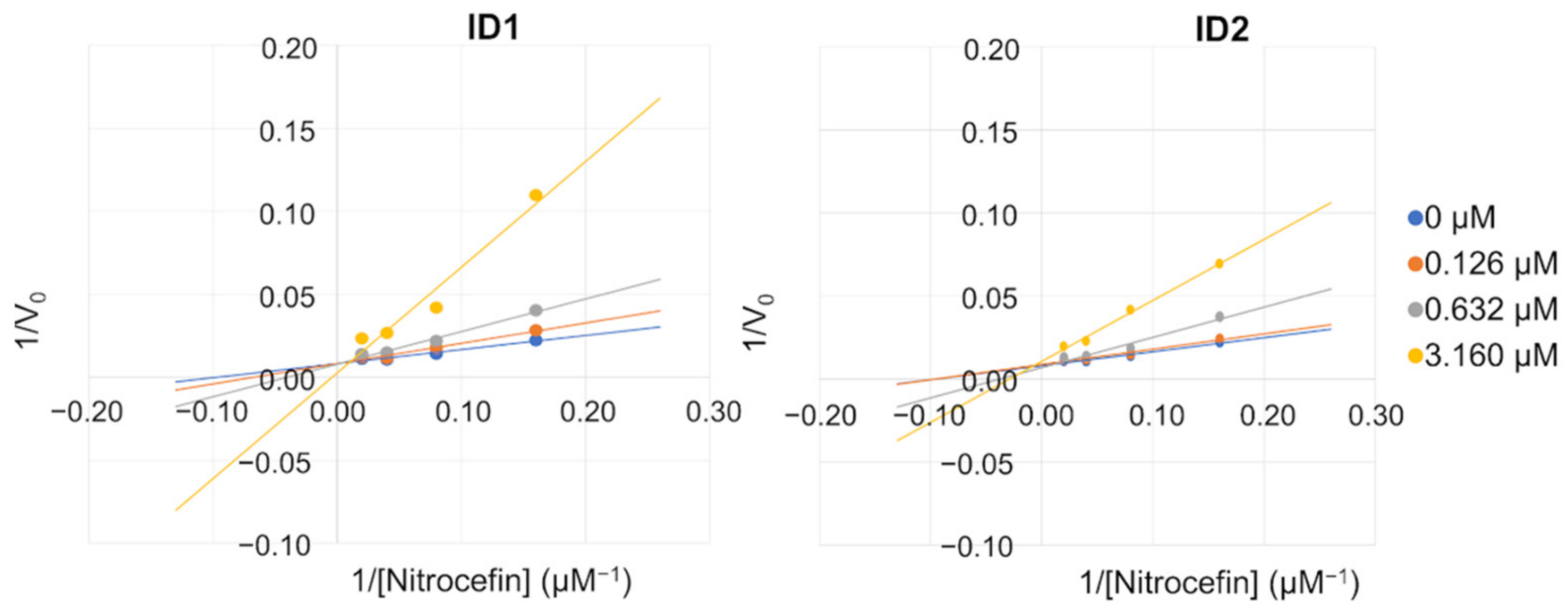
3.8.3. Determination of the Apparent Km and Vmax of Nitrocefin in the Presence of AC30, AC50 or AC70 of the Selected Inhibitors
3.8.4. Determination of the Ki and a Values
3.9. MICs Assays
3.10. Aqueous Solubility Assay (PBS, pH 7.4)
3.11. Cytotoxicity Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Bush, K.; Bradford, P.A. beta-Lactams and beta-Lactamase Inhibitors: An Overview. Cold Spring Harb. Perspect. Med. 2016, 6, a025247. [Google Scholar] [CrossRef] [PubMed]
- Tooke, C.L.; Hinchliffe, P.; Bragginton, E.C.; Colenso, C.K.; Hirvonen, V.H.A.; Takebayashi, Y.; Spencer, J. β-Lactamases and β-Lactamase Inhibitors in the 21st Century. J. Mol. Biol. 2019, 431, 3472–3500. [Google Scholar] [CrossRef] [PubMed]
- Abraham, E.P.; Chain, E. An enzyme from bacteria able to destroy penicillin. 1940. Rev. Infect. Dis. 1988, 10, 677–678. [Google Scholar]
- Wright, G.D.; Poinar, H. Antibiotic resistance is ancient: Implications for drug discovery. Trends Microbiol. 2012, 20, 157–159. [Google Scholar] [CrossRef] [PubMed]
- Bush, K. Bench-to-bedside review: The role of β-lactamases in antibiotic-resistant Gram-negative infections. Crit. Care 2010, 14, 224. [Google Scholar] [CrossRef] [Green Version]
- Bush, K.; Bradford, P.A. Interplay between β-lactamases and new β-lactamase inhibitors. Nat. Rev. Microbiol. 2019, 17, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Waxman, D.J.; Yocum, R.R.; Strominger, J.L. Penicillins and cephalosporins are active site-directed acylating agents: Evidence in support of the substrate analogue hypothesis. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1980, 289, 257–271. [Google Scholar] [CrossRef]
- Spratt, B.G.; Cromie, K.D. Penicillin-binding proteins of gram-negative bacteria. Rev. Infect. Dis. 1988, 10, 699–711. [Google Scholar] [CrossRef]
- Buynak, J.D. Cutting and stitching: The cross-linking of peptidoglycan in the assembly of the bacterial cell wall. ACS Chem. Biol. 2007, 2, 602–605. [Google Scholar] [CrossRef] [Green Version]
- Kitano, K.; Tomasz, A. Triggering of autolytic cell wall degradation in Escherichia coli by beta-lactam antibiotics. Antimicrob. Agents Chemother. 1979, 16, 838–848. [Google Scholar] [CrossRef] [Green Version]
- Naas, T.; Oueslati, S.; Bonnin, R.A.; Dabos, M.L.; Zavala, A.; Dortet, L.; Retailleau, P.; Iorga, B.I. Beta-lactamase database (BLDB) - structure and function. J. Enzyme Inhib. Med. Chem. 2017, 32, 917–919. [Google Scholar] [CrossRef]
- Ambler, R.P. The structure of beta-lactamases. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1980, 289, 321–331. [Google Scholar] [CrossRef]
- Bush, K.; Jacoby, G.A. Updated functional classification of beta-lactamases. Antimicrob. Agents Chemother. 2010, 54, 969–976. [Google Scholar] [CrossRef] [Green Version]
- Drawz, S.M.; Bonomo, R.A. Three Decades of β-Lactamase Inhibitors. Clin. Microbiol. Rev. 2010, 23, 160–201. [Google Scholar] [CrossRef] [Green Version]
- Docquier, J.D.; Mangani, S. An update on β-lactamase inhibitor discovery and development. Drug Resist. Updates 2018, 36, 13–29. [Google Scholar] [CrossRef] [PubMed]
- Zasowski, E.J.; Rybak, J.M.; Rybak, M.J. The β-Lactams Strike Back: Ceftazidime-Avibactam. Pharmacotherapy 2015, 35, 755–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, J.C.; Zmarlicka, M.T.; Shaeer, K.M.; Pardo, J. Meropenem/Vaborbactam, the First Carbapenem/β-Lactamase Inhibitor Combination. Ann. Pharmacother. 2018, 52, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Zhanel, G.G.; Lawrence, C.K.; Adam, H.; Schweizer, F.; Zelenitsky, S.; Zhanel, M.; Lagacé-Wiens, P.R.S.; Walkty, A.; Denisuik, A.; Golden, A.; et al. Imipenem-relebactam and meropenem-vaborbactam: Two Novel Carbapenem-β-Lactamase Inhibitor Combinations. Drugs 2018, 78, 65–98. [Google Scholar] [CrossRef]
- Smith, J.R.; Rybak, J.M.; Claeys, K.C. Imipenem-Cilastatin-Relebactam: A novel beta-lactam-beta-lactamase inhibitor combination for the treatment of multidrug-resistant gram-negative infections. Pharmacotherapy 2020, 40, 343–356. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Trout, R.E.L.; Chu, G.-H.; McGarry, D.; Jackson, R.W.; Hamrick, J.C.; Daigle, D.M.; Cusick, S.M.; Pozzi, C.; De Luca, F.; et al. Discovery of Taniborbactam (VNRX-5133): A Broad-Spectrum Serine- and Metallo-β-lactamase Inhibitor for Carbapenem-Resistant Bacterial Infections. J. Med. Chem. 2020, 63, 2789–2801. [Google Scholar] [CrossRef] [Green Version]
- Reading, C.; Cole, M. Clavulanic acid: A beta-lactamase-inhiting beta-lactam from Streptomyces clavuligerus. Antimicrob. Agents Chemother. 1977, 11, 852–857. [Google Scholar] [CrossRef] [Green Version]
- Ehmann, D.E.; Jahic, H.; Ross, P.L.; Gu, R.F.; Hu, J.; Durand-Réville, T.F.; Lahiri, S.; Thresher, J.; Livchak, S.; Gao, N.; et al. Kinetics of avibactam inhibition against Class A, C, and D β-lactamases. J. Biol. Chem. 2013, 288, 27960–27971. [Google Scholar] [CrossRef] [Green Version]
- Poirel, L.; Héritier, C.; Tolün, V.; Nordmann, P. Emergence of oxacillinase-mediated resistance to imipenem in Klebsiella pneumoniae. Antimicrob. Agents Chemother. 2004, 48, 15–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shields, R.K.; Chen, L.; Cheng, S.; Chavda, K.D.; Press, E.G.; Snyder, A.; Pandey, R.; Doi, Y.; Kreiswirth, B.N.; Nguyen, M.H.; et al. Emergence of Ceftazidime-Avibactam Resistance Due to Plasmid-Borne bla(KPC-3) Mutations during Treatment of Carbapenem-Resistant Klebsiella pneumoniae Infections. Antimicrob. Agents Chemother. 2017, 61, e02097-02016. [Google Scholar] [CrossRef] [Green Version]
- Frohlich, C.; Sorum, V.; Thomassen, A.M.; Johnsen, P.J.; Leiros, H.S.; Samuelsen, O. OXA-48-Mediated Ceftazidime-Avibactam Resistance Is Associated with Evolutionary Trade-Offs. mSphere 2019, 4. [Google Scholar] [CrossRef] [Green Version]
- Arca-Suárez, J.; Fraile-Ribot, P.; Vázquez-Ucha, J.C.; Cabot, G.; Martínez-Guitián, M.; Lence, E.; González-Bello, C.; Beceiro, A.; Rodríguez-Iglesias, M.; Galán-Sánchez, F.; et al. Challenging Antimicrobial Susceptibility and Evolution of Resistance (OXA-681) during Treatment of a Long-Term Nosocomial Infection Caused by“named-content genus-species” Pseudomonas aeruginosa ST175 Clone. Antimicrob. Agents Chemother. 2019, 63, e01110-19. [Google Scholar] [CrossRef] [Green Version]
- Fraile-Ribot, P.A.; Cabot, G.; Mulet, X.; Perianez, L.; Martin-Pena, M.L.; Juan, C.; Perez, J.L.; Oliver, A. Mechanisms leading to in vivo ceftolozane/tazobactam resistance development during the treatment of infections caused by MDR Pseudomonas aeruginosa. J. Antimicrob. Chemother. 2018, 73, 658–663. [Google Scholar] [CrossRef] [Green Version]
- Fraile-Ribot, P.A.; Mulet, X.; Cabot, G.; del Barrio-Tofiño, E.; Juan, C.; Pérez, J.L.; Oliver, A. In vivo emergence of resistance to novel cephalosporin–β-lactamase inhibitor combinations through the duplication of amino acid D149 from OXA-2 β-Lactamase (OXA-539) in Sequence Type 235 Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2017, 61, e01117-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akhter, S.; Lund, B.A.; Ismael, A.; Langer, M.; Isaksson, J.; Christopeit, T.; Leiros, H.S.; Bayer, A. A focused fragment library targeting the antibiotic resistance enzyme - Oxacillinase-48: Synthesis, structural evaluation and inhibitor design. Eur. J. Med. Chem. 2018, 145, 634–648. [Google Scholar] [CrossRef] [Green Version]
- Taylor, D.M.; Anglin, J.; Park, S.; Ucisik, M.N.; Faver, J.C.; Simmons, N.; Jin, Z.; Palaniappan, M.; Nyshadham, P.; Li, F.; et al. Identifying Oxacillinase-48 Carbapenemase Inhibitors Using DNA-Encoded Chemical Libraries. ACS Infect. Dis. 2020, 6, 1214–1227. [Google Scholar] [CrossRef]
- Lund, B.A.; Christopeit, T.; Guttormsen, Y.; Bayer, A.; Leiros, H.K. Screening and design of inhibitor scaffolds for the antibiotic resistance oxacillinase-48 (OXA-48) through surface plasmon resonance screening. J. Med. Chem. 2016, 59, 5542–5554. [Google Scholar] [CrossRef]
- Drawz, S.M.; Papp-Wallace, K.M.; Bonomo, R.A. New β-lactamase inhibitors: A therapeutic renaissance in an MDR world. Antimicrob. Agents Chemother. 2014, 58, 1835–1846. [Google Scholar] [CrossRef] [Green Version]
- Muegge, I. Selection criteria for drug-like compounds. Med. Res. Rev. 2003, 23, 302–321. [Google Scholar] [CrossRef]
- Metz, J.T.; Huth, J.R.; Hajduk, P.J. Enhancement of chemical rules for predicting compound reactivity towards protein thiol groups. J. Comput. Aided Mol. Des. 2007, 21, 139–144. [Google Scholar] [CrossRef]
- Walters, W.P.; Murcko, M.A. Prediction of ‘drug-likeness’. Adv. Drug Deliv. Rev. 2002, 54, 255–271. [Google Scholar] [CrossRef]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (pains) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [Green Version]
- Roche, O.; Schneider, P.; Zuegge, J.; Guba, W.; Kansy, M.; Alanine, A.; Bleicher, K.; Danel, F.; Gutknecht, E.-M.; Rogers-Evans, M.; et al. Development of a virtual screening method for identification of “frequent hitters” in compound libraries. J. Med. Chem. 2002, 45, 137–142. [Google Scholar] [CrossRef]
- Bemis, G.W.; Murcko, M.A. The properties of known drugs. 1. Molecular frameworks. J. Med. Chem. 1996, 39, 2887–2893. [Google Scholar] [CrossRef]
- Heller, S.R.; McNaught, A.; Pletnev, I.; Stein, S.; Tchekhovskoi, D. InChI, the IUPAC International chemical identifier. J. Cheminform. 2015, 7, 23. [Google Scholar] [CrossRef] [Green Version]
- Lipkin, M.J.; Stevens, A.P.; Livingstone, D.J.; Harris, C.J. How large does a compound screening collection need to be? Comb. Chem. High Throughput Screen. 2008, 11, 482–493. [Google Scholar] [CrossRef]
- Nilakantan, R.; Immermann, F.; Haraki, K. A novel approach to combinatorial library design. Comb. Chem. High Throughput Screen. 2002, 5, 105–110. [Google Scholar] [CrossRef]
- González-Medina, M.; Prieto-Martínez, F.D.; Owen, J.R.; Medina-Franco, J.L. Consensus Diversity Plots: A global diversity analysis of chemical libraries. J. Cheminform. 2016, 8, 63. [Google Scholar] [CrossRef] [Green Version]
- Lipkus, A.H.; Yuan, Q.; Lucas, K.A.; Funk, S.A.; Bartelt, W.F.; Schenck, R.J.; Trippe, A.J. Structural Diversity of Organic Chemistry. A Scaffold Analysis of the CAS Registry. J. Org. Chem. 2008, 73, 4443–4451. [Google Scholar] [CrossRef] [Green Version]
- Shang, J.; Sun, H.; Liu, H.; Chen, F.; Tian, S.; Pan, P.; Li, D.; Kong, D.; Hou, T. Comparative analyses of structural features and scaffold diversity for purchasable compound libraries. J. Cheminform. 2017, 9, 25. [Google Scholar] [CrossRef]
- Krier, M.; Bret, G.; Rognan, D. Assessing the scaffold diversity of screening libraries. J. Chem. Inf. Model. 2006, 46, 512–524. [Google Scholar] [CrossRef]
- Medina-Franco, J.L.; Martínez-Mayorga, K.; Bender, A.; Scior, T. Scaffold diversity analysis of compound data sets using an entropy-based measure. QSAR Comb. Sci. 2009, 28, 1551–1560. [Google Scholar] [CrossRef]
- Goktug, A.N.; Chai, S.C.; Chen, T. Data analysis approaches in high throughput screening. In Drug Discovery; IntechOpen Limited: London, UK, 2013; pp. 201–226. [Google Scholar]
- Epi-Absorbance Primary Biochemical High throughput Screening Assay to Identify Inhibitors of IMP-1 Metallo-Beta-Lactamase. Available online: https://pubchem.ncbi.nlm.nih.gov/bioassay/1556 (accessed on 14 June 2021).
- Minond, D.; Saldanha, S.A.; Spicer, T.; Qin, L.; Mercer, B.A.; Roush, W.R.; Hodder, P. HTS Assay for Discovery of Novel Metallo-Beta-lactamase (MBL) Inhibitors. In Probe Reports from the NIH Molecular Libraries Program; Bethesda: Rockville, MD, USA, 2010. [Google Scholar]
- Spicer, T.; Minond, D.; Enogieru, I.; Saldanha, S.A.; Allais, C.; Liu, Q.; Mercer, B.A.; Roush, W.R.; Hodder, P. ML302, a Novel Beta-lactamase (BLA) Inhibitor. In Probe Reports from the NIH Molecular Libraries Program; Bethesda: Rockville, MD, USA, 2010. [Google Scholar]
- Zhang, J.H.; Chung, T.D.; Oldenburg, K.R. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J. Biomol. Screen. 1999, 4, 67–73. [Google Scholar] [CrossRef]
- Hopkins, A.L.; Keserü, G.M.; Leeson, P.D.; Rees, D.C.; Reynolds, C.H. The role of ligand efficiency metrics in drug discovery. Nat. Rev. Drug Discov. 2014, 13, 105–121. [Google Scholar] [CrossRef]
- Gavara, L.; Verdirosa, F.; Legru, A.; Mercuri, P.S.; Nauton, L.; Sevaille, L.; Feller, G.; Berthomieu, D.; Sannio, F.; Marcoccia, F.; et al. 4-(N-Alkyl- and -Acyl-amino)-1,2,4-triazole-3-thione analogs as metallo-beta-lactamase inhibitors: Impact of 4-linker on potency and spectrum of inhibition. Biomolecules 2020, 10, 1094. [Google Scholar] [CrossRef]
- Linciano, P.; Gianquinto, E.; Montanari, M.; Maso, L.; Bellio, P.; Cebrian-Sastre, E.; Celenza, G.; Blazquez, J.; Cendron, L.; Spyrakis, F.; et al. 4-Amino-1,2,4-triazole-3-thione as a Promising Scaffold for the Inhibition of Serine and Metallo-beta-Lactamases. Pharmaceuticals 2020, 13, 52. [Google Scholar] [CrossRef] [Green Version]
- Olsen, L.; Jost, S.; Adolph, H.W.; Pettersson, I.; Hemmingsen, L.; Jørgensen, F.S. New leads of metallo-beta-lactamase inhibitors from structure-based pharmacophore design. Bioorg. Med. Chem. 2006, 14, 2627–2635. [Google Scholar] [CrossRef]
- Sevaille, L.; Gavara, L.; Bebrone, C.; De Luca, F.; Nauton, L.; Achard, M.; Mercuri, P.; Tanfoni, S.; Borgianni, L.; Guyon, C.; et al. 1,2,4-Triazole-3-thione Compounds as Inhibitors of Dizinc Metallo-β-lactamases. ChemMedChem 2017, 12, 972–985. [Google Scholar] [CrossRef]
- Craig, P.N. Interdependence between physical parameters and selection of substituent groups for correlation studies. J. Med. Chem. 1971, 14, 680–684. [Google Scholar] [CrossRef]
- Vercheval, L.; Bauvois, C.; di Paolo, A.; Borel, F.; Ferrer, J.L.; Sauvage, E.; Matagne, A.; Frère, J.M.; Charlier, P.; Galleni, M.; et al. Three factors that modulate the activity of class D β-lactamases and interfere with the post-translational carboxylation of Lys70. Biochem. J. 2010, 432, 495–504. [Google Scholar] [CrossRef]
- Golemi, D.; Maveyraud, L.; Vakulenko, S.; Samama, J.-P.; Mobashery, S. Critical involvement of a carbamylated lysine in catalytic function of class D β-lactamases. Proc. Natl. Acad. Sci. USA 2001, 98, 14280–14285. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.A.; Stewart, N.K.; Toth, M.; Vakulenko, S.B. Structural Insights into the Mechanism of Carbapenemase Activity of the OXA-48 β-Lactamase. Antimicrob. Agents Chemother. 2019, 63, e01202-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lahiri, S.D.; Mangani, S.; Jahić, H.; Benvenuti, M.; Durand-Reville, T.F.; De Luca, F.; Ehmann, D.E.; Rossolini, G.M.; Alm, R.A.; Docquier, J.D. Molecular basis of selective inhibition and slow reversibility of avibactam against class D carbapenemases: A structure-guided study of OXA-24 and OXA-48. ACS Chem. Biol. 2015, 10, 591–600. [Google Scholar] [CrossRef]
- Ombrato, R.; Garofalo, B.; Prati, F.; Magaro, G.; Buonfiglio, R. Preparation of 5- or 7-Azaindazoles as β-Lactamase Inhibitors. WO2020178316A1, 10 September 2020. [Google Scholar]
- Aktaş, Z.; Kayacan, C.; Oncul, O. In vitro activity of avibactam (NXL104) in combination with β-lactams against Gram-negative bacteria, including OXA-48 β-lactamase-producing Klebsiella pneumoniae. Int. J. Antimicrob. Agents 2012, 39, 86–89. [Google Scholar] [CrossRef] [PubMed]
- Docquier, J.-D.; Calderone, V.; De Luca, F.; Benvenuti, M.; Giuliani, F.; Bellucci, L.; Tafi, A.; Nordmann, P.; Botta, M.; Rossolini, G.M.; et al. Crystal Structure of the OXA-48 β-Lactamase Reveals Mechanistic Diversity among Class D Carbapenemases. Chem. Biol. 2009, 16, 540–547. [Google Scholar] [CrossRef]
- Clinical and Laboratory Standards Institute (2017). Performance Standards for Antimicrobial Susceptibility Testing; CLSI Twenty-Seventh Edition M100; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 1898. [Google Scholar]
- Clinical and Laboratory Standards Institute. Methods for Dilution of Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standard, 10th ed.; CLSI Document M07-A10; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2015. [Google Scholar]
- Zgurskaya, H.I.; Lopez, C.A.; Gnanakaran, S. Permeability Barrier of Gram-Negative Cell Envelopes and Approaches to Bypass It. ACS Infect. Dis. 2015, 1, 512–522. [Google Scholar] [CrossRef] [Green Version]
- O’Shea, R.; Moser, H.E. Physicochemical properties of antibacterial compounds: Implications for drug discovery. J. Med. Chem. 2008, 51, 2871–2878. [Google Scholar] [CrossRef]
- Nociari, M.M.; Shalev, A.; Benias, P.; Russo, C. A novel one-step, highly sensitive fluorometric assay to evaluate cell-mediated cytotoxicity. J. Immunol. Methods 1998, 213, 157–167. [Google Scholar] [CrossRef]
- Pipeline Pilot 9.5. Available online: http://accelrys.com/ (accessed on 5 April 2016).
- Bochevarov, A.D.; Harder, E.; Hughes, T.F.; Greenwood, J.R.; Braden, D.A.; Philipp, D.M.; Rinaldo, D.; Halls, M.D.; Zhang, J.; Friesner, R.A. Jaguar: A high-performance quantum chemistry software program with strengths in life and materials sciences. Int. J. Quantum Chem. 2013, 113, 2110–2142. [Google Scholar] [CrossRef]
- Schrödinger. LigPrep; Schrödinger: New York, NY, USA, 2019. [Google Scholar]
- Schrödinger. MacroModel; Schrödinger: New York, NY, USA, 2019. [Google Scholar]
- Miroux, B.; Walker, J.E. Over-production of proteins in Escherichia coli: Mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J. Mol. Biol. 1996, 260, 289–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Studier, F.W. Stable expression clones and auto-induction for protein production in E. coli. Methods Mol. Biol. 2014, 1091, 17–32. [Google Scholar] [CrossRef]
- Leslie, A.G.W. Joint CCP4 and ESF-EAMCB. Newsl. Protein Crystallogr. 1992, 26. [Google Scholar]
- Navaza, J. AMoRe: An automated package for molecular replacement. Acta Crystallogr. Sect. A Found. Adv. 1994, 50, 157–163. [Google Scholar] [CrossRef]
- Brünger, A.T.; Adams, P.D.; Clore, G.M.; DeLano, W.L.; Gros, P.; Grosse-Kunstleve, R.W.; Jiang, J.S.; Kuszewski, J.; Nilges, M.; Pannu, N.S.; et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. Sect. D Biol. Crystallogr. 1998, 54, 905–921. [Google Scholar] [CrossRef]
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef] [Green Version]
- Schrödinger. Epik; Schrödinger LLC: New York, NY, USA, 2017. [Google Scholar]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Zuck, P.; O’Donnell, G.T.; Cassaday, J.; Chase, P.; Hodder, P.; Strulovici, B.; Ferrer, M. Miniaturization of absorbance assays using the fluorescent properties of white microplates. Anal. Biochem. 2005, 342, 254–259. [Google Scholar] [CrossRef] [PubMed]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Scaffold | Confirmed Hit | Tot Tested Cmpd a | Hit Rate b | AC50 (µM) c | |||||
|---|---|---|---|---|---|---|---|---|---|
| Most Active | ≤5 | 5–10 | 10–30 | 30–60 | >60 | ||||
| SC_2 | 38 | 287 | 13.2% | 0.72 | 4 | 15 | 11 | 8 | |
| SC_4 | 5 | 76 | 6.6% | 45.80 | 1 | 4 | |||
| SC_5 | 2 | 136 | 1.5% | 69.40 | 2 | ||||
| SC_7 | 2 | 37 | 5.4% | 0.99 | 1 | 1 | |||
| SC_9 | 8 | 206 | 3.9% | 31.90 | 4 | 4 | |||
| SC_11 | 6 | 174 | 3.4% | 35.10 | 1 | 5 | |||
| SC_12 | 11 | 24 | 45.8% | 37.10 | 5 | 6 | |||
| SC_15 | 22 | 133 | 16.5% | 29.30 | 1 | 11 | 10 | ||
| SC_16 | 3 | 41 | 7.3% | 62.40 | 3 | ||||
| SC_17 | 24 | 77 | 31.2% | 13.40 | 6 | 5 | 13 | ||
| SC_18 | 10 | 45 | 22.2% | 9.15 | 3 | 3 | 1 | 3 | |
| SC_19 | 4 | 76 | 5.3% | 26.60 | 1 | 1 | 2 | ||
| SC_20 | 4 | 14 | 28.6% | 4.14 | 1 | 1 | 1 | 1 | |
| SC_21 | 2 | 156 | 1.3% | 3.21 | 1 | 1 | |||
| SC_22 | 3 | 401 | 0.7% | 1.81 | 2 | 1 | |||
| SC_23 | 18 | 233 | 7.7% | 20.60 | 2 | 4 | 12 | ||
| SC_24 | 1 | 13 | 7.7% | 373.85 | 1 | ||||
| SC_26 | 1 | 33 | 3.0% | 651.84 | 1 | ||||
| SC_30 | 8 | 105 | 7.6% | 79.50 | 8 | ||||
| SC_31 | 4 | 42 | 9.5% | 21.40 | 1 | 3 | |||
| Compound | Structure | Time [Min] a | OXA-48 AC50 (µM) | |
|---|---|---|---|---|
| Standard Preincubation b | High Dose Preincubation c | |||
| ID1 |  | 0 | 1.73 | 0.63 |
| 60 | 1.93 | 1.09 | ||
| ID2 |  | 0 | 2.31 | 1.01 |
| 60 | 2.44 | 1.45 | ||
| tazobactam | 0 | 0.82 | 0.79 | |
| 90 | 0.26 | 0.097 | ||
| Conc (µM) | ID1 | ID2 | ||
|---|---|---|---|---|
| Vmax ± SE a | Km ± SE a | Vmax ± SE a | Km ± SE a | |
| 0 | 116.8 ± 4.62 | 9.6 ± 1.06 | 116.8 ± 4.62 | 9.6 ± 1.06 |
| 0.126 | 113.4 ± 8.72 | 12.7 ± 2.51 | 110.8 ± 7.80 | 10.7 ± 2.04 |
| 0.632 | 104.0 ± 7.50 | 17.3 ± 2.91 | 106.5 ± 7.88 | 15.0 ± 2.70 |
| 3.16 | 71.9 ± 12.50 | 29.2 ± 10.00 | 84.3 ± 10.91 | 29.2 ± 7.44 |
 | ||||||
| Compound | R1 | R2 | OXA-48 Activity% ± SE a | OXA-48 AC50 (μM) b | Lower 95% CL | Upper 95% CL |
| ID1 |  |  | −92.19 ± 3.86 | 1.14 | 1.03 | 1.27 |
| ID3 | |  | −94.10 ± 0.48 | 0.72 | 0.65 | 0.80 |
| ID4 | |  | −85.80 ± 0.09 | 3.17 | 2.86 | 3.51 |
| ID5 | |  | −88.29 ± 2.58 | 2.88 | 2.57 | 3.24 |
| ID6 | |  | −61.30 ± 0.08 | 15.70 | 14.01 | 17.60 |
| ID7 |  | | −1.24 ± 0.05 | N.A. c | ||
| ID8 | | | −1.78 ± 0.268 | N.A. c | ||
| ID9 |  | | −23.84 ± 1.45 | 35.60 | 32.03 | 39.64 |
| ID10 | | | −6.64 ± 1.05 | N.A. c | ||
| ID11 |  | | −58.40 ± 0.96 | 12.00 | 9.82 | 14.60 |
| ID12 |  | | −1.33 ± 1.30 | N.A. c | ||
| ID13 | | | 0.02 ± 1.18 | N.A. c | ||
| ID14 |  | | −9.43 ±1.35 | N.A. c | ||
| ID15 |  | | 9.13 ± 5.58 | N.A. c | ||
| ID16 |  | | −0.68 ± 2.27 | N.A. c | ||
| ID17 |  | | −6.76 ± 0.02 | N.A. c | ||
| ID18 |  | | −5.55 ± 0.73 | N.A. c | ||
| ID19 | | | −18.85 ± 1.53 | N.A. c | ||
| ID20 |  | | −15.40 ± 1.77 | N.A. c | ||
| ID21 | | | −6.90 ± 0.43 | N.A. c | ||
| ID22 | | | −5.12 ± 0.84 | N.A. c | ||
| ID23 |  | | −29.46 ± 3.40 | 24.80 | 22.26 | 27.57 |
| ID24 |  | | −2.71 ± 0.78 | N.A. c | ||
| ID25 | | | 1.40 ± 1.52 | N.A. c | ||
| ID26 | | | −9.29 ± 1.75 | N.A. c | ||
| ID27 |  | | 1.32 ± 1.03 | N.A. c | ||
| ID28 | | | 1.10 ± 1.56 | N.A.c | ||
| ID29 | | | 0.08 ± 0.16 | N.A. c | ||
| tazobactam | 0.84 | 0.78 | 0.91 | |||
 | ||||||
| Compound | R1 | R2 | OXA-48 Activity% ± SE a | OXA-48 AC50 (μM) b | Lower 95% CL | Upper 95% CL |
| ID2 |  |  | −93.92 ± 0.54 | 0.99 | 0.80 | 1.22 |
| ID30 | | | −13.17 ± 1.07 | 248.59 | 176.58 | 349.95 |
| ID31 | |  | −25.86 ± 0.57 | N.C. c | ||
| 1 | |  | −93.15 ± 0.46 | 0.92 | 0.86 | 0.99 |
| 2 | |  | −86.38 ± 0.35 | 1.48 | 1.35 | 1.62 |
| 3 | |  | −49.91 ± 1.58 | 16.79 | 15.41 | 18.29 |
| 4 | |  | −87.36 ± 0.60 | 4.78 | 4.46 | 5.13 |
| 5 | |  | −55.90 ± 0.64 | 16.21 | 14.64 | 17.95 |
| 6 | |  | −46.92 ± 1.73 | 24.38 | 21.88 | 27.15 |
| 7 | |  | −28.26 ± 0.31 | 51.21 | 45.08 | 58.17 |
| 8 | |  | −64.34 ± 1.55 | 13.69 | 12.23 | 15.33 |
| 9 | |  | −14.72 ± 2.19 | 63.77 | 58.40 | 69.63 |
| 10 |  | | −10.57 ± 1.45 | >100 | ||
| 11 |  | | −12.66 ± 0.87 | >100 | ||
| 12 |  | | −4.62 ± 1.13 | >100 | ||
| 13 |  | | −9.28 ± 1.06 | >100 | ||
| 14 |  | | −0.74 ± 0.97 | N.C. c | ||
| tazobactam | 0.84 | 0.78 | 0.91 | |||
| Bacterial Strain | Strain Code | β-lactam | MIC β-lactam (µg/mL) | MIC (µg/mL) (β-lactam + BLI) | |||
|---|---|---|---|---|---|---|---|
| Avibactam (4 µg/mL) | Tazobactam (32 µg/mL) | ID2 (32 µg/mL) | ID3 (32 µg/mL) | ||||
| E.coli | ATCC25922 | AMP | 4 | 2 | 4 | 4 | 8 |
| IMP | 0.12 | 0.06 | 0.12 | 0.12 | 0.12 | ||
| no | - | - | - | >32 | >32 | ||
| E.coli | ATCC BAA-2523 | AMP | >512 | 16 | 256 | >512 | >512 |
| IMP | 2 | ≤0.03 | 0.12 | 0.5 | 1 | ||
| no | - | - | >32 | >32 | >32 | ||
| K.pneumoniae | ATCC BAA-2524 | AMP | >512 | 128 | >512 | >512 | >512 |
| IMP | 4 | 0.5 | 1 | 2 | 2 | ||
| no | - | - | >32 | >32 | >32 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garofalo, B.; Prati, F.; Buonfiglio, R.; Coletta, I.; D’Atanasio, N.; Molteni, A.; Carettoni, D.; Wanke, V.; Pochetti, G.; Montanari, R.; et al. Discovery of Novel Chemical Series of OXA-48 β-Lactamase Inhibitors by High-Throughput Screening. Pharmaceuticals 2021, 14, 612. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14070612
Garofalo B, Prati F, Buonfiglio R, Coletta I, D’Atanasio N, Molteni A, Carettoni D, Wanke V, Pochetti G, Montanari R, et al. Discovery of Novel Chemical Series of OXA-48 β-Lactamase Inhibitors by High-Throughput Screening. Pharmaceuticals. 2021; 14(7):612. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14070612
Chicago/Turabian StyleGarofalo, Barbara, Federica Prati, Rosa Buonfiglio, Isabella Coletta, Noemi D’Atanasio, Angela Molteni, Daniele Carettoni, Valeria Wanke, Giorgio Pochetti, Roberta Montanari, and et al. 2021. "Discovery of Novel Chemical Series of OXA-48 β-Lactamase Inhibitors by High-Throughput Screening" Pharmaceuticals 14, no. 7: 612. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14070612