
Dysregulation of Blood-Brain Barrier and Exacerbated Inflammatory Response in Cx47-Deficient Mice after Induction of EAE

Abstract
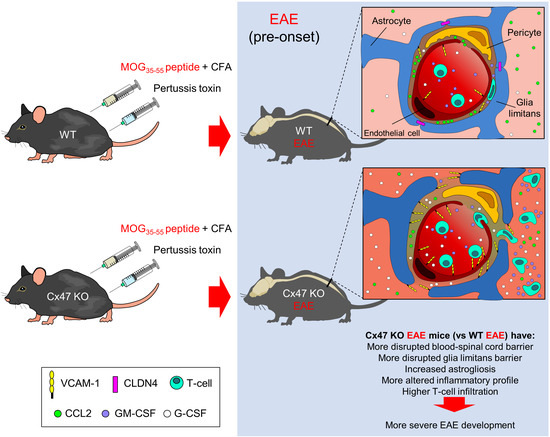
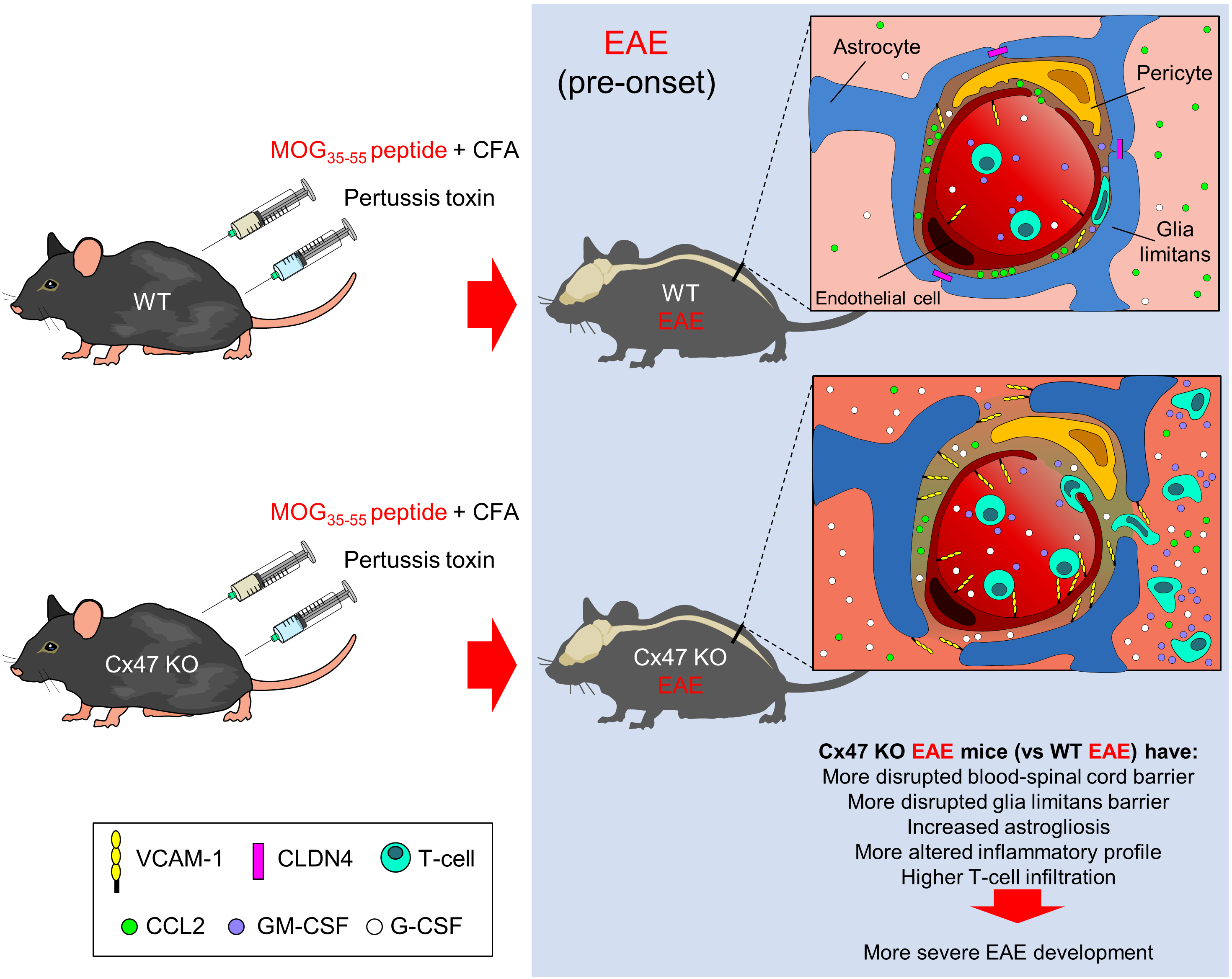
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
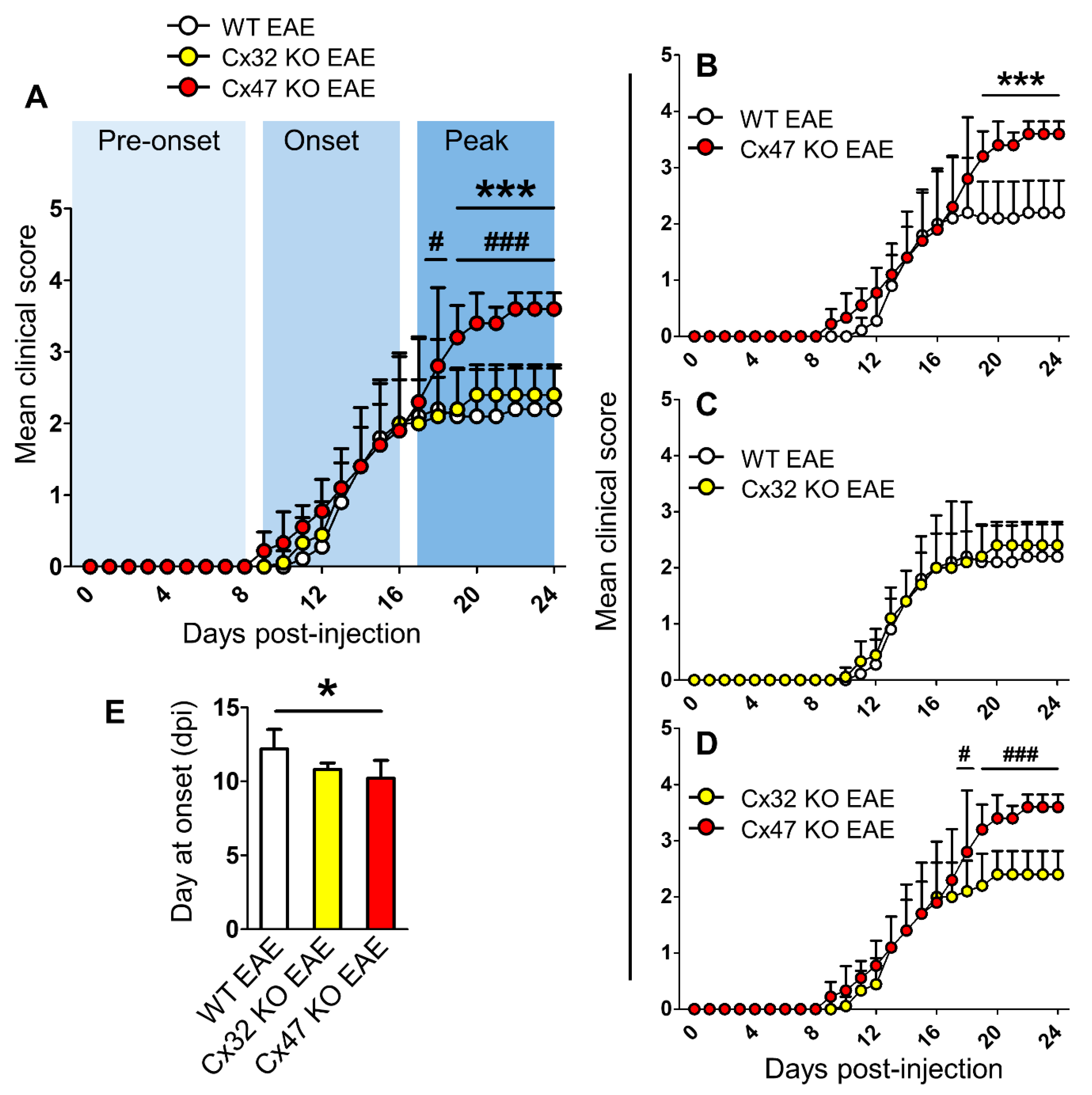
2.1. Cx47 KO EAE Mice Experience an Earlier EAE Onset and a More Severe EAE Disease Course
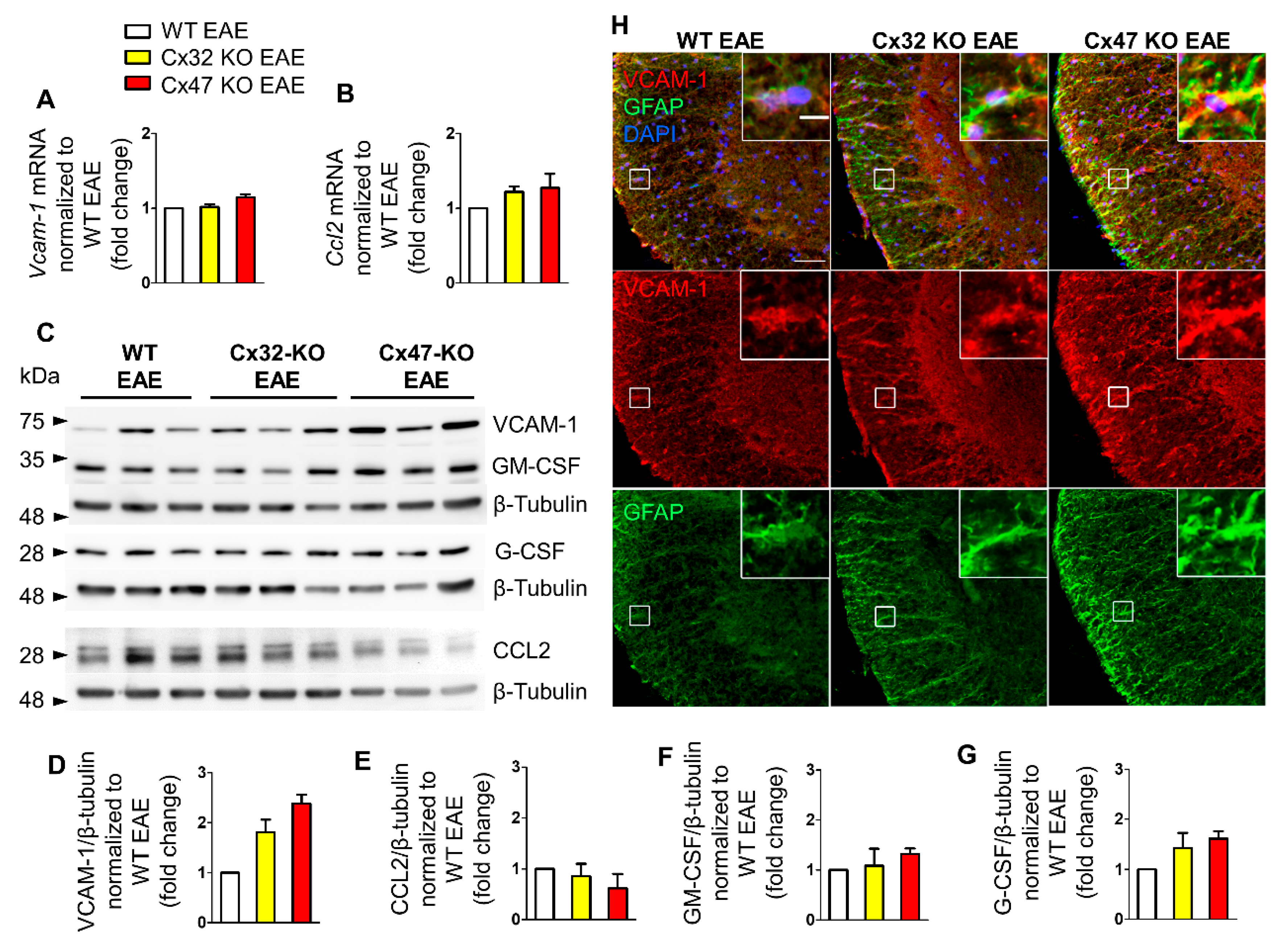
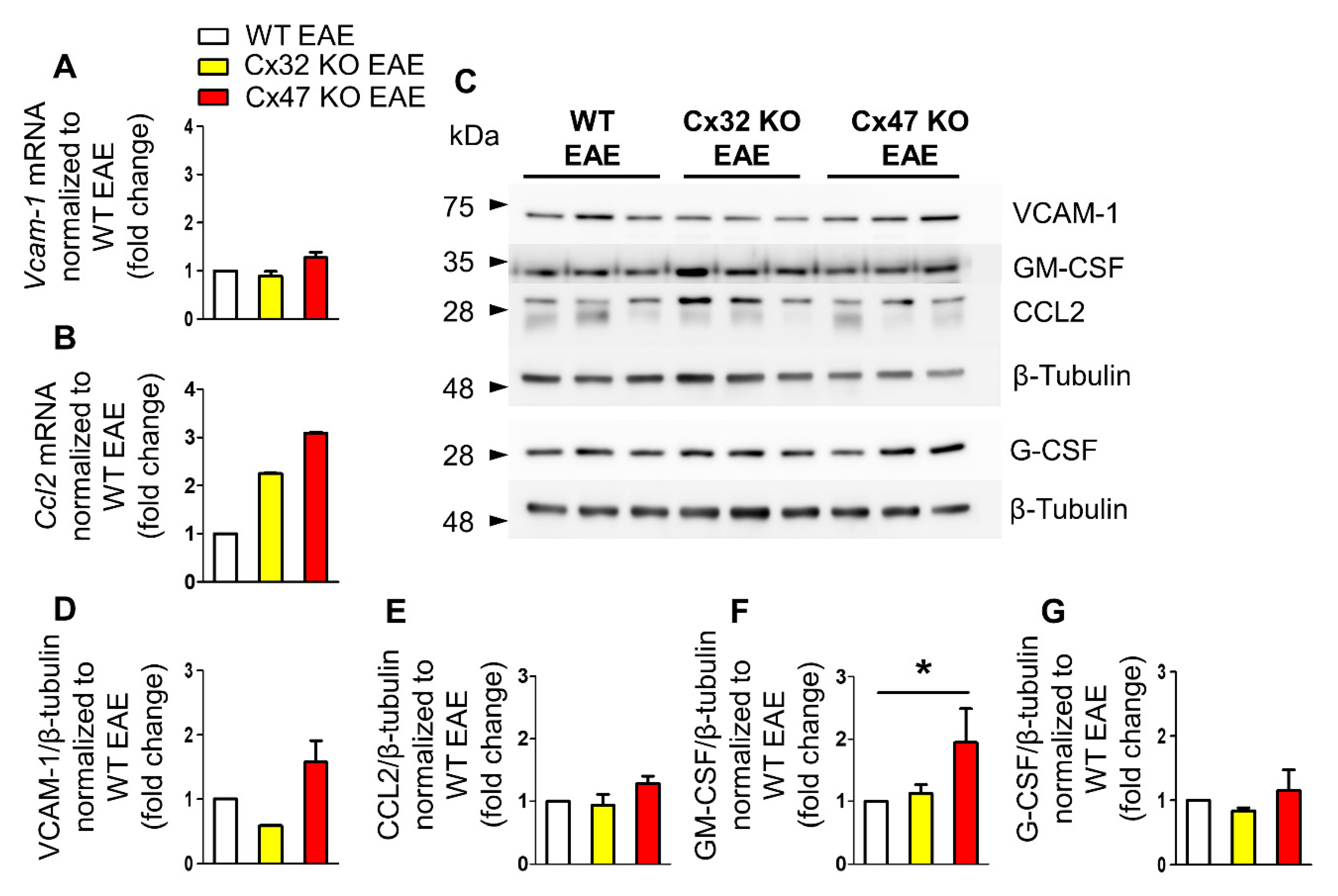
2.2. Cx47 KO EAE Mice Display a Trend of Increased Vcam-1 Translation at 7 dpi
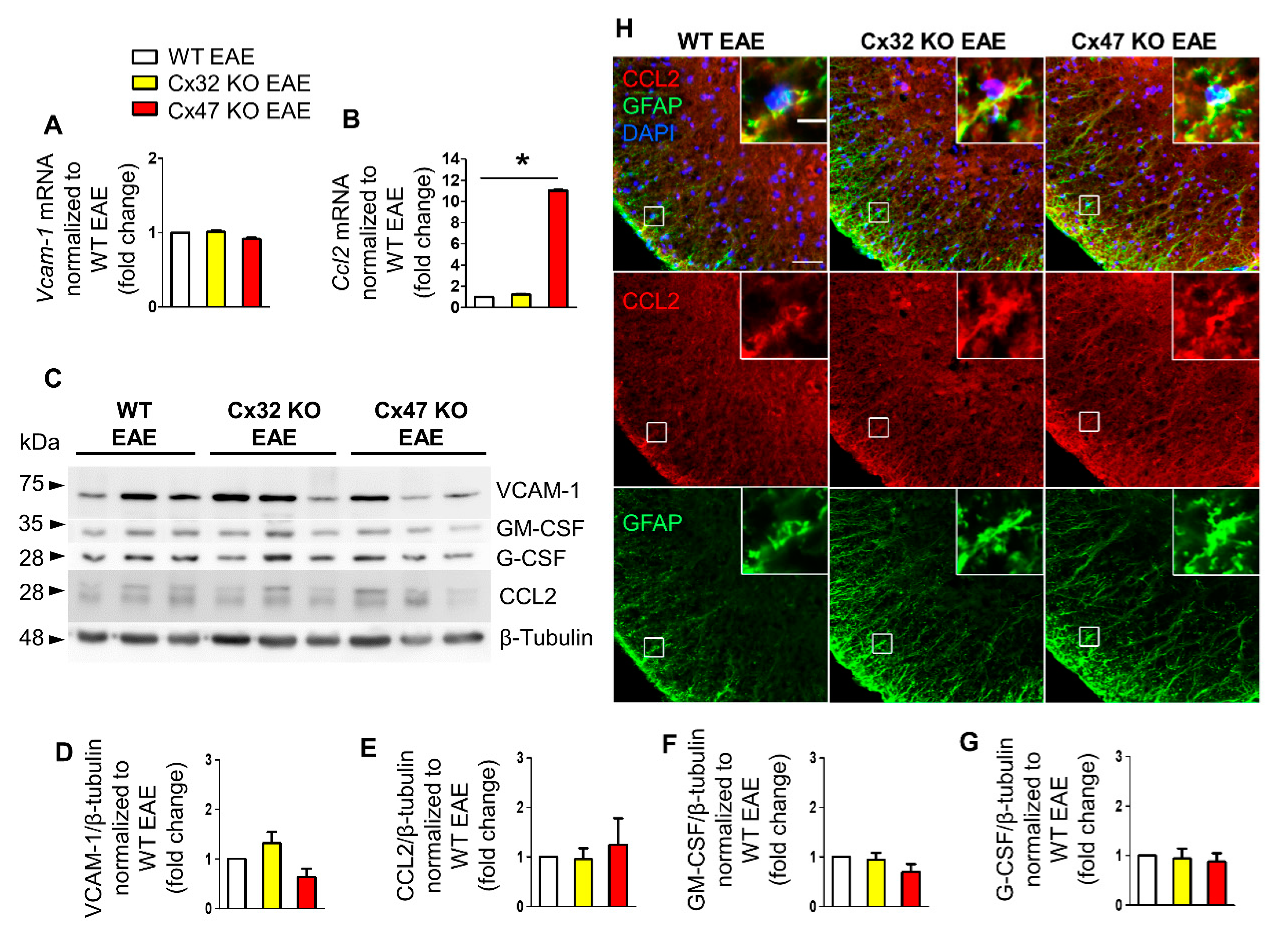
2.3. Increased Ccl2 Transcription in Cx47 KO EAE Mice at 12 dpi
2.4. Increased Gm-Csf Translation in Cx47 KO EAE Mice at 24 dpi
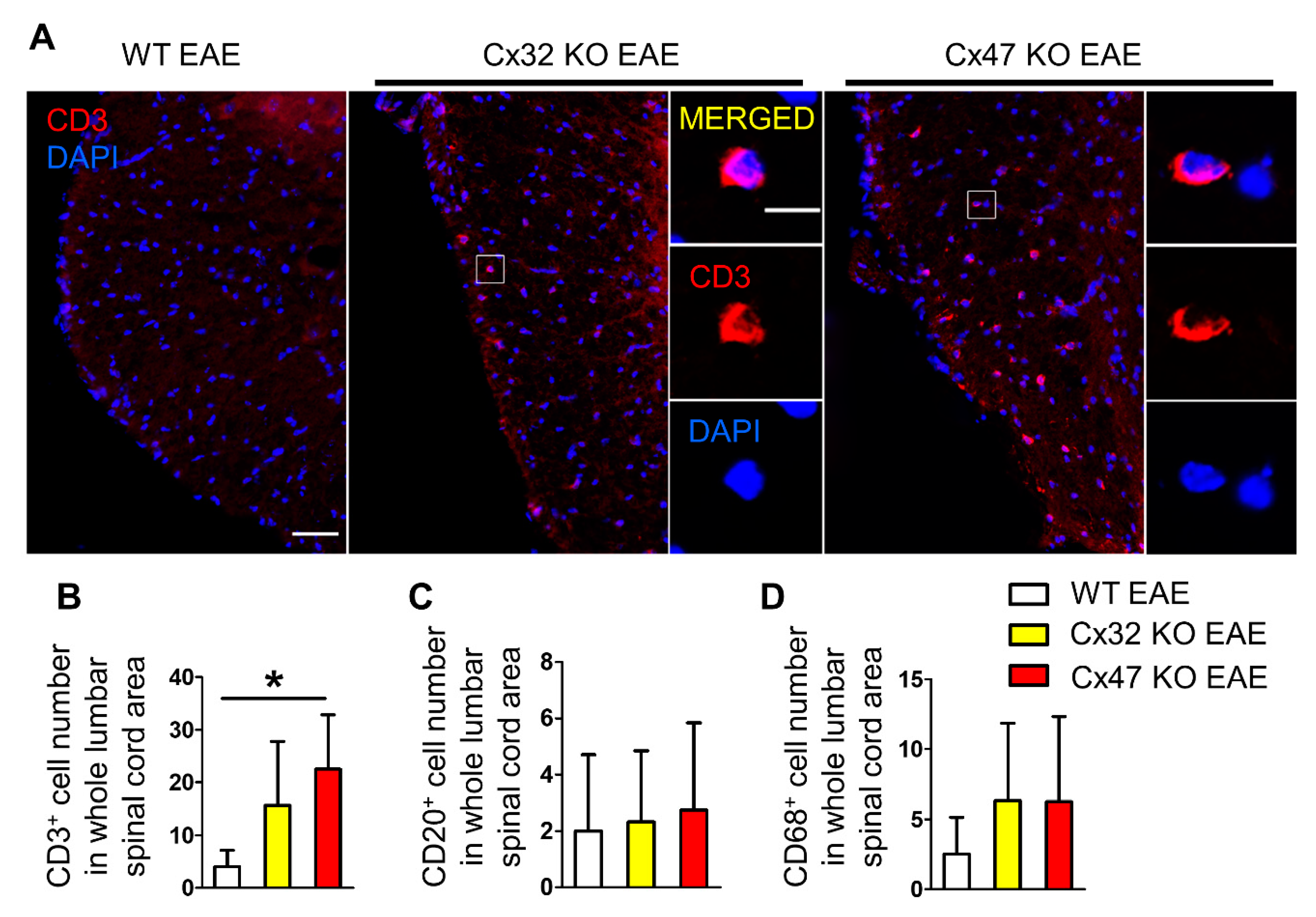
2.5. T-Cell Infiltration Is Higher in the Spinal Cords of 7 dpi Cx47 KO EAE Mice
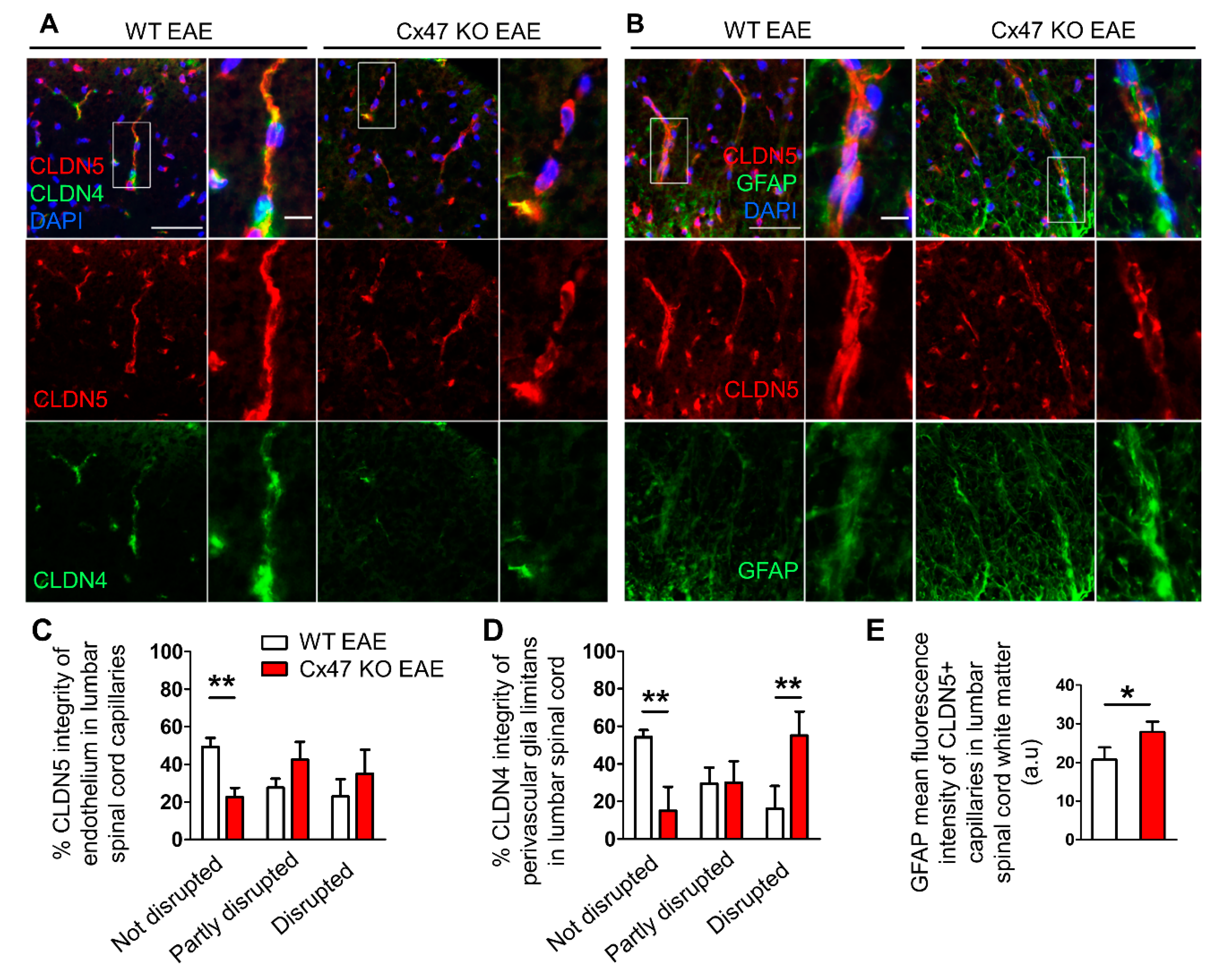
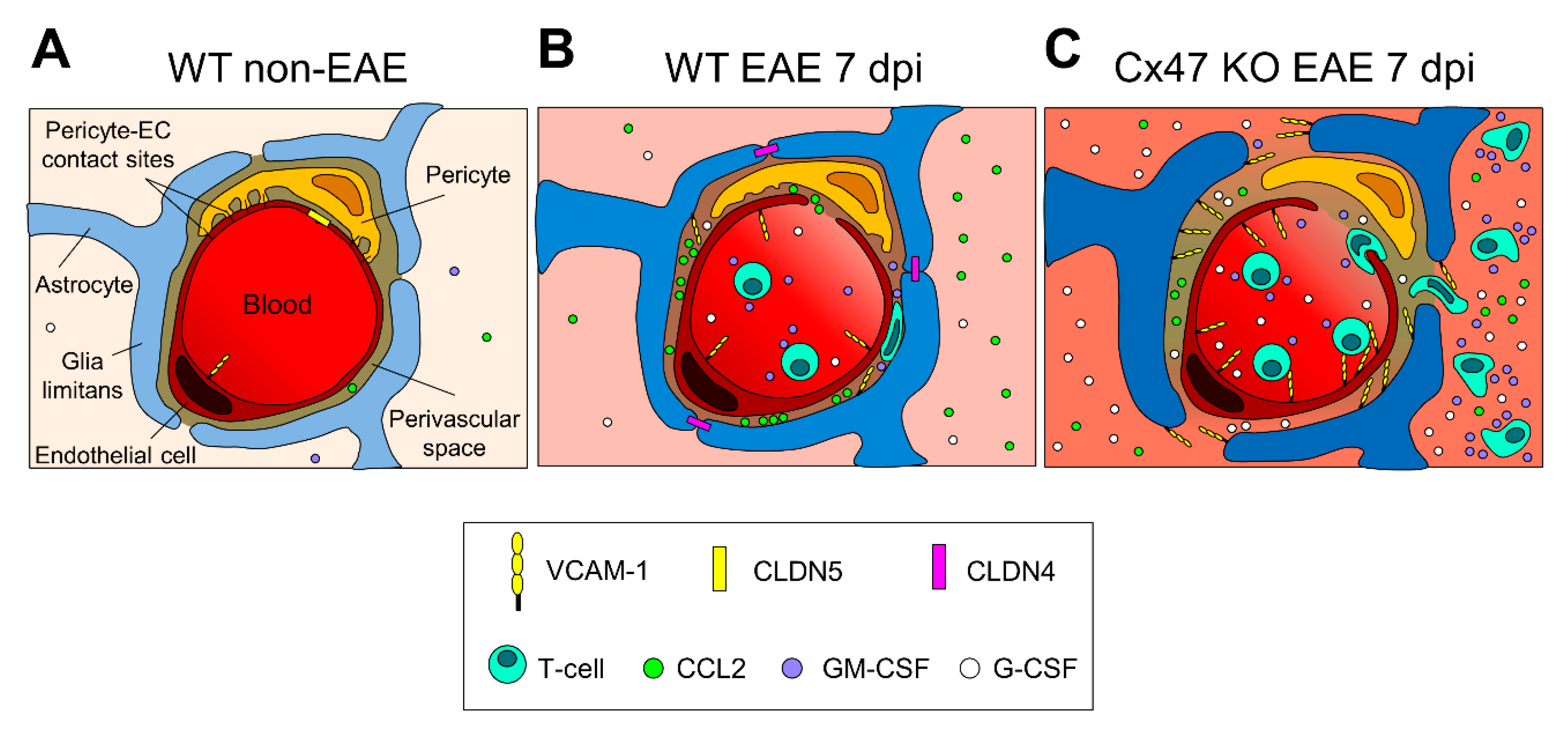
2.6. More Severe BSCB and Glia Limitans Disruption in 7 dpi Cx47 KO EAE Mice
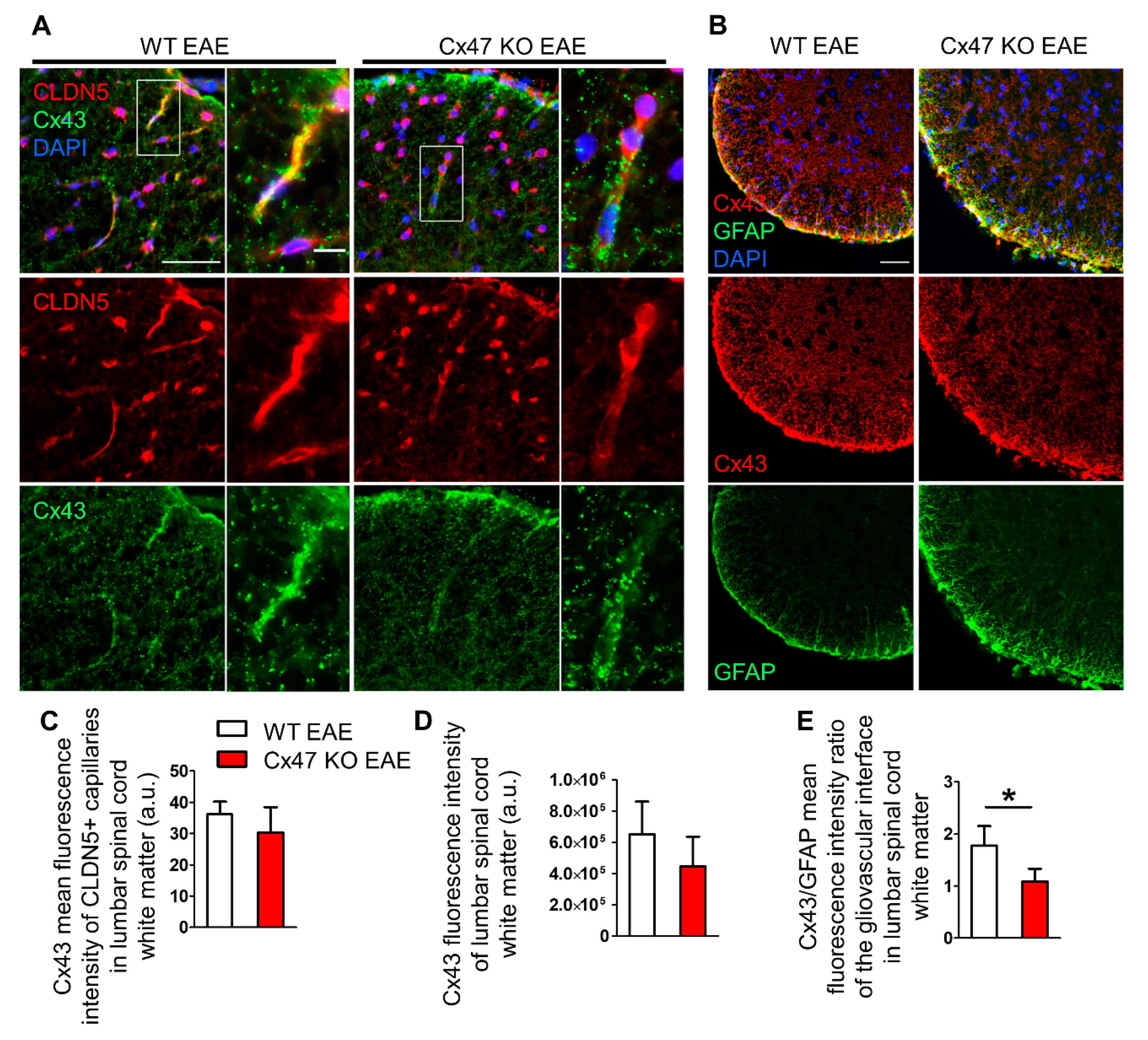
2.7. Cx47 KO EAE Mice Experience a Non-Significant Reduction in Cx43 at 7 dpi
3. Discussion
4. Materials and Methods
4.1. Experimental Animals
4.2. EAE Induction and Disease Assessment
4.3. Tissue Processing and Immunofluorescence Staining
4.4. Immunoblot Analysis
4.5. RNA Extraction and Reverse Transcription PCR (RT-qPCR)
4.6. Statistical Analyses
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wallin, M.T.; Culpepper, W.J.; Nichols, E.; Bhutta, Z.A.; Gebrehiwot, T.T.; Hay, S.I.; Khalil, I.A.; Krohn, K.J.; Liang, X.; Naghavi, M.; et al. Global, regional, and national burden of multiple sclerosis 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 269–285. [Google Scholar] [CrossRef] [Green Version]
- Robinson, A.P.; Harp, C.T.; Noronha, A.; Miller, S.D. The experimental autoimmune encephalomyelitis (EAE) model of MS: Utility for understanding disease pathophysiology and treatment. Handb. Clin. Neurol. 2014, 122, 173–189. [Google Scholar]
- Simmons, S.B.; Pierson, E.R.; Lee, S.Y.; Goverman, J.M. Modeling the heterogeneity of multiple sclerosis in animals. Trends Immunol. 2013, 34, 410–422. [Google Scholar] [CrossRef] [Green Version]
- Constantinescu, C.S.; Farooqi, N.; O’Brien, K.; Gran, B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br. J. Pharmacol. 2011, 164, 1079–1106. [Google Scholar] [CrossRef]
- Mei, F.; Lehmann-Horn, K.; Shen, Y.A.; Rankin, K.A.; Stebbins, K.J.; Lorrain, D.S.; Pekarek, K.; Sagan, S.A.; Xiao, L.; Teuscher, C.; et al. Accelerated remyelination during inflammatory demyelination prevents axonal loss and improves functional recovery. eLife 2016, 5, e18246. [Google Scholar] [CrossRef]
- Yandamuri, S.S.; Lane, T.E. Imaging Axonal Degeneration and Repair in Preclinical Animal Models of Multiple Sclerosis. Front. Immunol. 2016, 7, 189. [Google Scholar] [CrossRef] [Green Version]
- Ortiz, G.G.; Pacheco-Moises, F.P.; Macias-Islas, M.A.; Flores-Alvarado, L.J.; Mireles-Ramirez, M.A.; Gonzalez-Renovato, E.D.; Hernandez-Navarro, V.E.; Sanchez-Lopez, A.L.; Alatorre-Jimenez, M.A. Role of the blood-brain barrier in multiple sclerosis. Arch. Med. Res. 2014, 45, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Floris, S.; Blezer, E.L.; Schreibelt, G.; Dopp, E.; Van der Pol, S.M.; Schadee-Eestermans, I.L.; Nicolay, K.; Dijkstra, C.D.; De Vries, H.E. Blood-brain barrier permeability and monocyte infiltration in experimental allergic encephalomyelitis: A quantitative MRI study. Brain 2004, 127, 616–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maglione, M.; Tress, O.; Haas, B.; Karram, K.; Trotter, J.; Willecke, K.; Kettenmann, H. Oligodendrocytes in mouse corpus callosum are coupled via gap junction channels formed by connexin47 and connexin32. Glia 2010, 58, 1104–1117. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Gloor, G.B.; Bai, D. The distribution and functional properties of Pelizaeus-Merzbacher-like disease-linked Cx47 mutations on Cx47/Cx47 homotypic and Cx47/Cx43 heterotypic gap junctions. Biochem. J. 2013, 452, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Kamasawa, N.; Sik, A.; Morita, M.; Yasumura, T.; Davidson, K.G.; Nagy, J.I.; Rash, J.E. Connexin-47 and connexin-32 in gap junctions of oligodendrocyte somata, myelin sheaths, paranodal loops and Schmidt-Lanterman incisures: Implications for ionic homeostasis and potassium siphoning. Neuroscience 2005, 136, 65–86. [Google Scholar] [CrossRef] [Green Version]
- Kleopa, K.A. The role of gap junctions in Charcot-Marie-Tooth disease. J. Neurosci. 2011, 31, 17753–17760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wasseff, S.K.; Scherer, S.S. Cx32 and Cx47 mediate oligodendrocyte:astrocyte and oligodendrocyte:oligodendrocyte gap junction coupling. Neurobiol. Dis. 2011, 42, 506–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srinivas, M.; Verselis, V.K.; White, T.W. Human diseases associated with connexin mutations. Biochim. Biophys. Acta 2018, 1860, 192–201. [Google Scholar] [CrossRef]
- Markoullis, K.; Sargiannidou, I.; Schiza, N.; Hadjisavvas, A.; Roncaroli, F.; Reynolds, R.; Kleopa, K.A. Gap junction pathology in multiple sclerosis lesions and normal-appearing white matter. Acta Neuropathol. 2012, 123, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Menichella, D.M.; Goodenough, D.A.; Sirkowski, E.; Scherer, S.S.; Paul, D.L. Connexins are critical for normal myelination in the CNS. J. Neurosci. 2003, 23, 5963–5973. [Google Scholar] [CrossRef]
- Nelles, E.; Butzler, C.; Jung, D.; Temme, A.; Gabriel, H.D.; Dahl, U.; Traub, O.; Stumpel, F.; Jungermann, K.; Zielasek, J.; et al. Defective propagation of signals generated by sympathetic nerve stimulation in the liver of connexin32-deficient mice. Proc. Natl. Acad. Sci. USA 1996, 93, 9565–9570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papaneophytou, C.P.; Georgiou, E.; Karaiskos, C.; Sargiannidou, I.; Markoullis, K.; Freidin, M.M.; Abrams, C.K.; Kleopa, K.A. Regulatory role of oligodendrocyte gap junctions in inflammatory demyelination. Glia 2018, 66, 2589–2603. [Google Scholar] [CrossRef]
- Gibson-Corley, K.N.; Boyden, A.W.; Leidinger, M.R.; Lambertz, A.M.; Ofori-Amanfo, G.P.; Naumann, W.; Goeken, J.A.; Karandikar, N.J. A method for histopathological study of the multifocal nature of spinal cord lesions in murine experimental autoimmune encephalomyelitis. PeerJ 2016, 4, e1600. [Google Scholar] [CrossRef] [Green Version]
- Caravagna, C.; Jaouen, A.; Desplat-Jego, S.; Fenrich, K.K.; Bergot, E.; Luche, H.; Grenot, P.; Rougon, G.; Malissen, M.; Debarbieux, F. Diversity of innate immune cell subsets across spatial and temporal scales in an EAE mouse model. Sci. Rep. 2018, 8, 5146. [Google Scholar] [CrossRef]
- Argaw, A.T.; Gurfein, B.T.; Zhang, Y.; Zameer, A.; John, G.R. VEGF-mediated disruption of endothelial CLN-5 promotes blood-brain barrier breakdown. Proc. Natl. Acad. Sci. USA 2009, 106, 1977–1982. [Google Scholar] [CrossRef] [Green Version]
- Nitta, T.; Hata, M.; Gotoh, S.; Seo, Y.; Sasaki, H.; Hashimoto, N.; Furuse, M.; Tsukita, S. Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J. Cell Biol. 2003, 161, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Horng, S.; Therattil, A.; Moyon, S.; Gordon, A.; Kim, K.; Argaw, A.T.; Hara, Y.; Mariani, J.N.; Sawai, S.; Flodby, P.; et al. Astrocytic tight junctions control inflammatory CNS lesion pathogenesis. J. Clin. Investig. 2017, 127, 3136–3151. [Google Scholar] [CrossRef]
- Mora, P.; Hollier, P.L.; Guimbal, S.; Abelanet, A.; Diop, A.; Cornuault, L.; Couffinhal, T.; Horng, S.; Gadeau, A.P.; Renault, M.A.; et al. Blood-brain barrier genetic disruption leads to protective barrier formation at the Glia Limitans. PLoS Biol. 2020, 18, e3000946. [Google Scholar] [CrossRef]
- Konietzko, U.; Muller, C.M. Astrocytic dye coupling in rat hippocampus: Topography, developmental onset, and modulation by protein kinase C. Hippocampus 1994, 4, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Brand-Schieber, E.; Werner, P.; Iacobas, D.A.; Iacobas, S.; Beelitz, M.; Lowery, S.L.; Spray, D.C.; Scemes, E. Connexin43, the major gap junction protein of astrocytes, is down-regulated in inflamed white matter in an animal model of multiple sclerosis. J. Neurosci. Res. 2005, 80, 798–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simard, M.; Arcuino, G.; Takano, T.; Liu, Q.S.; Nedergaard, M. Signaling at the Gliovascular Interface. J. Neurosci. 2003, 23, 9254. [Google Scholar] [CrossRef]
- Miyamoto, Y.; Torii, T.; Tanoue, A.; Yamauchi, J. VCAM1 acts in parallel with CD69 and is required for the initiation of oligodendrocyte myelination. Nat. Commun. 2016, 7, 13478. [Google Scholar] [CrossRef] [Green Version]
- Carlos, T.M.; Schwartz, B.R.; Kovach, N.L.; Yee, E.; Rosa, M.; Osborn, L.; Chi-Rosso, G.; Newman, B.; Lobb, R.; Rosso, M. Vascular cell adhesion molecule-1 mediates lymphocyte adherence to cytokine-activated cultured human endothelial cells. Blood 1990, 76, 965–970. [Google Scholar] [CrossRef] [PubMed]
- Gimenez, M.A.; Sim, J.E.; Russell, J.H. TNFR1-dependent VCAM-1 expression by astrocytes exposes the CNS to destructive inflammation. J. Neuroimmunol. 2004, 151, 116–125. [Google Scholar] [CrossRef]
- Baron, J.L.; Madri, J.A.; Ruddle, N.H.; Hashim, G.; Janeway, C.A. Surface expression of alpha 4 integrin by CD4 T cells is required for their entry into brain parenchyma. J. Exp. Med. 1993, 177, 57–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, J.W.; Bo, L.; Mork, S.; Chang, A.; Ransohoff, R.M.; Trapp, B.D. VCAM-1-positive microglia target oligodendrocytes at the border of multiple sclerosis lesions. J. Neuropathol. Exp. Neurol. 2002, 61, 539–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitroulis, I.; Alexaki, V.I.; Kourtzelis, I.; Ziogas, A.; Hajishengallis, G.; Chavakis, T. Leukocyte integrins: Role in leukocyte recruitment and as therapeutic targets in inflammatory disease. Pharmacol. Ther. 2015, 147, 123–135. [Google Scholar] [CrossRef] [Green Version]
- Engelhardt, B.; Laschinger, M.; Schulz, M.; Samulowitz, U.; Vestweber, D.; Hoch, G. The development of experimental autoimmune encephalomyelitis in the mouse requires alpha4-integrin but not alpha4beta7-integrin. J. Clin. Investig. 1998, 102, 2096–2105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garton, K.J.; Gough, P.J.; Philalay, J.; Wille, P.T.; Blobel, C.P.; Whitehead, R.H.; Dempsey, P.J.; Raines, E.W. Stimulated shedding of vascular cell adhesion molecule 1 (VCAM-1) is mediated by tumor necrosis factor-alpha-converting enzyme (ADAM 17). J. Biol. Chem. 2003, 278, 37459–37464. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.J.; Mason, J.C.; Lidington, E.A.; Edwards, D.R.; Nuttall, R.K.; Khokha, R.; Knauper, V.; Murphy, G.; Gavrilovic, J. Cytokine stimulated vascular cell adhesion molecule-1 (VCAM-1) ectodomain release is regulated by TIMP-3. Cardiovasc. Res. 2005, 67, 39–49. [Google Scholar] [CrossRef] [Green Version]
- Cook-Mills, J.M.; Marchese, M.E.; Abdala-Valencia, H. Vascular cell adhesion molecule-1 expression and signaling during disease: Regulation by reactive oxygen species and antioxidants. Antioxid. Redox Signal. 2011, 15, 1607–1638. [Google Scholar] [CrossRef] [Green Version]
- Ueki, S.; Kihara, J.; Kato, H.; Ito, W.; Takeda, M.; Kobayashi, Y.; Kayaba, H.; Chihara, J. Soluble vascular cell adhesion molecule-1 induces human eosinophil migration. Allergy 2009, 64, 718–724. [Google Scholar] [CrossRef]
- Tchalla, A.E.; Wellenius, G.A.; Travison, T.G.; Gagnon, M.; Iloputaife, I.; Dantoine, T.; Sorond, F.A.; Lipsitz, L.A. Circulating vascular cell adhesion molecule-1 is associated with cerebral blood flow dysregulation, mobility impairment, and falls in older adults. Hypertension 2015, 66, 340–346. [Google Scholar] [CrossRef] [Green Version]
- Fife, B.T.; Huffnagle, G.B.; Kuziel, W.A.; Karpus, W.J. CC chemokine receptor 2 is critical for induction of experimental autoimmune encephalomyelitis. J. Exp. Med. 2000, 192, 899–905. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, K.J.; Strieter, R.M.; Kunkel, S.L.; Lukacs, N.W.; Karpus, W.J. Acute and relapsing experimental autoimmune encephalomyelitis are regulated by differential expression of the CC chemokines macrophage inflammatory protein-1alpha and monocyte chemotactic protein-1. J. Neuroimmunol. 1998, 92, 98–108. [Google Scholar] [CrossRef]
- Paul, D.; Ge, S.; Lemire, Y.; Jellison, E.R.; Serwanski, D.R.; Ruddle, N.H.; Pachter, J.S. Cell-selective knockout and 3D confocal image analysis reveals separate roles for astrocyte-and endothelial-derived CCL2 in neuroinflammation. J. Neuroinflamm. 2014, 11, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahad, D.; Callahan, M.K.; Williams, K.A.; Ubogu, E.E.; Kivisakk, P.; Tucky, B.; Kidd, G.; Kingsbury, G.A.; Chang, A.; Fox, R.J.; et al. Modulating CCR2 and CCL2 at the blood-brain barrier: Relevance for multiple sclerosis pathogenesis. Brain 2006, 129, 212–223. [Google Scholar] [CrossRef]
- Zhong, L.; Simard, M.J.; Huot, J. Endothelial microRNAs regulating the NF-kappaB pathway and cell adhesion molecules during inflammation. FASEB J. 2018, 32, 4070–4084. [Google Scholar] [CrossRef] [Green Version]
- Khyzha, N.; Khor, M.; DiStefano, P.V.; Wang, L.; Matic, L.; Hedin, U.; Wilson, M.D.; Maegdefessel, L.; Fish, J.E. Regulation of CCL2 expression in human vascular endothelial cells by a neighboring divergently transcribed long noncoding RNA. Proc. Natl. Acad. Sci. USA 2019, 116, 16410–16419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duncker, P.C.; Stoolman, J.S.; Huber, A.K.; Segal, B.M. GM-CSF Promotes Chronic Disability in Experimental Autoimmune Encephalomyelitis by Altering the Composition of Central Nervous System-Infiltrating Cells, but Is Dispensable for Disease Induction. J. Immunol. 2018, 200, 966–973. [Google Scholar] [CrossRef]
- Ponomarev, E.D.; Shriver, L.P.; Maresz, K.; Pedras-Vasconcelos, J.; Verthelyi, D.; Dittel, B.N. GM-CSF production by autoreactive T cells is required for the activation of microglial cells and the onset of experimental autoimmune encephalomyelitis. J. Immunol. 2007, 178, 39–48. [Google Scholar] [CrossRef] [Green Version]
- Rumble, J.M.; Huber, A.K.; Krishnamoorthy, G.; Srinivasan, A.; Giles, D.A.; Zhang, X.; Wang, L.; Segal, B.M. Neutrophil-related factors as biomarkers in EAE and MS. J. Exp. Med. 2015, 212, 23–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, W. G-CSF treatment promotes apoptosis of autoreactive T cells to restrict the inflammatory cascade and accelerate recovery in experimental allergic encephalomyelitis. Exp. Neurol. 2017, 289, 73–84. [Google Scholar] [CrossRef]
- Diederich, K.; Sevimli, S.; Dorr, H.; Kosters, E.; Hoppen, M.; Lewejohann, L.; Klocke, R.; Minnerup, J.; Knecht, S.; Nikol, S.; et al. The role of granulocyte-colony stimulating factor (G-CSF) in the healthy brain: A characterization of G-CSF-deficient mice. J. Neurosci. 2009, 29, 11572–11581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, A.; Kruger, C.; Steigleder, T.; Weber, D.; Pitzer, C.; Laage, R.; Aronowski, J.; Maurer, M.H.; Gassler, N.; Mier, W.; et al. The hematopoietic factor G-CSF is a neuronal ligand that counteracts programmed cell death and drives neurogenesis. J. Clin. Investig. 2005, 115, 2083–2098. [Google Scholar] [CrossRef] [Green Version]
- Aloisi, F.; Carè, A.; Borsellino, G.; Gallo, P.; Rosa, S.; Bassani, A.; Cabibbo, A.; Testa, U.; Levi, G.; Peschle, C. Production of hemolymphopoietic cytokines (IL-6, IL-8, colony-stimulating factors) by normal human astrocytes in response to IL-1 beta and tumor necrosis factor-alpha. J. Immunol. 1992, 149, 2358–2366. [Google Scholar] [PubMed]
- Zhao, L.R.; Navalitloha, Y.; Singhal, S.; Mehta, J.; Piao, C.S.; Guo, W.P.; Kessler, J.A.; Groothuis, D.R. Hematopoietic growth factors pass through the blood-brain barrier in intact rats. Exp. Neurol. 2007, 204, 569–573. [Google Scholar] [CrossRef] [Green Version]
- D’Amelio, F.E.; Smith, M.E.; Eng, L.F. Sequence of tissue responses in the early stages of experimental allergic encephalomyelitis (EAE): Immunohistochemical, light microscopic, and ultrastructural observations in the spinal cord. Glia 1990, 3, 229–240. [Google Scholar] [CrossRef] [Green Version]
- Eng, L.F.; D’Amelio, F.E.; Smith, M.E. Dissociation of GFAP intermediate filaments in EAE: Observations in the lumbar spinal cord. Glia 1989, 2, 308–317. [Google Scholar] [CrossRef]
- Fletcher, J.M.; Lalor, S.J.; Sweeney, C.M.; Tubridy, N.; Mills, K.H. T cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Clin. Exp. Immunol. 2010, 162, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Brown, L.S.; Foster, C.G.; Courtney, J.M.; King, N.E.; Howells, D.W.; Sutherland, B.A. Pericytes and Neurovascular Function in the Healthy and Diseased Brain. Front. Cell Neurosci. 2019, 13, 282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kloc, M.; Kubiak, J.Z.; Li, X.C.; Ghobrial, R.M. Pericytes, microvasular dysfunction, and chronic rejection. Transplantation 2015, 99, 658–667. [Google Scholar] [CrossRef] [Green Version]
- Duz, B.; Oztas, E.; Erginay, T.; Erdogan, E.; Gonul, E. The effect of moderate hypothermia in acute ischemic stroke on pericyte migration: An ultrastructural study. Cryobiology 2007, 55, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Dore-Duffy, P.; Owen, C.; Balabanov, R.; Murphy, S.; Beaumont, T.; Rafols, J.A. Pericyte migration from the vascular wall in response to traumatic brain injury. Microvasc. Res. 2000, 60, 55–69. [Google Scholar] [CrossRef] [PubMed]
- Markoullis, K.; Sargiannidou, I.; Gardner, C.; Hadjisavvas, A.; Reynolds, R.; Kleopa, K.A. Disruption of oligodendrocyte gap junctions in experimental autoimmune encephalomyelitis. Glia 2012, 60, 1053–1066. [Google Scholar] [CrossRef]
- Watanabe, M.; Masaki, K.; Yamasaki, R.; Kawanokuchi, J.; Takeuchi, H.; Matsushita, T.; Suzumura, A.; Kira, J.I. Th1 cells downregulate connexin 43 gap junctions in astrocytes via microglial activation. Sci. Rep. 2016, 6, 38387. [Google Scholar] [CrossRef] [PubMed]
- De Pina-Benabou, M.H.; Szostak, V.; Kyrozis, A.; Rempe, D.; Uziel, D.; Urban-Maldonado, M.; Benabou, S.; Spray, D.C.; Federoff, H.J.; Stanton, P.K.; et al. Blockade of gap junctions in vivo provides neuroprotection after perinatal global ischemia. Stroke 2005, 36, 2232–2237. [Google Scholar] [CrossRef] [Green Version]
- Frantseva, M.V.; Kokarovtseva, L.; Naus, C.G.; Carlen, P.L.; MacFabe, D.; Perez, J.L. Velazquez Specific gap junctions enhance the neuronal vulnerability to brain traumatic injury. J. Neurosci. 2002, 22, 644–653. [Google Scholar] [CrossRef] [PubMed]
- Tress, O.; Maglione, M.; Zlomuzica, A.; May, D.; Dicke, N.; Degen, J.; Dere, E.; Kettenmann, H.; Hartmann, D.; Willecke, K. Pathologic and phenotypic alterations in a mouse expressing a connexin47 missense mutation that causes Pelizaeus-Merzbacher-like disease in humans. PLoS Genet. 2011, 7, e1002146. [Google Scholar] [CrossRef] [PubMed]
- Odermatt, B.; Wellershaus, K.; Wallraff, A.; Seifert, G.; Degen, J.; Euwens, C.; Fuss, B.; Bussow, H.; Schilling, K.; Steinhauser, C.; et al. Connexin 47 (Cx47)-deficient mice with enhanced green fluorescent protein reporter gene reveal predominant oligodendrocytic expression of Cx47 and display vacuolized myelin in the CNS. J. Neurosci. 2003, 23, 4549–4559. [Google Scholar] [CrossRef] [Green Version]
- Seo, J.H.; Maki, T.; Maeda, M.; Miyamoto, N.; Liang, A.C.; Hayakawa, K.; Pham, L.D.; Suwa, F.; Taguchi, A.; Matsuyama, T.; et al. Oligodendrocyte precursor cells support blood-brain barrier integrity via TGF-beta signaling. PLoS ONE 2014, 9, e103174. [Google Scholar] [CrossRef] [Green Version]
- Niu, J.; Li, T.; Yi, C.; Huang, N.; Koulakoff, A.; Weng, C.; Li, C.; Zhao, C.J.; Giaume, C.; Xiao, L. Connexin-based channels contribute to metabolic pathways in the oligodendroglial lineage. J. Cell Sci. 2016, 129, 1902–1914. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Yamasaki, R.; Yamaguchi, H.; Nagata, S.; Une, H.; Cui, Y.; Masaki, K.; Nakamuta, Y.; Iinuma, K.; Watanabe, M.; et al. Oligodendroglial connexin 47 regulates neuroinflammation upon autoimmune demyelination in a novel mouse model of multiple sclerosis. Proc. Natl. Acad. Sci. USA 2020, 117, 2160–2169. [Google Scholar] [CrossRef]
- Meens, M.J.; Kutkut, I.; Rochemont, V.; Dubrot, J.; Kaladji, F.R.; Sabine, A.; Lyons, O.; Hendrikx, S.; Bernier-Latmani, J.; Kiefer, F.; et al. Cx47 fine-tunes the handling of serum lipids but is dispensable for lymphatic vascular function. PLoS ONE 2017, 12, e0181476. [Google Scholar] [CrossRef]
- Munger, S.J.; Davis, M.J.; Simon, A.M. Defective lymphatic valve development and chylothorax in mice with a lymphatic-specific deletion of Connexin43. Dev. Biol. 2017, 421, 204–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, M.V.; Nguyen, T.T.; Deboy, C.A.; Griffin, J.W.; Whartenby, K.A.; Kerr, D.A.; Calabresi, P.A. Behavioral and pathological outcomes in MOG 35–55 experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2008, 199, 83–93. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stavropoulos, F.; Georgiou, E.; Sargiannidou, I.; Kleopa, K.A. Dysregulation of Blood-Brain Barrier and Exacerbated Inflammatory Response in Cx47-Deficient Mice after Induction of EAE. Pharmaceuticals 2021, 14, 621. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14070621
Stavropoulos F, Georgiou E, Sargiannidou I, Kleopa KA. Dysregulation of Blood-Brain Barrier and Exacerbated Inflammatory Response in Cx47-Deficient Mice after Induction of EAE. Pharmaceuticals. 2021; 14(7):621. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14070621
Chicago/Turabian StyleStavropoulos, Filippos, Elena Georgiou, Irene Sargiannidou, and Kleopas A. Kleopa. 2021. "Dysregulation of Blood-Brain Barrier and Exacerbated Inflammatory Response in Cx47-Deficient Mice after Induction of EAE" Pharmaceuticals 14, no. 7: 621. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14070621