
Bioactive Compounds from Euphorbia usambarica Pax. with HIV-1 Latency Reversal Activity

 , ,
, ,  , , , ,
, , , ,
Abstract
:1. Introduction
2. Results
2.1. Structure Elucidation of New Compounds
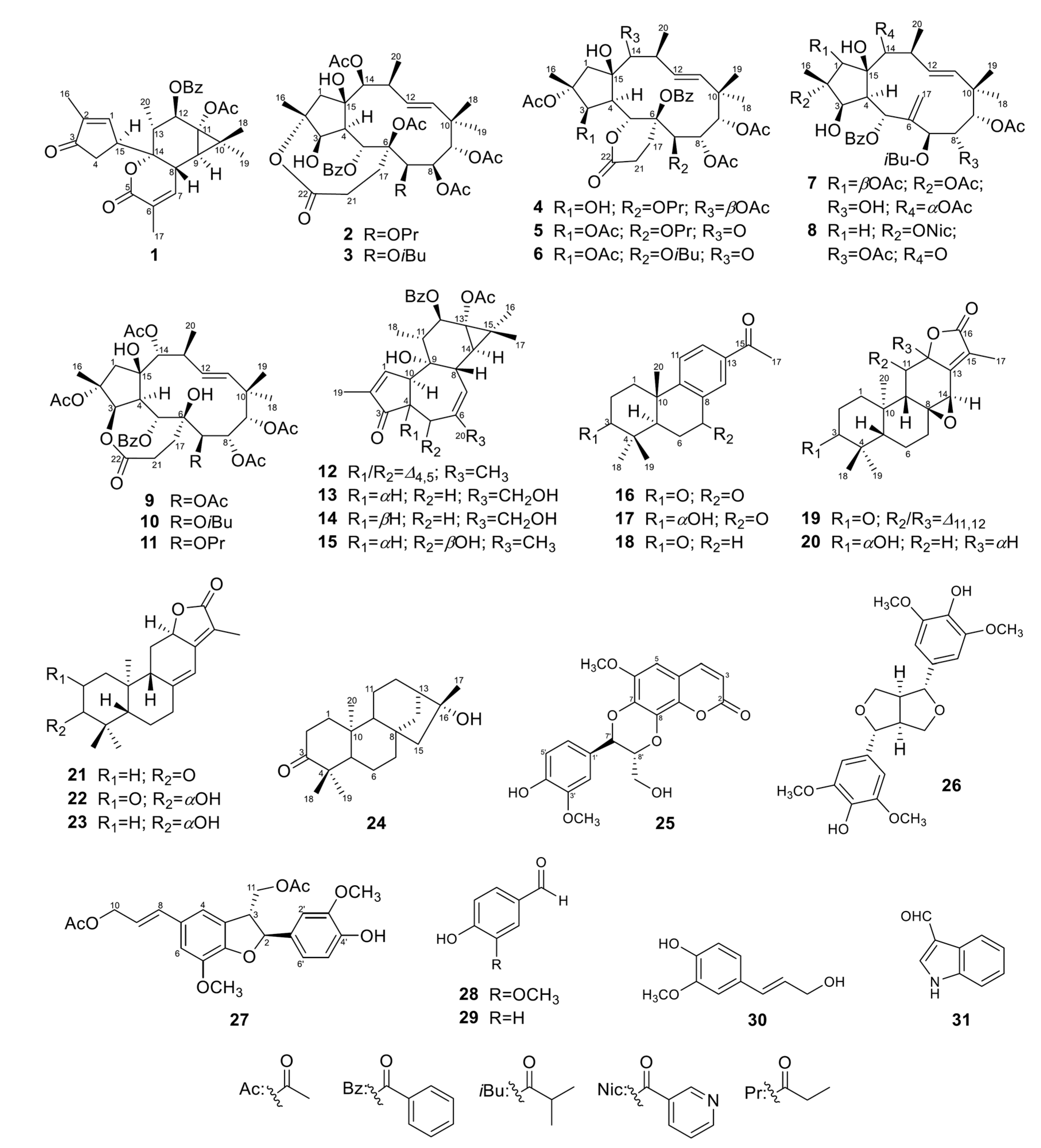
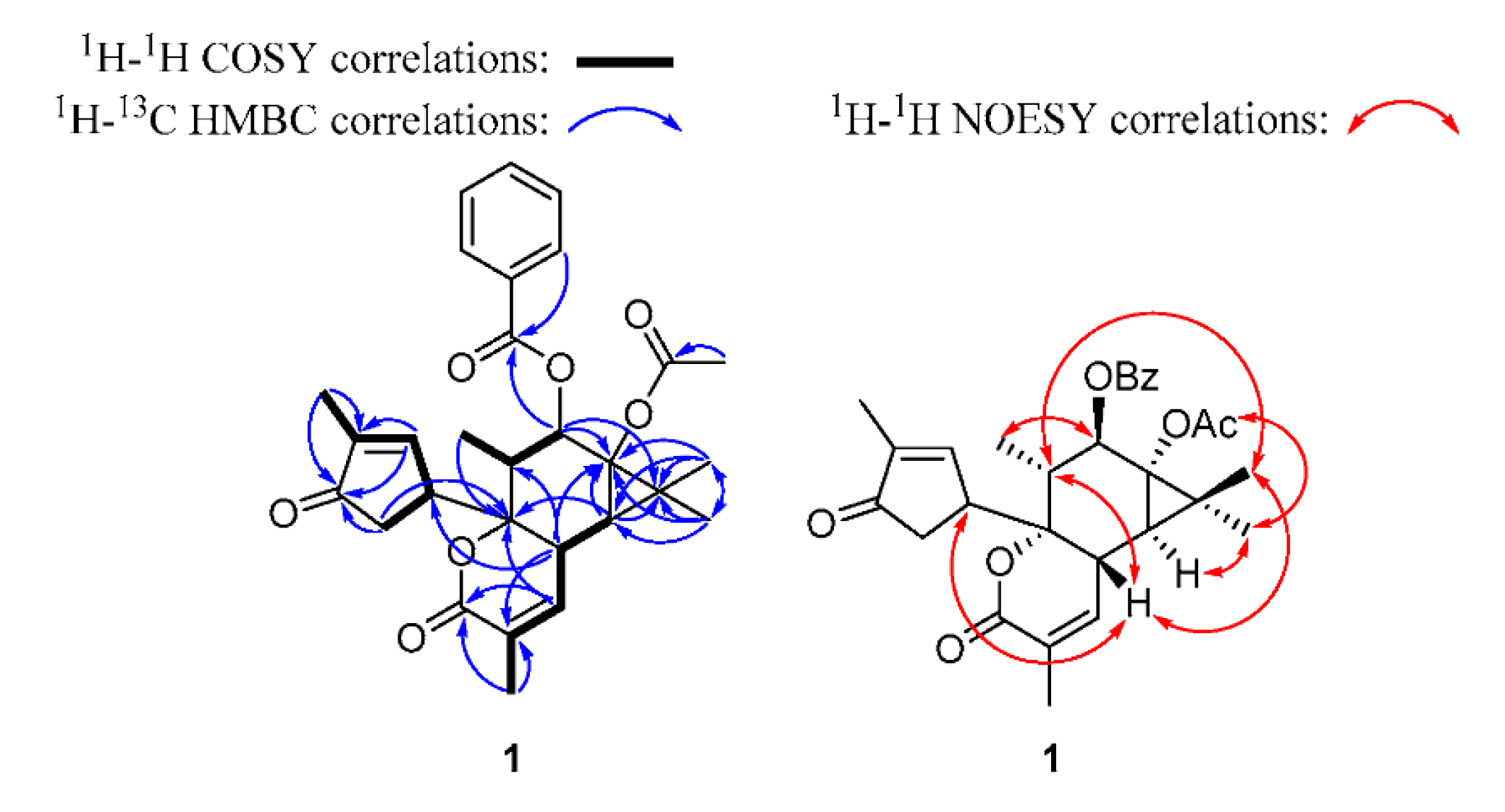
2.1.1. Euphordraculoate C (1)
2.1.2. Usambariphane A (2)
2.1.3. Usambariphane B (3)
2.1.4. Usambariphane C (4)
2.1.5. Usambariphane D (5)
2.1.6. Usambariphane E (6)
2.1.7. Usambariphane F (7)
2.1.8. Usambariphane G (8)
2.1.9. Isoterracinolides C (9)
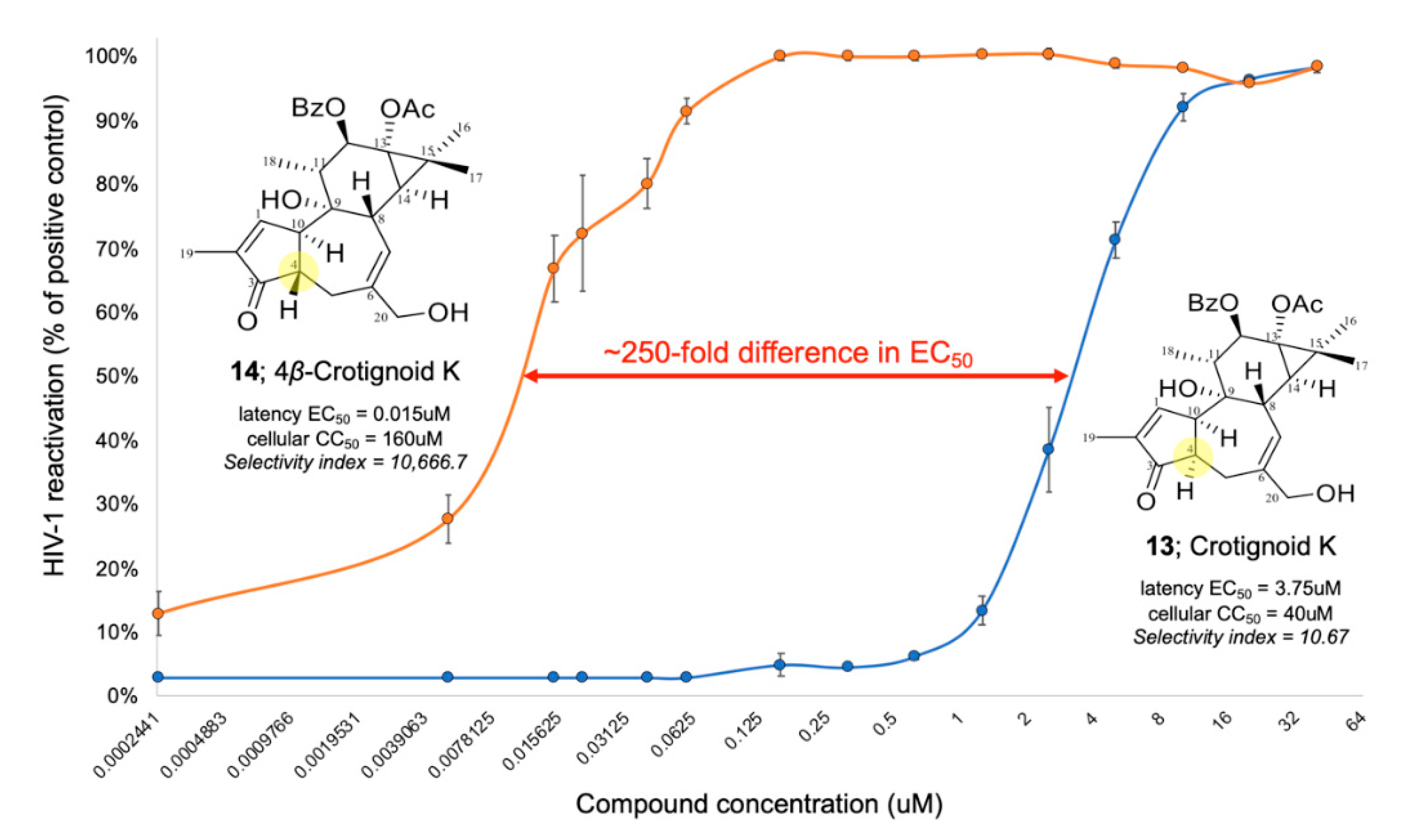
2.1.10. 4β-Crotignoid K (14)
2.1.11. Euphodendriane B (15)
2.1.12. 16-Nor-abieta-8,11,13-trien-3,7,15-trione (16)
2.1.13. 16-Nor-3β-hydroxy-abieta-8,11,13-trien-7,15-dione (17)
2.1.14. ent-8β,14β-Epoxyabieta-3-one-11,13(15)-dien-16,12-olide (19)
2.1.15. ent-8β,14β-Epoxyabieta-3α-hydroxy-13(15)-en-16,12-olide (20)
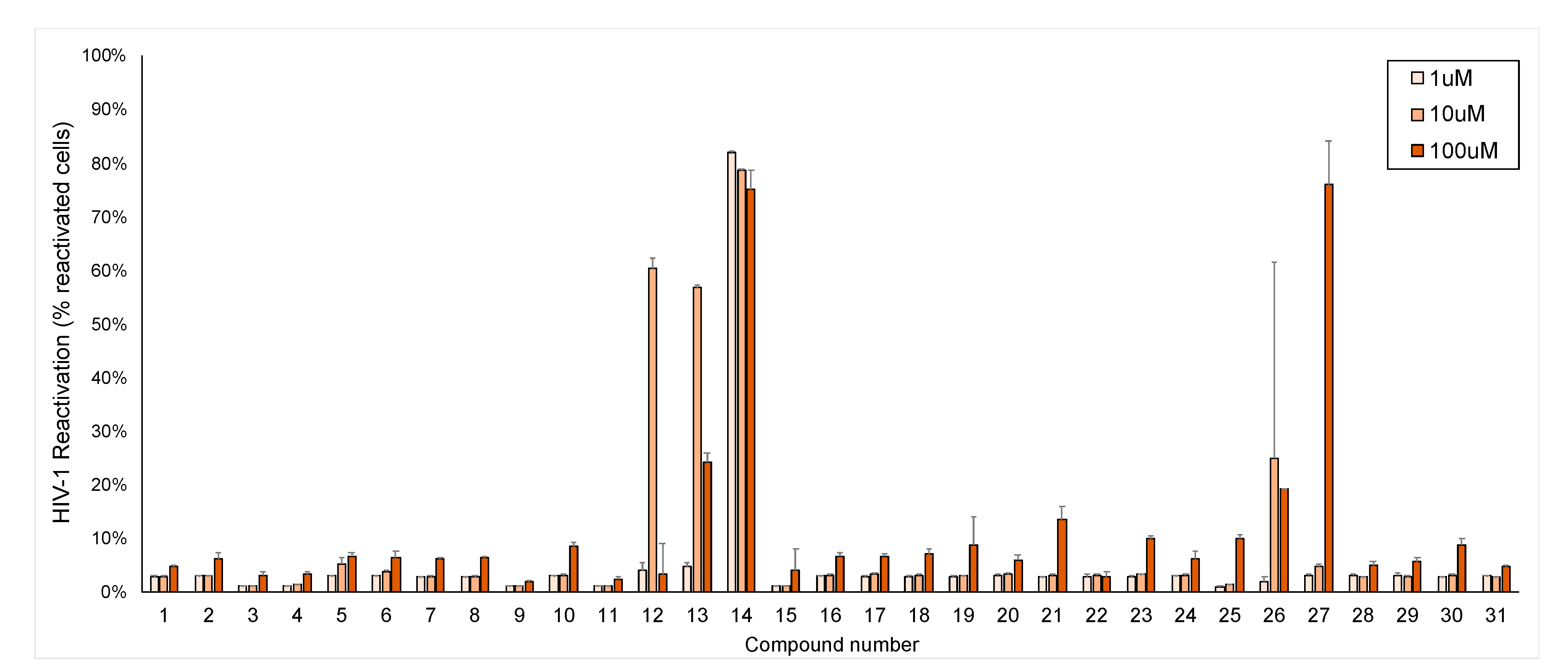
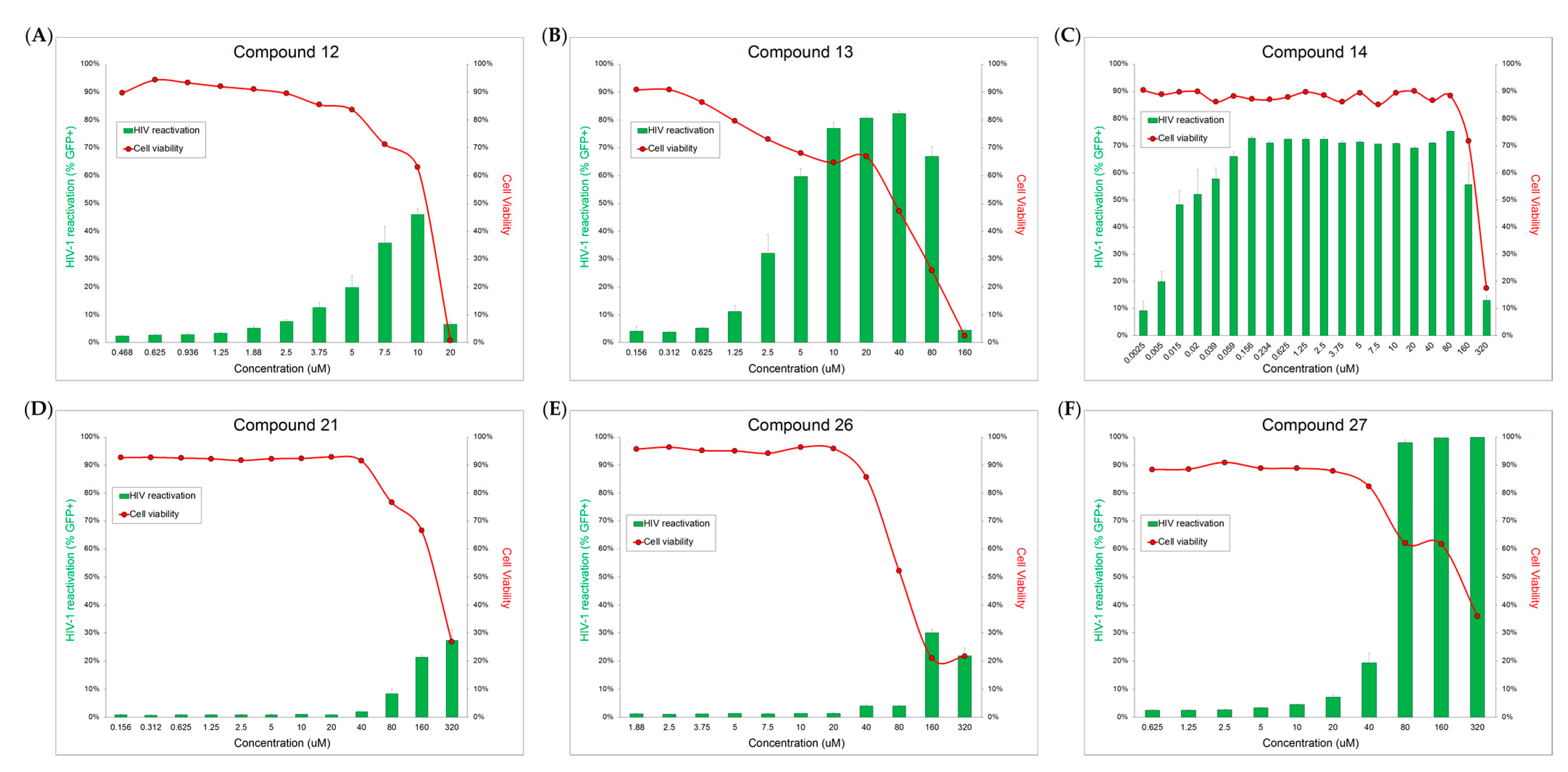
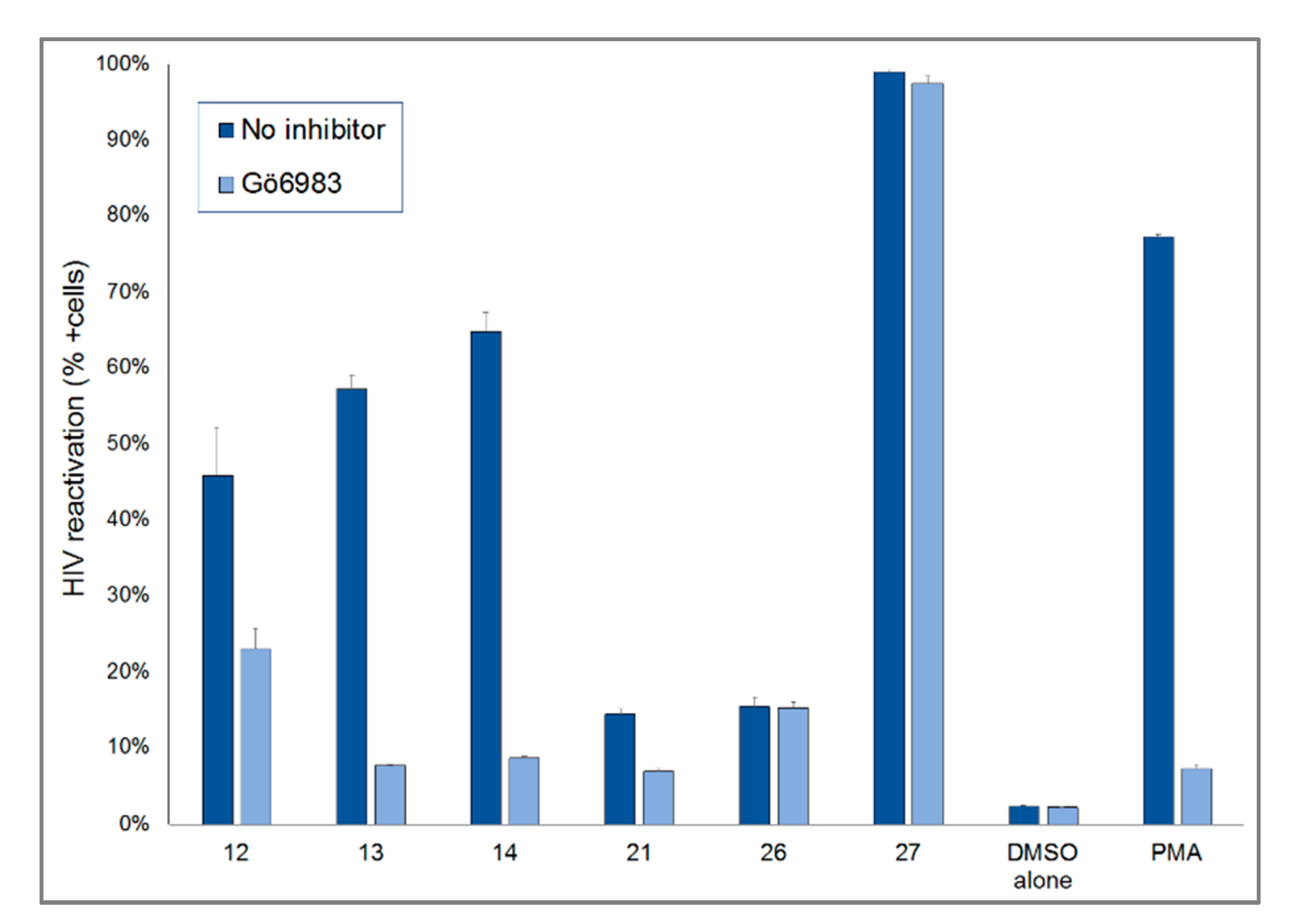
2.2. HIV-1 Latency Reversal Activity of Isolated Compounds in Vitro
3. Discussion
4. Materials and Methods
4.1. General Experimental Procedures
4.2. Plant Material
4.3. Extraction and Isolation
4.4. Physical Characteristic of New Compounds
4.5. Cell Isolation and Culture
4.6. Flow Cytometry
4.7. Compound Screening
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Spivak, A.M.; Planelles, V. HIV-1 eradication: Early trials (and tribulations). Trends Mol. Med. 2016, 22, 10–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, R.J.; Ntie-Kang, F.; Tietjen, I. Natural product-derived compounds in HIV suppression, remission, and eradication strategies. Antivir. Res. 2018, 158, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Cary, D.C.; Peterlin, B.M. Natural products and HIV/AIDS. AIDS Res. Hum. Retrovir. 2018, 34, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Salehi, B.; Kumar, N.V.A.; Sener, B.; Sharifi-Rad, M.; Kilic, M.; Mahady, G.B.; Vlaisavljevic, S.; Iriti, M.; Kobarfard, F.; Setzer, W.N.; et al. Medicinal plants used in the treatment of human immunodeficiency virus. Int. J. Mol. Sci. 2018, 19, 1459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mwine, J.T.; Van Damme, P. Why do Euphorbiaceae tick as medicinal plants? A review of Euphorbiaceae family and its medicinal features. J. Med. Plants Res. 2011, 5, 652–662. [Google Scholar]
- Shi, Q.W.; Su, X.H.; Kiyota, H. Chemical and pharmacological research of the plants in genus Euphorbia. Chem. Rev. 2008, 108, 4295–4327. [Google Scholar] [CrossRef]
- Vasas, A.; Hohmann, J. Euphorbia diterpenes: Isolation, structure, biological activity, and synthesis (2008–2012). Chem. Rev. 2014, 114, 8579–8612. [Google Scholar] [CrossRef] [Green Version]
- Patil, S.B.; Naikwade, N.S.; Magdum, C.S. Review on phytochemistry and pharmacological aspects of Euphorbia hirta Linn. Asian J. Pharm. Res. Health Care 2009, 1, 113–133. [Google Scholar]
- Kumar, S.; Malhotra, R.; Kumar, D. Euphorbia hirta: Its chemistry, traditional and medicinal uses, and pharmacological activities. Pharmacogn. Rev. 2010, 4, 58–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Özbilgin, S.; Saltan Çitoğlu, G. Uses of some Euphorbia species in traditional medicine in turkey and their biological activities. Turk. J. Pharm. Sci. 2012, 9, 241–255. [Google Scholar]
- Lee, J.W.; Jin, Q.; Jang, H.; Lee, D.; Han, S.B.; Kim, Y.; Hong, J.T.; Lee, M.K.; Hwang, B.Y. Jatrophane and ingenane-type diterpenoids from Euphorbia kansui inhibit the LPS-induced NO production in RAW 264.7 cells. Bioorg. Med. Chem. Lett. 2016, 26, 3351–3354. [Google Scholar] [CrossRef] [PubMed]
- Redei, D.; Forgo, P.; Molnar, J.; Szabo, P.; Zorig, T.; Hohmann, J. Jatrophane diterpenoids with multidrug resistance-modulating activity from Euphorbia mongolica Prokh. Tetrahedron 2012, 68, 8403–8407. [Google Scholar] [CrossRef]
- Corea, G.; Di Pietro, A.; Dumontet, C.; Fattorusso, E.; Lanzotti, V. Jatrophane diterpenes from Euphorbia spp. as modulators of multidrug resistance in cancer therapy. Phytochem. Rev. 2009, 8, 431–447. [Google Scholar] [CrossRef]
- Nothias-Scaglia, L.F.; Retailleau, P.; Paolini, J.; Pannecouque, C.; Neyts, J.; Dumontet, V.; Roussi, F.; Leyssen, P.; Costa, J.; Litaudon, M. Jatrophane diterpenes as inhibitors of chikungunya virus replication: Structure-activity relationship and discovery of a potent lead. J. Nat. Prod. 2014, 77, 1505–1512. [Google Scholar] [CrossRef] [PubMed]
- Remy, S.; Litaudon, M. Macrocyclic diterpenoids from Euphorbiaceae as a source of potent and selective inhibitors of chikungunya virus replication. Molecules 2019, 24, 2336. [Google Scholar] [CrossRef] [Green Version]
- Islam, M.T. Diterpenes and their derivatives as potential anticancer agents. Phytother. Res. 2017, 31, 691–712. [Google Scholar] [CrossRef] [PubMed]
- Fattahian, M.; Ghanadian, M.; Ali, Z.; Khan, I.A. Jatrophane and rearranged jatrophane-type diterpenes: Biogenesis, structure, isolation, biological activity and SARs (1984–2019). Phytochem. Rev. 2020, 19, 265–336. [Google Scholar] [CrossRef]
- Kusz, N.; Orvos, P.; Bereczki, L.; Fertey, P.; Bombicz, P.; Csorba, A.; Talosi, L.; Jakab, G.; Hohmann, J.; Redei, D. Diterpenoids from Euphorbia dulcis with potassium ion channel inhibitory activity with selective G protein-activated inwardly rectifying ion channel (GIRK) blocking effect. J. Nat. Prod. 2018, 81, 2483–2492. [Google Scholar] [CrossRef]
- Rawal, M.K.; Shokoohinia, Y.; Chianese, G.; Zolfaghari, B.; Appendino, G.; Taglialatela-Scafati, O.; Prasad, R.; Di Pietro, A. Jatrophanes from Euphorbia squamosa as potent inhibitors of Candida albicans multidrug transporters. J. Nat. Prod. 2014, 77, 2700–2706. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.Y.; Redei, D.; Forgo, P.; Li, Y.; Vasas, A.; Hohmann, J.; Wu, C.C. Isolation of phorbol esters from Euphorbia grandicornis and evaluation of protein kinase c and human platelet-activating effects of Euphorbiaceae. Diterpenes. J. Nat. Prod. 2016, 79, 2658–2666. [Google Scholar] [CrossRef]
- Park, K.H.; Koh, D.; Lee, S.; Jung, I.; Kim, K.H.; Lee, C.H.; Kim, K.H.; Lim, Y. Anti-allergic and anti-asthmatic activity of helioscopinin-A, a polyphenol compound, isolated from Euphorbia helioscopia. J. Microbiol. Biotechnol. 2001, 11, 138–142. [Google Scholar]
- Islam, N.U.; Khan, I.; Rauf, A.; Muhammad, N.; Shahid, M.; Shah, M.R. Antinociceptive, muscle relaxant and sedative activities of gold nanoparticles generated by methanolic extract of Euphorbia milii. BMC Complementary Altern. Med. 2015, 15, 160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spivak, A.M.; Planelles, V. Novel latency reversal agents for HIV-1 cure. Annu. Rev. Med. 2018, 69, 421–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wonderlich, E.R.; Subramanian, K.; Cox, B.; Wiegand, A.; Lackman-Smith, C.; Bale, M.J.; Stone, M.; Hoh, R.; Kearney, M.F.; Maldarelli, F.; et al. Effector memory differentiation increases detection of replication-competent HIV-l in resting CD4+ T cells from virally suppressed individuals. PLoS Pathog. 2019, 15, e1008074. [Google Scholar] [CrossRef] [PubMed]
- Cary, D.C.; Fujinaga, K.; Peterlin, B.M. Euphorbia kansui reactivates latent HIV. PLoS ONE 2016, 11, e0168027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pisano, M.B.; Cosentino, S.; Viale, S.; Spano, D.; Corona, A.; Esposito, F.; Tramontano, E.; Montoro, P.; Tuberoso, C.I.; Medda, R.; et al. Biological activities of aerial parts extracts of Euphorbia characias. Biomed. Res. Int. 2016, 2016, 1538703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasmussen, T.A.; Tolstrup, M.; Sogaard, O.S. Reversal of latency as part of a cure for HIV-1. Trends Microbiol. 2016, 24, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Li, W.; Huang, L.; Asada, Y.; Morris-Natschke, S.L.; Chen, C.H.; Lee, K.H.; Koike, K. Identification, structural modification, and dichotomous effects on human immunodeficiency virus type 1 (HIV-1) replication of ingenane esters from Euphorbia kansui. Eur. J. Med. Chem. 2018, 156, 618–627. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.L.; Li, Y.H.; Chen, X.Q.; Liu, D.; Chen, C.H.; Li, R.T. Diterpenes from the stem bark of Euphorbia neriifolia and their in vitro anti-HIV activity. Phytochemistry 2018, 145, 40–47. [Google Scholar] [CrossRef]
- Valadão, A.L.C.; Pezzuto, P.; Silva, V.A.O.; Gonçalves, B.S.; Rossi, Á.D.; Cunha, R.D.; Siani, A.C.; Tostes, J.B.F.; Trovó, M.; Damasco, P.; et al. Reactivation of latent HIV-1 in vitro using an ethanolic extract from Euphorbia umbellata (Euphorbiaceae) latex. PLoS ONE 2018, 13, e0207664. [Google Scholar] [CrossRef] [Green Version]
- Thiselton-Dyer W.T. Euphorbia usambarica Pax. In Flora of Tropical Africa; L. Reeve & Co. Ltd.: London, UK, 1913; Volume 6, pp. 538–539. [Google Scholar]
- Schlage, C.; Mabula, C.; Mahunnah, R.L.A.; Heinrich, M. Medicinal plants of the Washambaa (Tanzania): Documentation and ethnopharmacological evaluation. Plant Biol. 2000, 2, 83–92. [Google Scholar] [CrossRef]
- Chrispin, F.S.; Innocent, J.E.Z.; Patrick, L.P.M.; Matti, N. Use of medicinal plants in the eastern arc mountains with special reference to the hehe ethnic group in the Udzungwa mountains, Tanzania. J. East Afr. Nat. Hist. 2008, 97, 225–254. [Google Scholar]
- Wang, L.; Yang, J.; Kong, L.M.; Deng, J.; Xiong, Z.J.; Huang, J.P.; Luo, J.P.; Yan, Y.J.; Hu, Y.K.; Li, X.N.; et al. Natural and semisynthetic tigliane diterpenoids with new carbon skeletons from Euphorbia dracunculoides as a Wnt signaling pathway inhibitor. Org. Lett. 2017, 19, 3911–3914. [Google Scholar] [CrossRef]
- Aljancic, I.S.; Pesic, M.; Milosavljevic, S.M.; Todorovic, N.M.; Jadranin, M.; Miosavljevic, G.; Povrenovic, D.; Bankovic, J.; Tanic, N.; Markovic, I.D.; et al. Isolation and biological evaluation of jatrophane diterpenoids from Euphorbia dendroides. J. Nat. Prod. 2011, 74, 1613–1620. [Google Scholar] [CrossRef]
- Marco, J.A.; Sanz-Cervera, J.F.; Yuste, A.; Jakupovic, J. Isoterracinolides A and B, novel bishomoditerpene lactones from Euphorbia terracina. J. Nat. Prod. 1999, 62, 110–113. [Google Scholar] [CrossRef]
- Huang, Y.; Aisa, H.A. Three new diterpenoids from Euphorbia sororia L. Helv. Chim. Acta 2010, 93, 1156–1161. [Google Scholar] [CrossRef]
- Hu, R.; Gao, J.; Rozimamat, R.; Aisa, H.A. Jatrophane diterpenoids from Euphorbia sororia as potent modulators against P-glycoprotein-based multidrug resistance. Eur. J. Med. Chem. 2018, 146, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Corea, G.; Fattorusso, E.; Lanzotti, V.; Taglialatela-Scafati, O.; Appendino, G.; Ballero, M.; Sirnon, P.N.L.; Dumontet, C.; Di Pietro, A. Modified jatrophane diterpenes as modulators of multidrug resistance from Euphorbia dendroides L. Bioorgan. Med. Chem. 2003, 11, 5221–5227. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.L.; Liu, Y.Q.; Aisa, H.A. Jatrophane diterpenoid esters from Euphorbia sororia serving as multidrug resistance reversal agents. Fitoterapia 2014, 92, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Marco, J.A.; Sanz-Cervera, J.F.; Yuste, A.; Jakupovic, J.; Jeske, F. Jatrophane derivatives and a rearranged jatrophane from Euphorbia terracina. Phytochemistry 1998, 47, 1621–1630. [Google Scholar] [CrossRef]
- Zhang, D.D.; Zhou, B.; Yu, J.H.; Xu, C.H.; Ding, J.; Zhang, H.; Yue, J.M. Cytotoxic tigliane-type diterpenoids from Croton tiglium. Tetrahedron 2015, 71, 9638–9644. [Google Scholar] [CrossRef]
- Marco, J.A.; Sanz-Cervera, J.F.; Checa, J.; Palomares, E.; Fraga, B.M. Jatrophane and tigliane diterpenes from the latex of Euphorbia obtusifolia. Phytochemistry 1999, 52, 479–485. [Google Scholar] [CrossRef]
- Ara, I.; Siddiqui, B.S.; Faizi, S.; Siddiqui, S. Tricyclic diterpenoids from root bark of Azadirachta Indica. Phytochemistry 1990, 29, 911–914. [Google Scholar] [CrossRef]
- Corral, J.M.M.D.; Gordaliza, M.; Salinero, M.A.; Feliciano, A.S. 13C NMR data for abieta-8,11,13-triene diterpenoids. Magn. Reson. Chem. 1994, 32, 774–781. [Google Scholar] [CrossRef]
- Seca, A.M.L.; Silva, A.M.S.; Bazzocchi, I.L.; Jimenez, I.A. Diterpene constituents of leaves from Juniperus brevifolia. Phytochemistry 2008, 69, 498–505. [Google Scholar] [CrossRef]
- Lal, A.R.; Cambie, R.C.; Rutledge, P.S.; Woodgate, P.D. Ent-pimarane and ent-abietane diterpenes from Euphorbia fidjiana. Phytochemistry 1990, 29, 2239–2246. [Google Scholar] [CrossRef]
- Talapatra, S.K.; Das, G.; Talapatra, B. Stereostructures and molecular conformations of six diterpene lactones from Gelonium multiflorum. Phytochemistry 1989, 28, 1181–1185. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, X.F.; Cai, X.H.; Ma, Y.B.; Luo, X.D. Three new diterpenoids from Euphorbia wallichii. Chin. J. Chem. 2004, 22, 199–202. [Google Scholar] [CrossRef]
- Choudhary, M.I.; Gondal, H.Y.; Abbaskhan, A.; Jahan, I.A.; Parvez, M.; Nahar, N.; Rahman, A. Revisiting diterpene lactones of Suregada multiflora. Tetrahedron 2004, 60, 7933–7941. [Google Scholar] [CrossRef]
- Song, Q.Q.; Rao, Y.; Tang, G.H.; Sun, Z.H.; Zhang, J.S.; Huang, Z.S.; Yin, S. Tigliane diterpenoids as a new type of antiadipogenic agents inhibit GR alpha-Dexras1 axis in adipocytes. J. Med. Chem. 2019, 62, 2060–2075. [Google Scholar] [CrossRef]
- Yin, S.; Huang, Z.; Rao, Y.; Tang, G.; Song, Q. Tigliane-type diterpenoid compounds and preparation method and application thereof. China Patent CN108689851A, 28 May 2018. [Google Scholar]
- Guo, K.; Liu, Y.C.; Liu, Y.; Luo, S.H.; Li, W.Y.; Li, X.N.; Li, S.H. Diversified abietane family diterpenoids from the leaves of Leucosceptrurn canum and their cytotoxic activity. Phytochemistry 2019, 157, 43–52. [Google Scholar] [CrossRef]
- Borghi, D.; Baumer, L.; Ballabio, M.; Arlandini, E.; Perellino, N.C.; Minghetti, A.; Vincieri, F.F. Structure elucidation of helioscopinolides D and E from Euphorbia calyptrata cell cultures. J. Nat. Prod. 1991, 54, 1503–1508. [Google Scholar] [CrossRef]
- Tori, M.; Arbiyanti, H.; Taira, Z.; Asakawa, Y. Terpenoids of the liverwort Frullanoides densifolia and Trocholejeunea sandvicensis. Phytochemistry 1993, 32, 335–348. [Google Scholar] [CrossRef]
- Arisawa, M.; Handa, S.S.; McPherson, D.D.; Lankin, D.C.; Cordell, G.A.; Fong, H.H.S.; Farnsworth, N.R. Plant anticancer agents XXIX. Cleomiscosin A from Simaba multiflora, Soulamea soulameoides, and Matayba arborescens. J. Nat. Prod. 1984, 47, 300–307. [Google Scholar] [CrossRef]
- Min, Y.D.; Choi, S.U.; Lee, K.R. Aporphine alkaloids and their reversal activity of multidrug resistance (MDR) from the stems and rhizomes of Sinomenium acutum. Arch. Pharm. Res. 2006, 29, 627–632. [Google Scholar] [CrossRef] [PubMed]
- Valcic, S.; Montenegro, G.; Timmermann, B.N. Lignans from Chilean propolis. J. Nat. Prod. 1998, 61, 771–775. [Google Scholar] [CrossRef] [PubMed]
- Bao, K.; Fan, A.X.; Dai, Y.; Zhang, L.; Zhang, W.G.; Cheng, M.S.; Yao, X.S. Selective demethylation and debenzylation of aryl ethers by magnesium iodide under solvent-free conditions and its application to the total synthesis of natural products. Org. Biomol. Chem. 2009, 7, 5084–5090. [Google Scholar] [CrossRef]
- Kim, H.; Ralph, J.; Lu, F.C.; Ralph, S.A.; Boudet, A.M.; MacKay, J.J.; Sederoff, R.R.; Ito, T.; Kawai, S.; Ohashi, H.; et al. NMR analysis of lignins in CAD-deficient plants. Part 1. Incorporation of hydroxycinnamaldehydes and hydroxybenzaldehydes into lignins. Org. Biomol. Chem. 2003, 1, 268–281. [Google Scholar] [CrossRef]
- Yao, C.S.; Lin, M.; Wang, L. Isolation and biomimetic synthesis of anti-inflammatory stilbenolignans from Gnetum cleistostachyum. Chem. Pharm. Bull. 2006, 54, 1053–1057. [Google Scholar] [CrossRef] [Green Version]
- Duarte, N.; Ferreira, M.J.U. Lagaspholones A and B: Two new jatropholane-type diterpenes from Euphorbia lagascae. Org. Lett. 2007, 9, 489–492. [Google Scholar] [CrossRef] [PubMed]
- De la Torre-Tarazona, H.E.; Jimenez, R.; Bueno, P.; Camarero, S.; Roman, L.; Fernandez-Garcia, J.L.; Beltran, M.; Nothias, L.F.; Cachet, X.; Paolini, J.; et al. 4-Deoxyphorbol inhibits HIV-1 infection in synergism with antiretroviral drugs and reactivates viral reservoirs through PKC/MEK activation synergizing with vorinostat. Biochem. Pharmacol. 2020, 177, 113937. [Google Scholar] [CrossRef]
- Hammadi, R.; Kúsz, N.; Dávid, C.Z.; Behány, Z.; Papp, L.; Kemény, L.; Hohmann, J.; Lakatos, L.; Vasas, A. Ingol and ingenol-type diterpenes from Euphorbia trigona Miller with keratinocyte inhibitory activity. Plants 2021, 10, 1206. [Google Scholar] [CrossRef]
- Jiang, G.; Maverakis, E.; Cheng, M.Y.; Elsheikh, M.M.; Deleage, C.; Mendez-Lagares, G.; Shimoda, M.; Yukl, S.A.; Hartigan-O’Connor, D.J.; Thompson, G.R.; et al. Disruption of latent HIV in vivo during the clearance of actinic keratosis by ingenol mebutate. JCI Insight 2019, 4, e126027. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| 1 | 7.52, br s | a: 2.87, d (16.5) | a: 2.89, d (17.0) | a: 2.87, d (16.5) | a: 2.76, d (16.0) | a: 2.75, d (16.0) |
| b: 2.09, d (16.5) | b: 2.08, d (17.0) | b: 2.18, d (16.5) | b: 2.04, d (16.0) | b: 2.06, d (16.0) | ||
| 3 | 4.26, dd (9.5, 3.5) | 4.25, dd (9.5, 3.5) | 4.52, dd (12.5, 4.0) | 5.80, dd (4.5, 1.0) | 5.82, dd (4.0, 1.0) | |
| 4 | a: 2.52, dd (18.5, 6.5) | 2.53, m | 2.54, m | 2.63, m | 3.92, m | 3.92, m |
| b: 2.24, dd (18.5, 3.5) | ||||||
| 5 | 6.53, d (2.0) | 6.55, d (2.0) | 6.09, m | 5.70, d (10.0) | 5.70, d (10.0) | |
| 7 | 6.68, dd (6.5, 1.5) | 5.39, s | 5.38, s | 5.40, s | 6.39, s | 6.39, s |
| 8 | 2.37, br d (6.5) | 5.73, d (4.5) | 5.75, d (4.5) | 5.77, d (5.0) | 5.72, s | 5.75, s |
| 9 | 1.02, d (2.0) | 4.88, d (4.5) | 4.91, d (4.5) | 4.83, d (5.0) | 4.99, s | 5.01, s |
| 11 | 5.49, d (16.0) | 5.48, d (16.0) | 5.41, d (16.0) | 6.16, d (16.0) | 6.16, d (16.0) | |
| 12 | 5.86, d (10.5) | 5.83, dd (16.0, 10.0) | 5.85, dd (16.0, 9.5) | 5.72, dd (16.0, 10.0) | 5.43, dd (16.0, 10.0) | 5.41, dd (16.0, 10.0) |
| 13 | 2.10, dq (10.5, 6.5) | 2.46, m | 2.46, m | 2.55, m | 3.97, m | 3.98, m |
| 14 | 4.89, s | 4.89, s | 5.18, s | |||
| 15 | 3.80, m | |||||
| 16 | 1.74, dd (2.5, 1.5) | 1.73, s | 1.73, s | 1.72, s | 1.56, s | 1.57, s |
| 17 | 2.02, br s | a: 3.31, m | a: 3.33, m | a: 3.09, ddd (15.5, 7.5, 2.5) | a: 2.72, m | a: 2.69, m |
| b: 2.12, m | b: 2.06, m | b: 2.40, m | b: 2.01, m | b: 1.98, m | ||
| 18 | 1.43, s | 0.93, s | 0.93, s | 0.93, s | 0.98, s | 0.97, s |
| 19 | 1.16, s | 1.13, s | 1.12, s | 0.96, s | 1.44, s | 1.43, s |
| 20 | 0.91, d (6.5) | 1.14, d (6.5) | 1.14, d (7.0) | 1.15, d (7.0) | 1.23, d (7.0) | 1.23, d (6.5) |
| 21 | a: 2.36, m | a: 2.35, dd (11.5, 7.5) | a: 2.65, m | a: 3.43, m | a: 3.53, m | |
| b: 2.27, m | b: 2.28, dd (11.5, 5.0) | b: 2.16, m | b: 2.50, m | b: 2.52, m | ||
| 2-OAc | 2.13, s | 2.26, s | 2.27, s | |||
| 3-OAc | 2.05, s | 2.05, s | ||||
| 3-OH | 3.45, d (9.5) | 3.44, d (9.5) | 3.03, d (12.5) | |||
| 5-OBz | 7.93, m 7.52, m 7.39, m | 7.93, m 7.53, m 7.39, m | ||||
| 6-OAc | 2.14, s | 2.15, s | ||||
| 6-OBz | 7.92, m 7.58, m 7.41, m | 7.88, m 7.65, m 7.51, m | 7.90, m 7.67, m 7.51, m | |||
| 7-OiBu | 2.61, h (7.0) 1.21, d (7.0) 1.20, d (7.0) | 2.63, h (7.0) 1.26, d (7.0) 1.22, d (7.0) | ||||
| 7-OPr | 2.41, q (7.5) 1.16, t (7.5) | 2.57, m 2.50, m 1.22, t (7.5) | 2.49, m 2.31, m 1.23, m | |||
| 8-OAc | 2.08, s | 2.09, s | 1.35, s | 2.00, s | 2.02, s | |
| 9-OAc | 2.17, s | 2.17, s | 2.07, s | 2.03, s | 2.07, s | |
| 11-OAc | 1.97, s | |||||
| 12-OBz | 8.02, dd (8.5, 1.5) 7.58, m 7.46, dd (8.5, 7.5) | |||||
| 14-OAc | 2.56, s | 2.56, s | 2.45, s | |||
| 15-OH | 3.07, s | 3.05, s | 4.61, s | 4.04, s | 4.08, s |
| Position | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| 1 | 160.2 | 49.1 | 49 | 45.7 | 52.5 | 52.5 |
| 2 | 141.3 | 89.5 | 89.5 | 90.1 | 87.7 | 87.7 |
| 3 | 206.8 | 80.5 | 80.4 | 80.7 | 80 | 79.8 |
| 4 | 37.9 | 43.3 | 43.1 | 44.9 | 45.2 | 45.1 |
| 5 | 163.4 | 72.8 | 73.1 | 85 | 73.2 | 72.9 |
| 6 | 127.8 | 92.5 | 92.4 | 84.4 | 81.1 | 81.6 |
| 7 | 141.4 | 70.8 | 70.5 | 69.1 e | 68.2 | 68.1 |
| 8 | 35.2 | 68.9 | 69 | 69.2 e | 68.1 | 68.1 |
| 9 | 34 | 79.4 | 78.9 | 78.8 | 81.7 | 81.7 |
| 10 | 24.9 | 41.2 | 41.2 | 40.6 | 40.3 | 40.4 |
| 11 | 63.4 | 134.6 | 134.6 | 135.3 | 137.4 | 137.3 |
| 12 | 76.2 | 133 | 132.9 | 134.1 | 128.9 | 128.9 |
| 13 | 38.9 | 37.4 | 37.4 | 39 | 43.9 | 43.6 |
| 14 | 85.8 | 81.6 | 81.5 | 82.2 | 211.4 | 211.6 |
| 15 | 45.8 | 86.3 | 86.3 | 87.1 | 84.6 | 84.6 |
| 16 | 10.2 | 19.1 | 19 | 20 | 18.5 | 18.5 |
| 17 | 17.4 | 24.5 | 24 | 23.4 | 26.5 | 26.6 |
| 18 | 16.5 | 26 | 26 | 26.8 | 25.8 | 26 |
| 19 | 24.8 | 21.7 a | 21.2 c | 21.9 | 23.2 | 23.1 |
| 20 | 12.5 | 23.2 | 23.1 | 22.4 | 21.7 | 21.6 |
| 21 | 26 | 28.3 | 26 | 29.1 | 29.2 | |
| 22 | 175.1 | 174.9 | 168.1 | 172.7 | 172.7 | |
| 2-OAc | 169.7 f, 22.7 | 169.6 g, 22.4 | 169.6 i, 22.4 | |||
| 3-OAc | 169.1 g, 20.6 h | 169.2 i, 21.0 j | ||||
| 5-OBz | 164.5, 133.5, 129.7, 129.6, 128.8 | 164.3, 133.3, 129.6, 129.5, 128.7 | ||||
| 6-OAc | 169.8, 21.5 a | 169.6 d, 21.6 | ||||
| 6-OBz | 163.9, 133.8 130.1, 130.0 128.7 | 165.8, 133.81 30.6, 129.7 128.5 | 166.0, 133.8 130.7, 129.7 128.5 | |||
| 7-OiBu | 176.4, 34.2 18.8, 18.5 | 175.2, 34.5 19.0, 18.1 | ||||
| 7-OPr | 174.2 b, 27.7, 8.9 | 174.6, 27.5, 8.9 | 173.4, 27.6, 8.6 | |||
| 8-OAc | 170, 21.6 a | 169.9 d, 21.4 c | 169.9 f, 21.0 | 170.0 g, 21.2 h | 170.0 i, 21.1 j | |
| 9-OAc | 170.2, 22.7 | 169.8 d, 22.6 | 169.8 f, 21.5 | 169.9 g, 20.9 h | 170.1 i, 20.9 j | |
| 11-OAc | 170.4, 21.1 | |||||
| 12-OBz | 166.2, 133.2 130.4, 129.9 128.6 | |||||
| 14-OAc | 174.3 b, 21.4 a | 174.3, 21.3 c | 170.8, 20.8 |
| Position | 7 | 8 | 9 | |||
|---|---|---|---|---|---|---|
| δH, mult. (J in Hz) | δC | δH, mult. (J in Hz) | δC | δH, mult. (J in Hz) | δC | |
| 1 | 5.46, s | 79.8 | a: 2.95, d (15.5) b: 2.27, d (15.5) | 51.4 | a: 2.83, d (16.5) b: 2.24, d (16.5) | 52.0 |
| 2 | 90.6 | 92.0 | 88.8 | |||
| 3 | 4.36, dd (10.5, 5.5) | 78.0 | 4.67, dd (10.0, 4.5) | 79.1 | 5.48, d (4.0) | 84.7 |
| 4 | 2.76, m | 41.4 | 3.32, m | 47.9 | 2.97, dd (4.0, 3.5) | 44.2 |
| 5 | 6.00, br s | 71.0 | 5.67, br s | 69.2 | 6.58, d (3.5) | 77.9 |
| 6 | 144.4 | 144.9 | 81.8 | |||
| 7 | 5.21, s | 68.8 | 5.41, br s | 68.5 | 5.27, s | 68.4 |
| 8 | 4.30, d (11.0) | 70.2 | 5.18, s | 70.7 | 5.71, d (6.5) | 70.0 |
| 9 | 4.79, s | 86.6 | 4.96, s | 80.6 | 4.96, d (6.5) | 78.4 |
| 10 | 40.1 | 41.1 | 40.7 | |||
| 11 | 5.93, d (16.5) | 134.0 | 5.87, d (15.5) | 137.6 | 5.50, d (16.0) | 134.7 |
| 12 | 5.76, d (16.5) | 130.9 | 5.57, dd (15.5, 9.5) | 129.6 | 5.79, d (16.0) | 134.1 |
| 13 | 2.76, m | 36.9 | 3.75 | 44.4 | 2.69, m | 36.9 |
| 14 | 4.78, s | 76.9 | 211.2 | 5.04, s | 80.4 | |
| 15 | 84.8 | 89.0 | 85.4 | |||
| 16 | 1.55, s | 17.1 | 1.89, s | 20.9 | 1.75, s | 19.8 |
| 17 | a: 5.26, s; b: 5.10, s | 110.4 | a: 5.41, s; b: 5.16, s | 111.6 | a: 1.85, m; b: 1.73, m | 32.0 |
| 18 | 1.03, s | 27.6 | 0.91, s | 26.5 | 0.98, s | 26.4 |
| 19 | 1.40, s | 23.4 | 1.36, s | 23.2 | 1.04, s | 20.8 e |
| 20 | 1.06, d (7.0) | 23.9 | 1.24, d (6.5) | 19.6 | 1.11, d (7.0) | 22.4 |
| 21 | a: 3.21, m; b: 2.33, m | 28.1 | ||||
| 22 | 173.5 | |||||
| 1-OAc | 2.24, s | 170.1, 21.0 a | ||||
| 2-OAc | 2.12, s | 170.9, 22.5 | 2.19, s | 169.6, 22.9 | ||
| 2-ONic | 9.41, dd (2.0, 1.0) 8.79, dd (5.0, 2.0) 8.52, m; 7.39, m | 164.9, 153.4 151.5, 137.6 127.5, 123.2 | ||||
| 3-OH | 3.36, d (10.5) | 3.57, d (10.0) | ||||
| 5-OBz | 8.00, m 7.56, m 7.42, m | 165.4, 133.4 130.1, 129.7 128.8 | 8.06, m 7.56, m 7.44, m | 164.7, 133.4 131.1, 130.0 128.7 | 8.07, m 7.57, m 7.46, m | 168.3, 133.9 130.1, 128.8 128.6 |
| 6-OH | 3.57, s | |||||
| 7-OAc | 2.14, s | 171.0, 20.9 e | ||||
| 7-OiBu | 2.55, h (7.0) 1.19, d (7.0) 1.14, d (7.0) | 175.1, 34.0 19.6, 18.4 | 2.60, h (7.0) 1.23, d (7.0) 1.11, d (7.0) | 175.8, 34.0 19.7, 18.4 | ||
| 8-OAc | 2.00, s | 169.9 c, 20.8 d | 2.15, s | 171.2, 21.7 f | ||
| 8-OH | 3.15, d (11.0) | |||||
| 9-OAc | 2.06, s | 172.1 b, 20.9 a | 2.07, s | 169.7 c, 20.7 d | 2.16, s | 170.4, 21.4 f |
| 14-OAc | 1.70, s | 172.2 b, 20.4 | 2.36, s | 172.0, 20.7 e | ||
| 15-OH | 2.75, s | 4.34, s | 2.40, s |
| Position | 14 | 15 | 16 | 17 | 19 | 20 |
|---|---|---|---|---|---|---|
| 1 | 7.57, s | 7.06, br s | a: 2.68, m | a: 2.42, m | a: 2.05, m | a: 1.98, m |
| b: 2.05, m | b: 1.73, m | b: 1.75, m | b: 1.24, m | |||
| 2 | a: 2.91, m | a: 1.92, m | a: 2.65, ddd (15.5, 14.0, 6.0) | a: 1.73, m | ||
| b: 2.59, ddd (15.5, 5.5, 3.0) | b: 1.87, m | b: 2.37, ddd (15.5, 4.8, 3.2) | b: 1.62, m | |||
| 3 | 3.37, dd (11.5, 4.0) | 3.30, dd (12.0, 4.0) | ||||
| 4 | 2.52, m | 3.13, dd (6.5, 4.5) | ||||
| 5 | a: 2.87, dd (18.5,9.5) | 4.46, dd (11.5, 4.5) | 2.36, dd (14.0, 3.5) | 1.88, m | 1.67, m | 1.05, m |
| b: 2.19, dd (18.5,4.0) | ||||||
| 6 | a: 2.83, dd (17.5, 14.0) | a: 2.79, dd (18.0, 13.5) | a: 1.79, m | a: 1.79, m | ||
| b: 2.75, dd (17.5, 3.5) | b: 2.77, dd (18.0, 4.5) | b: 1.70, m | b: 1.52, m | |||
| 7 | 5.56, m | 4.88, br s | a: 2.17, m | a: 1.98, m | ||
| b: 1.68, m | b: 1.66, m | |||||
| 8 | 2.46, t (5.5) | 2.06, m | ||||
| 9 | 2.70, d (5.0) | 1.95, m | ||||
| 10 | 3.28, m | 3.65, m | ||||
| 11 | 1.75, m | 1.86, dd (10.5, 6.5) | 7.49, d (8.5) | 7.47, d (8.0) | 5.44, d (5.0) | a: 2.27, dd (13.5, 5.5) |
| b: 1.41, m | ||||||
| 12 | 5.68, d (10.0) | 5.73, d (10.5) | 8.17, dd (8.5, 2.5) | 8.14, dd (8.0, 2.0) | 4.99, ddd (13.0, 5.5, 2.0) | |
| 13 | ||||||
| 14 | 1.14, d (5.5) | 0.89, d (6.5) | 8.57, d (2.5) | 8.55, d (2.0) | 3.76, br s | 3.77, s |
| 15 | ||||||
| 16 | 1.21, s | 1.20, s | ||||
| 17 | 1.33, s | 1.33, s | 2.64, s | 2.63, s | 2.09, s | 1.97, d (2.0) |
| 18 | 0.98. d (6.5) | 1.16, d (6.5) | 1.17, s | 0.99, s | 1.17, s | 1.06, s |
| 19 | 1.73, dd (2.5, 1.0) | 1.83, br s | 1.23, s | 1.08, s | 1.09, s | 0.88, s |
| 20 | 4.05, m | 1.90, s | 1.48, s | 1.27, s | 0.95, s | 1.08, s |
| 5-OH | 5.92, d (11.5) | |||||
| 9-OH | 5.62, s | 5.95, s | ||||
| 12-OBz | 8.02, m 7.59, m 7.47, m | 8.06, m 7.61, m 7.49, m | ||||
| 13-OAc | 2.14, s | 2.11, s |
| Position | 14 | 15 | 16 | 17 | 19 | 20 |
|---|---|---|---|---|---|---|
| 1 | 159.7 | 154.6 | 36.8 | 35.9 | 38.1 | 38.7 |
| 2 | 136.7 | 144.3 | 34.6 | 27.5 | 34.2 | 27.3 |
| 3 | 208.7 | 207.5 | 214.0 | 78.0 | 215.0 | 78.6 |
| 4 | 44.4 | 56.3 | 47.6 | 39.1 | 48.1 | 39.0 |
| 5 | 29.8 | 71.1 | 49.2 | 48.3 | 54.1 | 53.6 |
| 6 | 142.3 | 138.0 | 36.5 | 36.1 | 21.7 | 20.8 |
| 7 | 126.6 | 125.5 | 197.4 a | 198.5 | 33.9 | 34.8 |
| 8 | 42.3 | 40.2 | 130.8 | 130.8 | 61.0 | 61.0 |
| 9 | 78.0 | 78.7 | 158.2 | 159.9 | 50.1 | 49.2 |
| 10 | 54.3 | 48.0 | 38.3 | 38.5 | 40.9 | 39.2 |
| 11 | 42.8 | 43.5 | 125.2 | 124.8 | 102.9 | 24.0 |
| 12 | 77.8 | 75.7 | 133.4 | 133.2 | 148.1 | 75.6 |
| 13 | 65.5 | 65.4 | 135.9 | 135.5 | 144.7 | 155.6 |
| 14 | 36.0 | 38.5 | 128.3 | 128.1 | 54.5 | 56.2 |
| 15 | 26.0 | 25.1 | 197.3 a | 197.4 | 126.2 | 128.9 |
| 16 | 23.9 | 24.3 | 170.5 | 174.1 | ||
| 17 | 17.1 | 16.7 | 26.9 | 26.8 | 9.0 | 8.9 |
| 18 | 15.3 | 11.9 | 25.2 | 27.6 | 25.9 | 29.1 |
| 19 | 10.3 | 10.6 | 21.7 | 15.2 | 22.4 | 16.1 |
| 20 | 67.6 | 27.2 | 22.8 | 23.3 | 15.0 | 19.3 |
| 12-OBz | 166.4 133.4 130.1 129.9 128.7 | 166.4 133.4 130.1 129.9 128.7 | ||||
| 13-OAc | 173.9 21.3 | 174.1 21.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsai, Y.-C.; Nell, R.A.; Buckendorf, J.E.; Kúsz, N.; Mwangi, P.W.; Berkecz, R.; Rédei, D.; Vasas, A.; Spivak, A.M.; Hohmann, J. Bioactive Compounds from Euphorbia usambarica Pax. with HIV-1 Latency Reversal Activity. Pharmaceuticals 2021, 14, 653. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14070653
Tsai Y-C, Nell RA, Buckendorf JE, Kúsz N, Mwangi PW, Berkecz R, Rédei D, Vasas A, Spivak AM, Hohmann J. Bioactive Compounds from Euphorbia usambarica Pax. with HIV-1 Latency Reversal Activity. Pharmaceuticals. 2021; 14(7):653. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14070653
Chicago/Turabian StyleTsai, Yu-Chi, Racheal A. Nell, Jonathan E. Buckendorf, Norbert Kúsz, Peter Waweru Mwangi, Róbert Berkecz, Dóra Rédei, Andrea Vasas, Adam M. Spivak, and Judit Hohmann. 2021. "Bioactive Compounds from Euphorbia usambarica Pax. with HIV-1 Latency Reversal Activity" Pharmaceuticals 14, no. 7: 653. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14070653