
Site-Specific Antibody Conjugation to Engineered Double Cysteine Residues

Abstract
:1. Introduction
2. Results
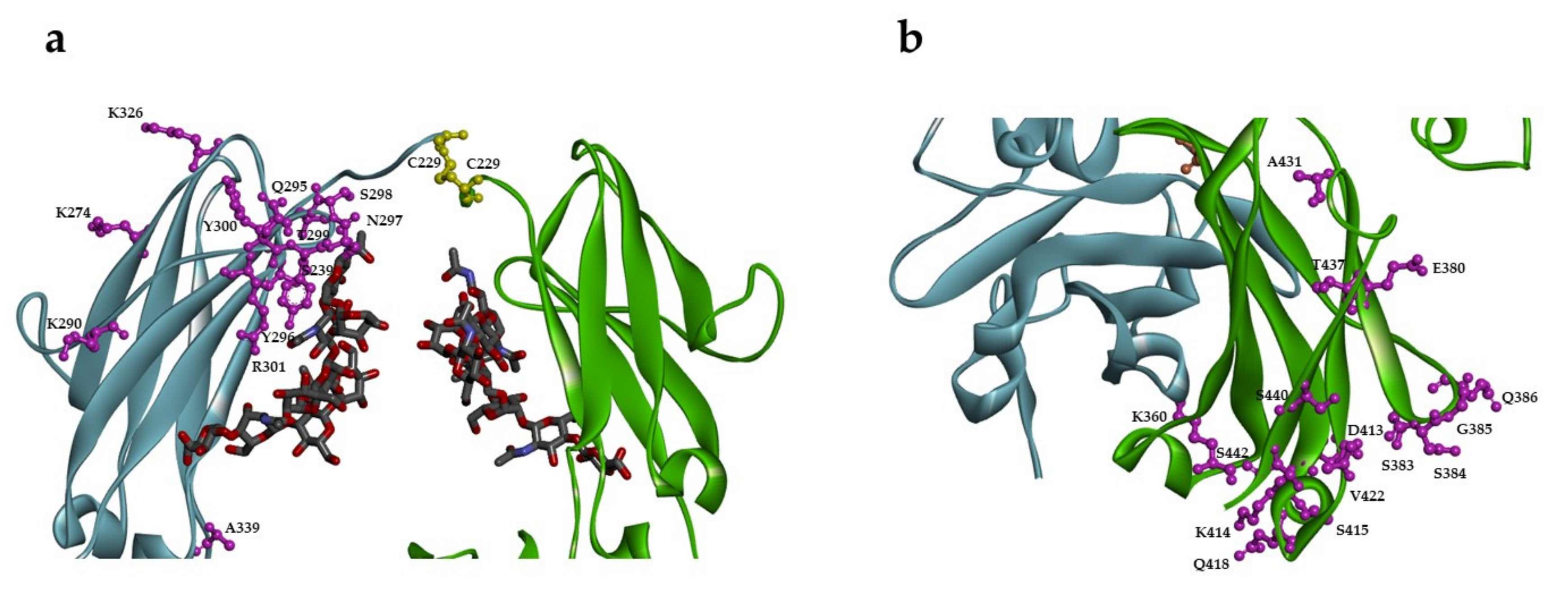
2.1. Design of Single Unpaired Cysteine Mutants
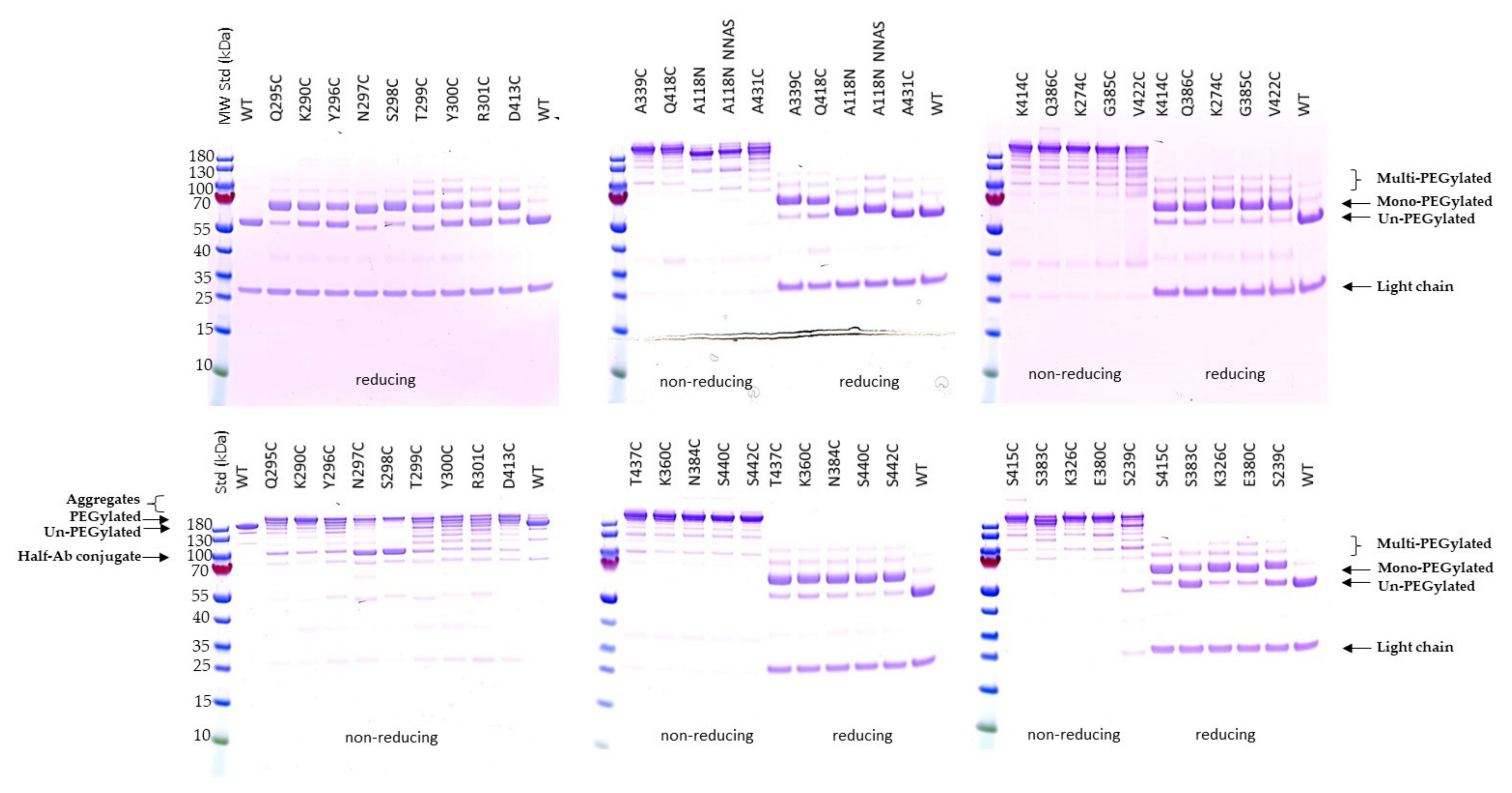
2.2. Expression and Characterization of Engineered Single Cysteine Mutants
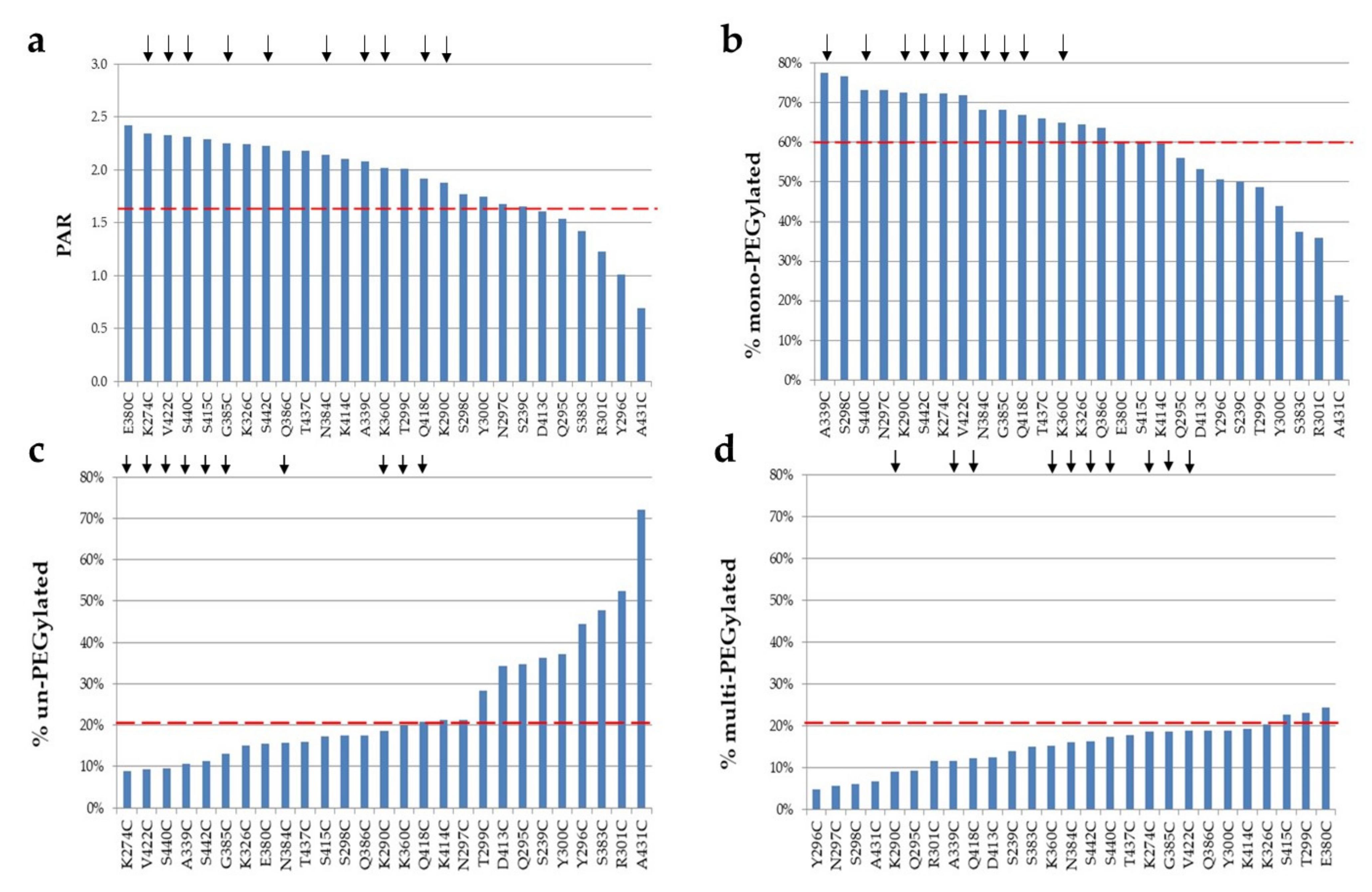
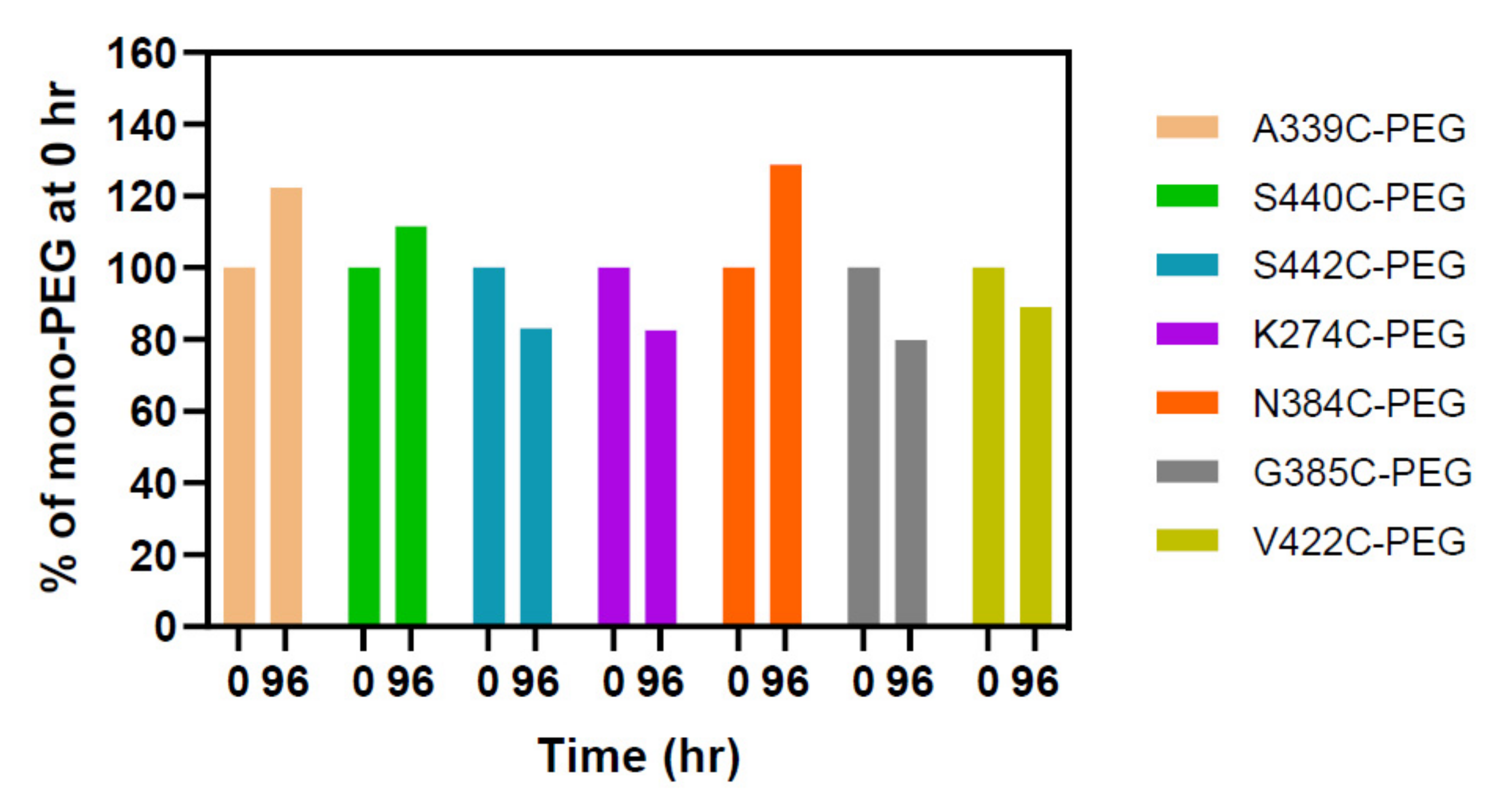
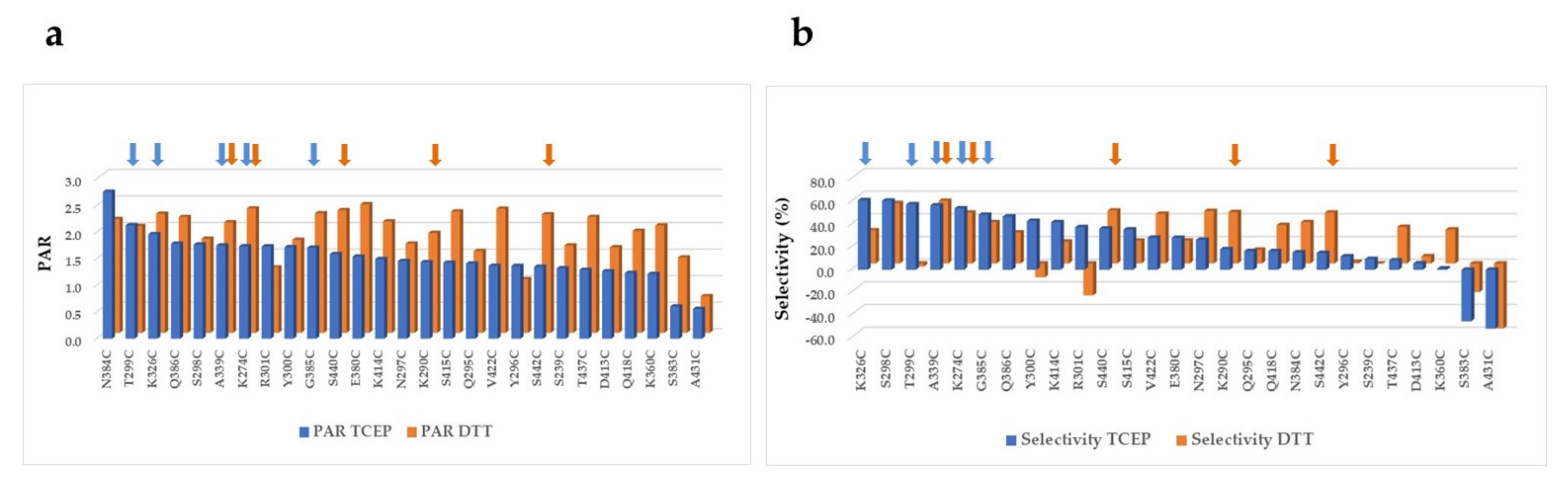
2.3. PEGylation Screening of Single Cysteine Mutants
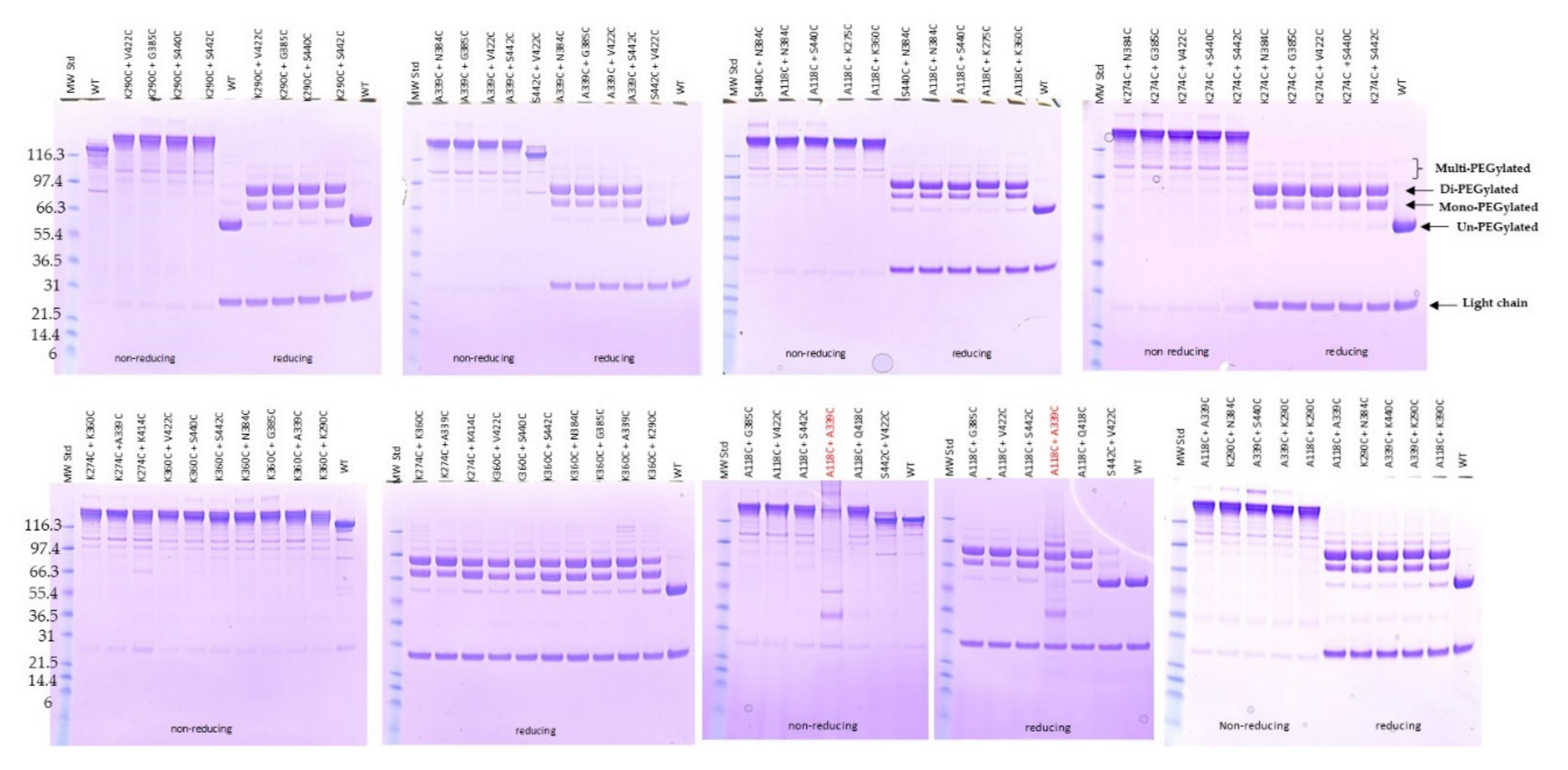
2.4. Expression and Characterization of Engineered Double Cysteine Mutants
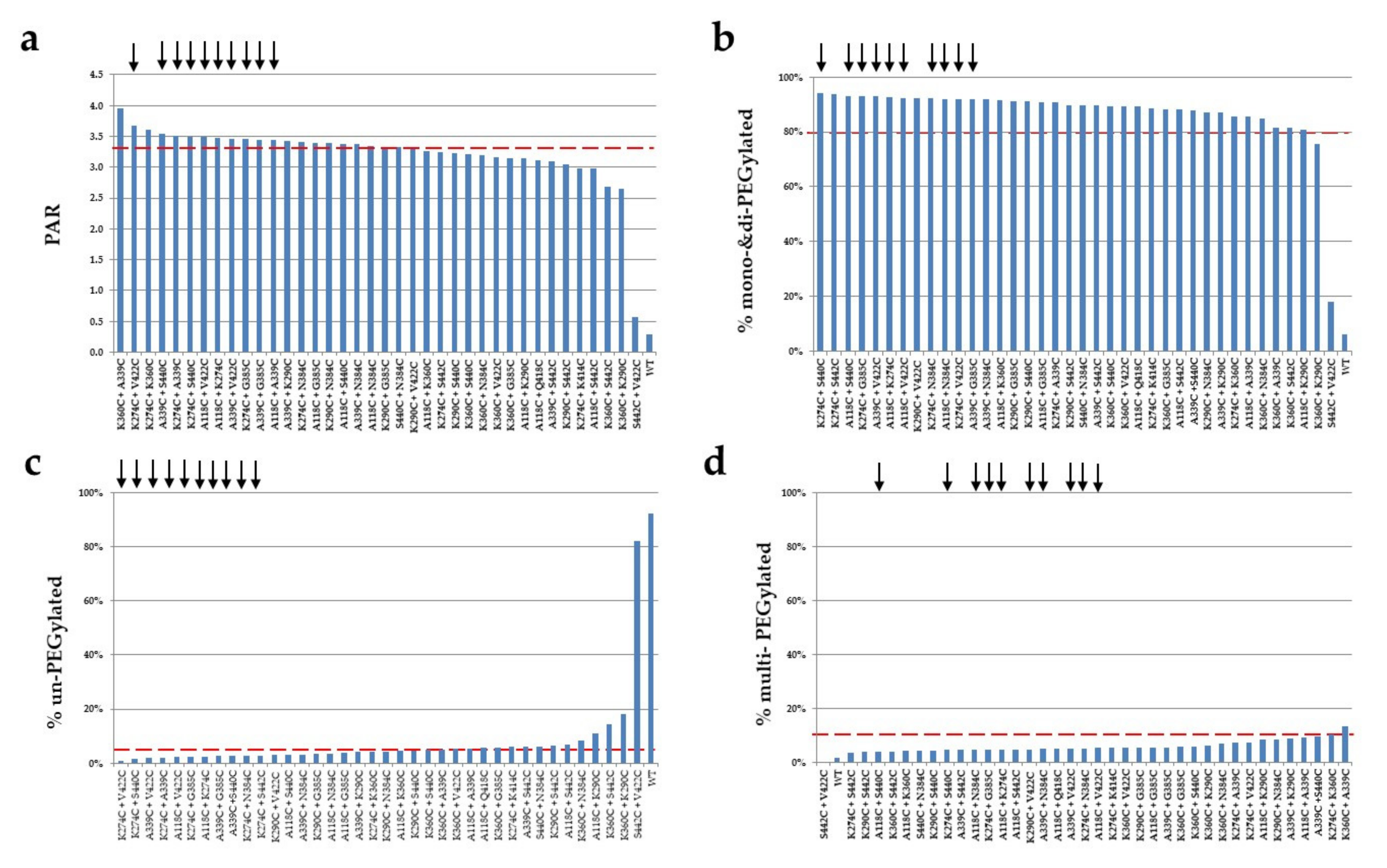
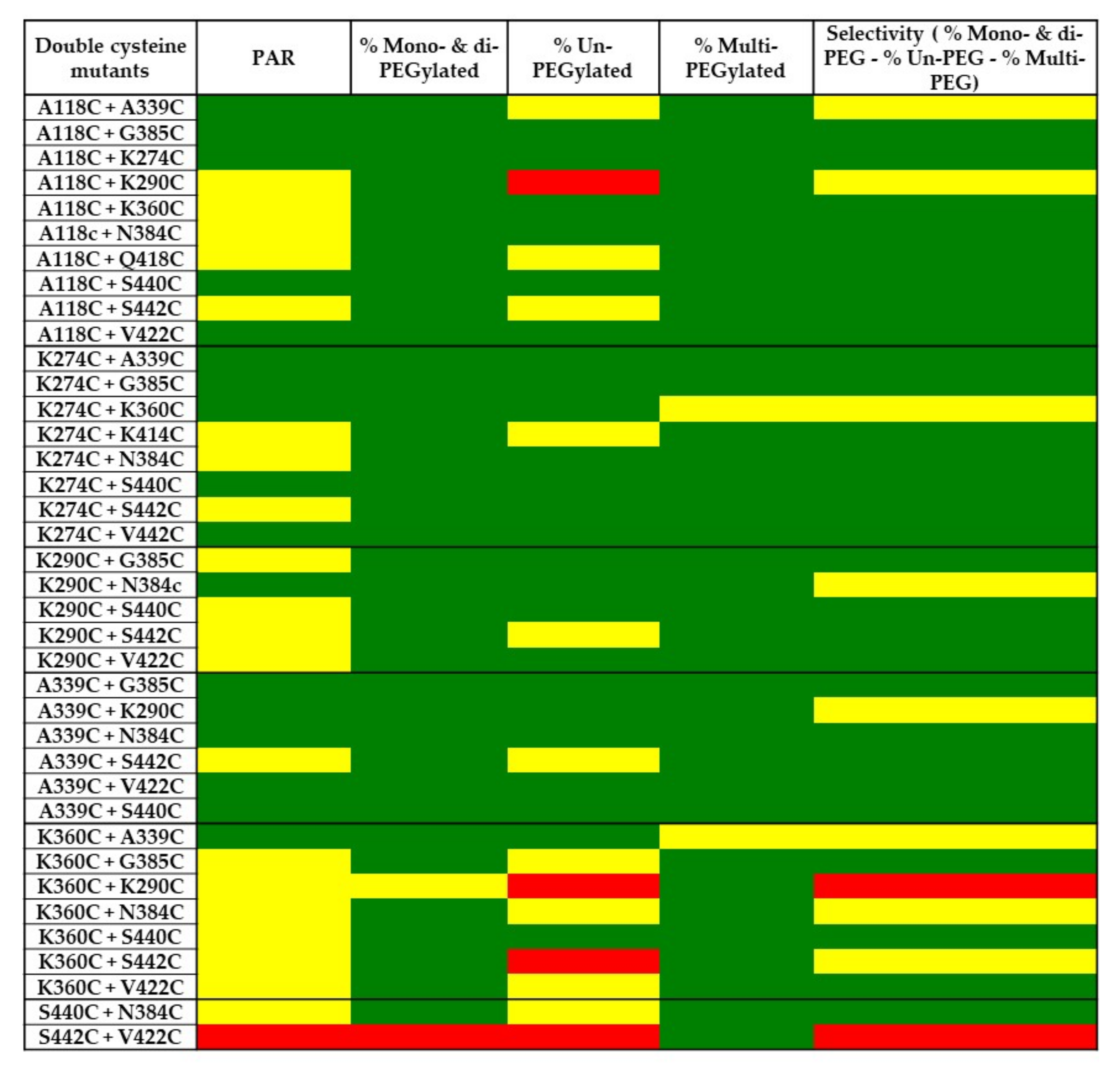
2.5. PEGylation Screening of Double Cysteine Mutants
3. Discussion
4. Methods
4.1. Site-Directed Mutagenesis
4.2. Transfection and Purification
4.3. SDS-PAGE Analysis
4.4. Plasma Stability Study
4.5. SEC-UPLC
4.6. Thermal Stability
4.7. Conjugation
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kaplon, H.; Reichert, J.M. Antibodies to watch in 2021. mAbs 2021, 13, 1860476. [Google Scholar] [CrossRef] [PubMed]
- Khongorzul, P.; Ling, C.J.; Khan, F.U.; Ihsan, A.U.; Zhang, J. Antibody–Drug Conjugates: A Comprehensive Review. Mol. Cancer Res. 2020, 18, 3–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrilli, R.; Pinheiro, D.P.; Oliveira, F.D.C.E.D.; Galvão, G.F.; Marques, L.G.A.; Lopez, R.F.V.; Pessoa, C.; Eloy, J.O. Immunoconjugates for Cancer Targeting: A Review of Antibody-Drug Conjugates and Antibody-Functionalized Nanoparticles. Curr. Med. Chem. 2021, 28, 2485–2520. [Google Scholar] [CrossRef] [PubMed]
- Chau, C.H.; Steeg, P.S.; Figg, W.D. Antibody–drug conjugates for cancer. Lancet 2019, 394, 793–804. [Google Scholar] [CrossRef] [PubMed]
- Von Minckwitz, G.; Huang, C.-S.; Mano, M.S.; Loibl, S.; Mamounas, E.P.; Untch, M.; Wolmark, N.; Rastogi, P.; Schneeweiss, A.; Redondo, A.; et al. Trastuzumab Emtansine for Residual Invasive HER2-Positive Breast Cancer. N. Engl. J. Med. 2019, 380, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaïa, N. Strategies and challenges for the next generation of antibody–drug conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef] [PubMed]
- Goulet, D.R.; Atkins, W.M. Considerations for the Design of Antibody-Based Therapeutics. J. Pharm. Sci. 2020, 109, 74–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, P.J.; Lazar, G.A. Next generation antibody drugs: Pursuit of the ‘high-hanging fruit’. Nat. Rev. Drug Discov. 2018, 17, 197–223. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q. Site-Specific Antibody Conjugation for ADC and Beyond. Biomedicines 2017, 5, 64. [Google Scholar] [CrossRef] [Green Version]
- Maneiro, M.; Forte, N.; Shchepinova, M.M.; Kounde, C.S.; Chudasama, V.; Baker, J.R.; Tate, E.W. Antibody–PROTAC Conjugates Enable HER2-Dependent Targeted Protein Degradation of BRD4. ACS Chem. Biol. 2020, 15, 1306–1312. [Google Scholar] [CrossRef]
- Lehar, S.M.; Pillow, T.; Xu, M.; Staben, L.; Kajihara, K.K.; Vandlen, R.; DePalatis, L.; Raab, H.; Hazenbos, W.L.; Morisaki, J.H.; et al. Novel antibody–antibiotic conjugate eliminates intracellular S. aureus. Nature 2015, 527, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Lacek, K.; Urbanowicz, R.A.; Troise, F.; De Lorenzo, C.; Severino, V.; Di Maro, A.; Tarr, A.; Ferrara, F.; Ploss, A.; Temperton, N.; et al. Dramatic Potentiation of the Antiviral Activity of HIV Antibodies by Cholesterol Conjugation. J. Biol. Chem. 2014, 289, 35015–35028. [Google Scholar] [CrossRef] [Green Version]
- Panowski, S.; Bhakta, S.; Raab, H.; Polakis, P.; Junutula, J.R. Site-specific antibody drug conjugates for cancer therapy. mAbs 2014, 6, 34–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagh, A.; Song, H.; Zeng, M.; Tao, L.; Das, T.K. Challenges and new frontiers in analytical characterization of antibody-drug conjugates. mAbs 2018, 10, 222–243. [Google Scholar] [CrossRef] [PubMed]
- Wakankar, A.; Chen, Y.; Gokarn, Y.; Jacobson, F.S. Analytical methods for physicochemical characterization of antibody drug conjugates. mAbs 2011, 3, 161–172. [Google Scholar] [CrossRef] [Green Version]
- Junutula, J.R.; Raab, H.; Clark, S.; Bhakta, S.; Leipold, D.D.; Weir, S.; Chen, Y.; Simpson, M.; Tsai, S.P.; Dennis, M.S.; et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat. Biotechnol. 2008, 26, 925–932. [Google Scholar] [CrossRef]
- Junutula, J.R.; Flagella, K.M.; Graham, R.A.; Parsons, K.L.; Ha, E.; Raab, H.; Bhakta, S.; Nguyen, T.; Dugger, D.L.; Li, G.; et al. Engineered Thio-Trastuzumab-DM1 Conjugate with an Improved Therapeutic Index to Target Human Epidermal Growth Factor Receptor 2–Positive Breast Cancer. Clin. Cancer Res. 2010, 16, 4769–4778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Junutula, J.R.; Bhakta, S.; Raab, H.; Ervin, K.E.; Eigenbrot, C.; Vandlen, R.; Scheller, R.H.; Lowman, H.B. Rapid identification of reactive cysteine residues for site-specific labeling of antibody-Fabs. J. Immunol. Methods 2008, 332, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, S.C.; Burke, P.J.; Lyon, R.P.; Meyer, D.W.; Sussman, D.; Anderson, M.; Hunter, J.H.; Leiske, C.I.; Miyamoto, J.B.; Nicholas, N.D.; et al. A Potent Anti-CD70 Antibody–Drug Conjugate Combining a Dimeric Pyrrolobenzodiazepine Drug with Site-Specific Conjugation Technology. Bioconjugate Chem. 2013, 24, 1256–1263. [Google Scholar] [CrossRef] [PubMed]
- Stimmel, J.B.; Merrill, B.M.; Kuyper, L.F.; Moxham, C.P.; Hutchins, J.T.; Fling, M.E.; Kull, F.C. Site-specific Conjugation on Serine → Cysteine Variant Monoclonal Antibodies. J. Biol. Chem. 2000, 275, 30445–30450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voynov, V.; Chennamsetty, N.; Kayser, V.; Wallny, H.-J.; Helk, B.; Trout, B.L. Design and Application of Antibody Cysteine Variants. Bioconjugate Chem. 2010, 21, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Tumey, L.N.; Li, F.; Rago, B.; Han, X.; Loganzo, F.; Musto, S.; Graziani, E.I.; Puthenveetil, S.; Casavant, J.; Marquette, K.; et al. Site Selection: A Case Study in the Identification of Optimal Cysteine Engineered Antibody Drug Conjugates. AAPS J. 2017, 19, 1123–1135. [Google Scholar] [CrossRef] [PubMed]
- Shinmi, D.; Taguchi, E.; Iwano, J.; Yamaguchi, T.; Masuda, K.; Enokizono, J.; Shiraishi, Y. One-Step Conjugation Method for Site-Specific Antibody–Drug Conjugates through Reactive Cysteine-Engineered Antibodies. Bioconjugate Chem. 2016, 27, 1324–1331. [Google Scholar] [CrossRef] [PubMed]
- Dragovich, P.S.; Pillow, T.H.; Blake, R.A.; Sadowsky, J.D.; Adaligil, E.; Adhikari, P.; Bhakta, S.; Blaquiere, N.; Chen, J.; Cruz-Chuh, J.D.; et al. Antibody-Mediated Delivery of Chimeric BRD4 Degraders. Part 1: Exploration of Antibody Linker, Payload Loading, and Payload Molecular Properties. J. Med. Chem. 2021, 64, 2534–2575. [Google Scholar] [CrossRef] [PubMed]
- Dragovich, P.S.; Pillow, T.H.; Blake, R.A.; Sadowsky, J.D.; Adaligil, E.; Adhikari, P.; Chen, J.; Corr, N.; Cruz-Chuh, J.D.; Del Rosario, G.; et al. Antibody-Mediated Delivery of Chimeric BRD4 Degraders. Part 2: Improvement of In Vitro Antiproliferation Activity and In Vivo Antitumor Efficacy. J. Med. Chem. 2021, 64, 2576–2607. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Teng, P.; Montgomery, N.T.; Li, X.; Tang, W. Development of Triantennary N-Acetylgalactosamine Conjugates as Degraders for Extracellular Proteins. ACS Central Sci. 2021, 7, 499–506. [Google Scholar] [CrossRef]
- Sondermann, P.; Huber, R.; Oosthuizen, V.; Jacob, U. The 3.2-Å crystal structure of the human IgG1 Fc fragment–FcγRIII complex. Nat. Cell Biol. 2000, 406, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Jaworski, J.; Zhou, Y.; Valente, D.; Cotton, J.; Honey, D.; Boudanova, E.; Beninga, J.; Rao, E.; Wei, R.; et al. Engineered Fc-glycosylation switch to eliminate antibody effector function. mAbs 2020, 12. [Google Scholar] [CrossRef]
- Katorcha, E.; Baskakov, I.V. Analyses of N-linked glycans of PrPScrevealed predominantly 2,6-linked sialic acid residues. FEBS J. 2017, 284, 3727–3738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, B.-Q.; Xu, K.; Liu, L.; Raab, H.; Bhakta, S.; Kenrick, M.; Parsons-Reponte, K.L.; Tien, J.; Yu, S.-F.; Mai, E.; et al. Conjugation site modulates the in vivo stability and therapeutic activity of antibody-drug conjugates. Nat. Biotechnol. 2012, 30, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Shui, W.; Carlson, B.L.; Hu, N.; Rabuka, D.; Lee, J.; Bertozzi, C.R. Site-specific chemical modification of recombinant proteins produced in mammalian cells by using the genetically encoded aldehyde tag. Proc. Natl. Acad. Sci. USA 2009, 106, 3000–3005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeglis, B.M.; Davis, C.B.; Abdel-Atti, D.; Carlin, S.; Chen, A.; Aggeler, R.; Agnew, B.J.; Lewis, J.S. Chemoenzymatic Strategy for the Synthesis of Site-Specifically Labeled Immunoconjugates for Multimodal PET and Optical Imaging. Bioconjugate Chem. 2014, 25, 2123–2128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Q.; Stefano, J.E.; Manning, C.; Kyazike, J.; Chen, B.; Gianolio, D.A.; Park, A.; Busch, M.; Bird, J.; Zheng, X.; et al. Site-Specific Antibody–Drug Conjugation through Glycoengineering. Bioconjugate Chem. 2014, 25, 510–520. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Ramakrishnan, B.; Li, J.; Wang, Y.; Feng, Y.; Prabakaran, P.; Colantonio, S.; A Dyba, M.A.; Qasba, P.K.; Dimitrov, D.S. Site-specific antibody-drug conjugation through an engineered glycotransferase and a chemically reactive sugar. mAbs 2014, 6, 1190–1200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dennler, P.; Chiotellis, A.; Fischer, E.; Brégeon, D.; Belmant, C.; Gauthier, L.; Lhospice, F.; Romagne, F.; Schibli, R. Transglutaminase-Based Chemo-Enzymatic Conjugation Approach Yields Homogeneous Antibody–Drug Conjugates. Bioconjugate Chem. 2014, 25, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Okeley, N.M.; Toki, B.E.; Zhang, X.; Jeffrey, S.C.; Burke, P.J.; Alley, S.C.; Senter, P.D. Metabolic Engineering of Monoclonal Antibody Carbohydrates for Antibody–Drug Conjugation. Bioconjugate Chem. 2013, 24, 1650–1655. [Google Scholar] [CrossRef] [PubMed]
- Strop, P.; Liu, S.-H.; Dorywalska, M.; Delaria, K.; Dushin, R.G.; Tran, T.-T.; Ho, W.-H.; Farias, S.; Casas, M.G.; Abdiche, Y.; et al. Location Matters: Site of Conjugation Modulates Stability and Pharmacokinetics of Antibody Drug Conjugates. Chem. Biol. 2013, 20, 161–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Axup, J.Y.; Bajjuri, K.M.; Ritland, M.; Hutchins, B.M.; Kim, C.H.; Kazane, S.A.; Halder, R.; Forsyth, J.S.; Santidrian, A.F.; Stafin, K.; et al. Synthesis of site-specific antibody-drug conjugates using unnatural amino acids. Proc. Natl. Acad. Sci. USA 2012, 109, 16101–16106. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Nelson, C.G.; Nair, R.R.; Hazlehurst, L.; Moroni, T.; Martinez-Acedo, P.; Nanna, A.R.; Hymel, D.; Burke, T.R.; Rader, C. Stable and Potent Selenomab-Drug Conjugates. Cell Chem. Biol. 2017, 24, 433–442.e6. [Google Scholar] [CrossRef] [Green Version]
- Rabuka, D.; Rush, J.S.; Dehart, G.W.; Wu, P.; Bertozzi, C.R. Site-specific chemical protein conjugation using genetically encoded aldehyde tags. Nat. Protoc. 2012, 7, 1052–1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmerman, E.S.; Heibeck, T.H.; Gill, A.; Li, X.; Murray, C.J.; Madlansacay, M.R.; Tran, C.; Uter, N.T.; Yin, G.; Rivers, P.J.; et al. Production of Site-Specific Antibody–Drug Conjugates Using Optimized Non-Natural Amino Acids in a Cell-Free Expression System. Bioconjugate Chem. 2014, 25, 351–361. [Google Scholar] [CrossRef] [PubMed]
- DiMasi, N.; Fleming, R.; Zhong, H.; Bezabeh, B.; Kinneer, K.; Christie, R.J.; Fazenbaker, C.; Wu, H.; Gao, C. Efficient Preparation of Site-Specific Antibody–Drug Conjugates Using Cysteine Insertion. Mol. Pharm. 2017, 14, 1501–1516. [Google Scholar] [CrossRef] [PubMed]
- Van Brunt, M.P.; Shanebeck, K.; Caldwell, Z.; Johnson, J.; Thompson, P.; Martin, T.; Dong, H.; Li, G.; Xu, H.; D’Hooge, F.; et al. Genetically Encoded Azide Containing Amino Acid in Mammalian Cells Enables Site-Specific Antibody–Drug Conjugates Using Click Cycloaddition Chemistry. Bioconjugate Chem. 2015, 26, 2249–2260. [Google Scholar] [CrossRef] [PubMed]
- Van Geel, R.; Wijdeven, M.A.; Heesbeen, R.; Verkade, J.M.M.; Wasiel, A.A.; Van Berkel, S.S.; Van Delft, F.L. Chemoenzymatic Conjugation of Toxic Payloads to the Globally Conserved N-Glycan of Native mAbs Provides Homogeneous and Highly Efficacious Antibody–Drug Conjugates. Bioconjugate Chem. 2015, 26, 2233–2242. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Y.; Perry, S.R.; Muniz-Medina, V.; Wang, X.; Wetzel, L.K.; Rebelatto, M.C.; Hinrichs, M.J.M.; Bezabeh, B.Z.; Fleming, R.L.; Dimasi, N.; et al. A Biparatopic HER2-Targeting Antibody-Drug Conjugate Induces Tumor Regression in Primary Models Refractory to or Ineligible for HER2-Targeted Therapy. Cancer Cell 2016, 29, 117–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behrens, C.R.; Ha, E.H.; Chinn, L.L.; Bowers, S.; Probst, G.; Fitch-Bruhns, M.; Monteon, J.; Valdiosera, A.; Bermudez, A.; Liao-Chan, S.; et al. Antibody–Drug Conjugates (ADCs) Derived from Interchain Cysteine Cross-Linking Demonstrate Improved Homogeneity and Other Pharmacological Properties over Conventional Heterogeneous ADCs. Mol. Pharm. 2015, 12, 3986–3998. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Ho, S.O.; Gassman, N.; Korlann, Y.; Landorf, E.V.; Collart, F.; Weiss, S. Efficient Site-Specific Labeling of Proteins via Cysteines. Bioconjugate Chem. 2008, 19, 786–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Fang, T.; Boons, G. Preparation of Well-Defined Antibody–Drug Conjugates through Glycan Remodeling and Strain-Promoted Azide–Alkyne Cycloadditions. Angew. Chem. Int. Ed. 2014, 53, 7179–7182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohri, R.; Bhakta, S.; Fourie-O’Donohue, A.; Cruz-Chuh, J.D.; Tsai, S.P.; Cook, R.; Wei, B.; Ng, C.; Wong, A.W.; Bos, A.B.; et al. High-Throughput Cysteine Scanning to Identify Stable Antibody Conjugation Sites for Maleimide- and Disulfide-Based Linkers. Bioconjugate Chem. 2018, 29, 473–485. [Google Scholar] [CrossRef] [PubMed]
- Puthenveetil, S.; Musto, S.; Loganzo, F.; Tumey, L.N.; O’Donnell, C.J.; Graziani, E. Development of Solid-Phase Site-Specific Conjugation and Its Application toward Generation of Dual Labeled Antibody and Fab Drug Conjugates. Bioconjugate Chem. 2016, 27, 1030–1039. [Google Scholar] [CrossRef] [PubMed]
- Durocher, Y. High-level and high-throughput recombinant protein production by transient transfection of suspension-growing human 293-EBNA1 cells. Nucleic Acids Res. 2002, 30, e9. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Zhong, K.; Tian, H.; Gao, X.; Xu, X.; Yin, X.; Yao, W. Characterization of a monoPEG20000-Endostar. Int. J. Biol. Macromol. 2010, 46, 331–336. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| mAb | Secondary Structure and Location of Mutation in Fc | mAb Concentration (µg/mL) * | Stability (Tm1, °C) ** | Half-Ab *** | Aggregates *** |
|---|---|---|---|---|---|
| S239C | Loop, CH2 | 180, 190 | 69.8 | ||
| K274C | β-sheet, CH2 | 210 | 71.9 | ||
| K290C | Loop, CH2 | 130, 250 | 72.2 | ||
| Q295C | Loop, CH2 | 240, 260 | 71.2 | ||
| Y296C | Loop, CH2 | 250, 270 | 70.6 | ||
| N297C | Loop, CH2 | 180, 260 | 57.6 | ++ | |
| S298C | Loop, CH2 | 190, 220 | 72.0 | ++ | |
| T299C | Loop, CH2 | 200, 260 | 61.2 | ||
| Y300C | Loop, CH2 | 170, 260 | 62.7 | ||
| R301C | Loop, CH2 | 240 | 62.3 | + | |
| K326C | Turn, CH2 | 170 | 66.5 | ||
| A339C | Loop, CH2 | 100 | 68.0 | ||
| K360C | Loop, CH3 | 190 | 70.1 | ||
| E380C | β-sheet, CH3 | 130, 200 | 63.3 | + | |
| S383C | Loop, CH3 | 190, 240 | 68.8 | ||
| N384C | Loop, CH3 | 160, 170 | 68.7 | + | |
| G385C | Loop, CH3 | 200, 210 | 69.7 | ++ | |
| Q386C | Loop, CH3 | 200 | 70.9 | ++ | |
| D413C | β-sheet, CH3 | 110 | 69.9 | ||
| K414C | α-helix, CH3 | 210 | 71.7 | ||
| S415C | α-helix, CH3 | 160, 160 | 68.7 | ||
| Q418C | α-helix, CH3 | 180 | 70.2 | + | |
| V422C | Loop, CH3 | 240 | 68.3 | + | |
| A431C | Loop, CH3 | 170 | 67.0 | ||
| T437C | β-sheet, CH3 | 190 | 67.9 | + | |
| S440C | β-sheet, CH3 | 150, 150 | 69.4 | ||
| S442C | Loop, CH3 | 170, 220 | 68.4 | ||
| Wild-type | 140, 230 | 68.8 |
| mAb | mAb Concentration (µg/mL) * | Stability (Tm1, °C) ** | % Monomer *** |
|---|---|---|---|
| A118C + N384C | 138 | 67.7 | 93.3 |
| A118C + G385C | 336 | 68.2 | 94.9 |
| A118C + V422C | 337 | 67.9 | 97.4 |
| A118C + S440C | 177 | 69.8 | 89.2 |
| A118C + S442C | 346 | 68.3 | 95.7 |
| A118C + K290C | 424 | ND | NA |
| A118C + K274C | 190 | 69.3 | 95.7 |
| A118C + A339C | 414 | 58.8 | 99.4 |
| A118C + K360C | 189 | 68.3 | 96.0 |
| A118C + Q418C | 395 | 69.0 | 93.9 |
| K274C + N384C | 300 | 69.6 | 87.0 |
| K274C + G385C | 248 | 70.8 | 84.9 |
| K274C + V422C | 499 | 69.9 | 94.3 |
| K274C + S440C | 361 | 71.7 | 85.4 |
| K274C + S442C | 346 | 69.5 | 94.3 |
| K274C + K360C | 381 | 70.4 | 97.4 |
| K274C + A339C | 296 | 66.4 | 97.9 |
| K274C + K414C | 332 | 72.3 | 96.5 |
| K290C + N384C | 146 | ND | 92.1 |
| K290C + G385C | 224 | 74.1 | 89.4 |
| K290C + V422C | 361 | 72.9 | 92.9 |
| K290C + S440C | 243 | 74.2 | 89.3 |
| K290C + S442C | 329 | 73.3 | 94.4 |
| A339C + N384C | 267 | 66.0 | 89.5 |
| A339C + G385C | 335 | 65.0 | 84.9 |
| A339C + V422C | 343 | 65.9 | 93.6 |
| A339C + S440C | 365 | ND | 77.0 |
| A339C + S442C | 366 | 65.5 | 94.0 |
| A339C + K290C | 372 | ND | 96.7 |
| K360C + V422C | 320 | 68.6 | 97.3 |
| K360C + S440C | 476 | 69.3 | 90.8 |
| K360C + S442C | 323 | 69.0 | 97.4 |
| K360C + N384C | 335 | 69.0 | 89.0 |
| K360C + G385C | 330 | 68.3 | 90.1 |
| K360C + A339C | 375 | 66.9 | 98.5 |
| K360C + K290C | 33 | 72.5 | 98.1 |
| S440C + N384C | 308 | 70.2 | 45.7 |
| S442C + V422C | 345 | 69.5 | 97.6 |
| Wild-type | 293 | 68.5 | 98.1 |
| Mutant | PAR | % Mono- and Di-PEGylated | % Multi-PEGylated | % Un-PEGylated | Selectivity (% Mono- and Di-PEG—Un-PEG—Multi-PEG) |
|---|---|---|---|---|---|
| A118C + A339C | 3.4 | 85.5% | 9.0% | 5.4% | 71% |
| A118C + G385C | 3.4 | 90.9% | 5.4% | 3.7% | 82% |
| A118C + K274C | 3.5 | 92.8% | 4.7% | 2.5% | 86% |
| A118C + N384C | 3.4 | 92.0% | 4.6% | 3.4% | 84% |
| A118C + S440C | 3.4 | 93.0% | 4.0% | 3.0% | 86% |
| A118C + V422C | 3.5 | 92.4% | 5.3% | 2.3% | 85% |
| K274C + A339C | 3.5 | 90.8% | 7.2% | 2.0% | 82% |
| K274C + G385C | 3.5 | 92.9% | 4.6% | 2.4% | 86% |
| K274C + N384C | 3.4 | 92.1% | 5.1% | 2.8% | 84% |
| K274C + S440C | 3.5 | 94.0% | 4.5% | 1.5% | 88% |
| K274C + V422C | 3.7 | 92.0% | 7.2% | 0.8% | 84% |
| K290C + N384C | 3.4 | 87.2% | 8.5% | 4.4% | 74% |
| A339C + G385C | 3.4 | 92.0% | 5.5% | 2.6% | 84% |
| A339C + K290C | 3.4 | 87.0% | 9.0% | 4.1% | 74% |
| A339C + N384C | 3.4 | 91.9% | 4.9% | 3.2% | 84% |
| A339C + V422C | 3.5 | 92.9% | 5.1% | 2.0% | 86% |
| A339C + S440C | 3.5 | 87.9% | 9.5% | 2.6% | 76% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Q.; Kyazike, J.; Boudanova, E.; Drzyzga, M.; Honey, D.; Cost, R.; Hou, L.; Duffieux, F.; Brun, M.-P.; Park, A.; et al. Site-Specific Antibody Conjugation to Engineered Double Cysteine Residues. Pharmaceuticals 2021, 14, 672. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14070672
Zhou Q, Kyazike J, Boudanova E, Drzyzga M, Honey D, Cost R, Hou L, Duffieux F, Brun M-P, Park A, et al. Site-Specific Antibody Conjugation to Engineered Double Cysteine Residues. Pharmaceuticals. 2021; 14(7):672. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14070672
Chicago/Turabian StyleZhou, Qun, Josephine Kyazike, Ekaterina Boudanova, Michael Drzyzga, Denise Honey, Robert Cost, Lihui Hou, Francis Duffieux, Marie-Priscille Brun, Anna Park, and et al. 2021. "Site-Specific Antibody Conjugation to Engineered Double Cysteine Residues" Pharmaceuticals 14, no. 7: 672. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14070672