
Retinoids Delivery Systems in Cancer: Liposomal Fenretinide for Neuroectodermal-Derived Tumors

 , ,
, ,  , , and
, , and
Abstract
:1. Introduction
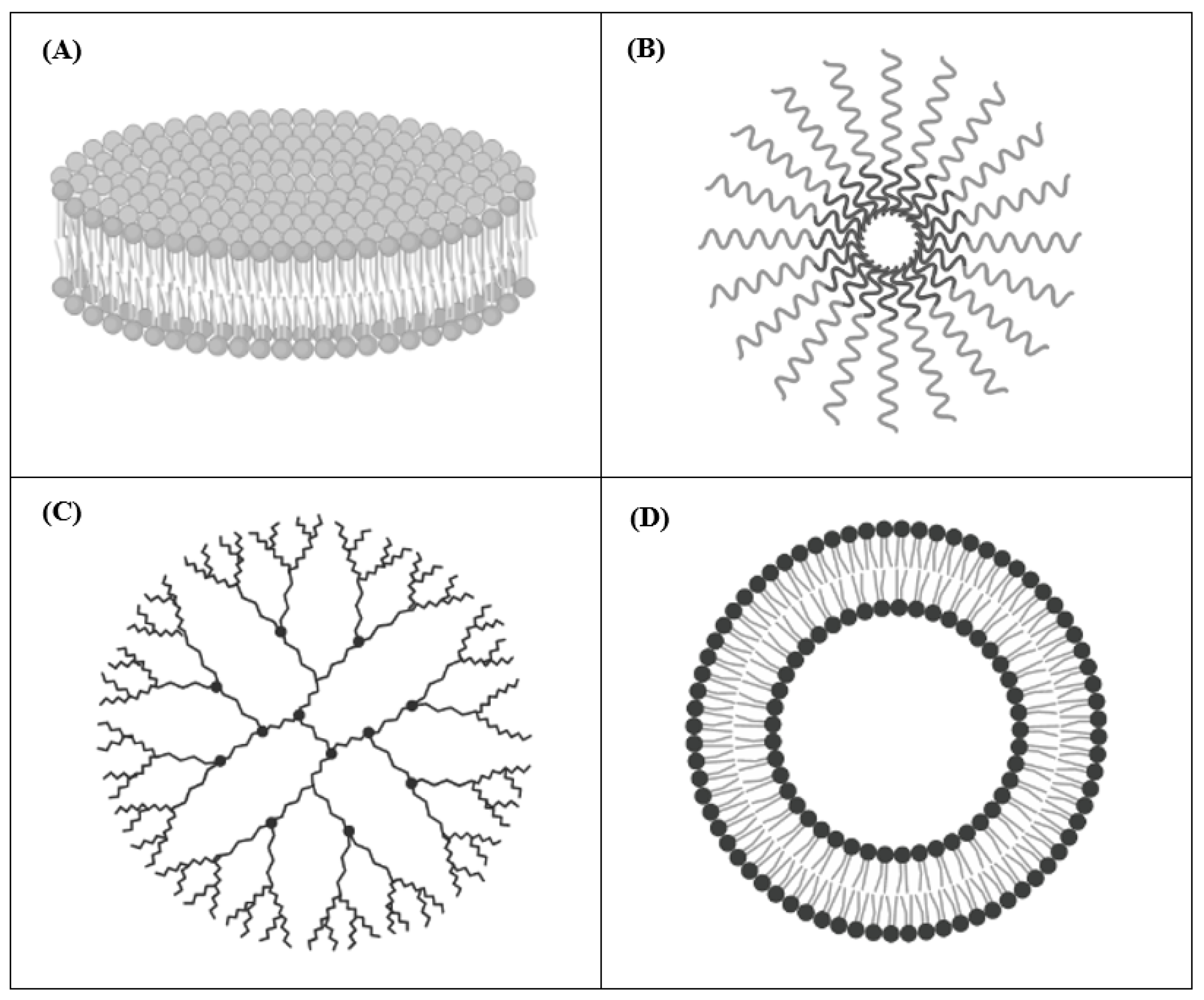
2. Drug Delivery Systems for Cancer Therapy
2.1. Nanodisks (NDs)
2.2. Polymeric Micelles
2.3. Dendrimers
2.4. Liposomes
2.4.1. Neuroectodermal-Derived Tumors
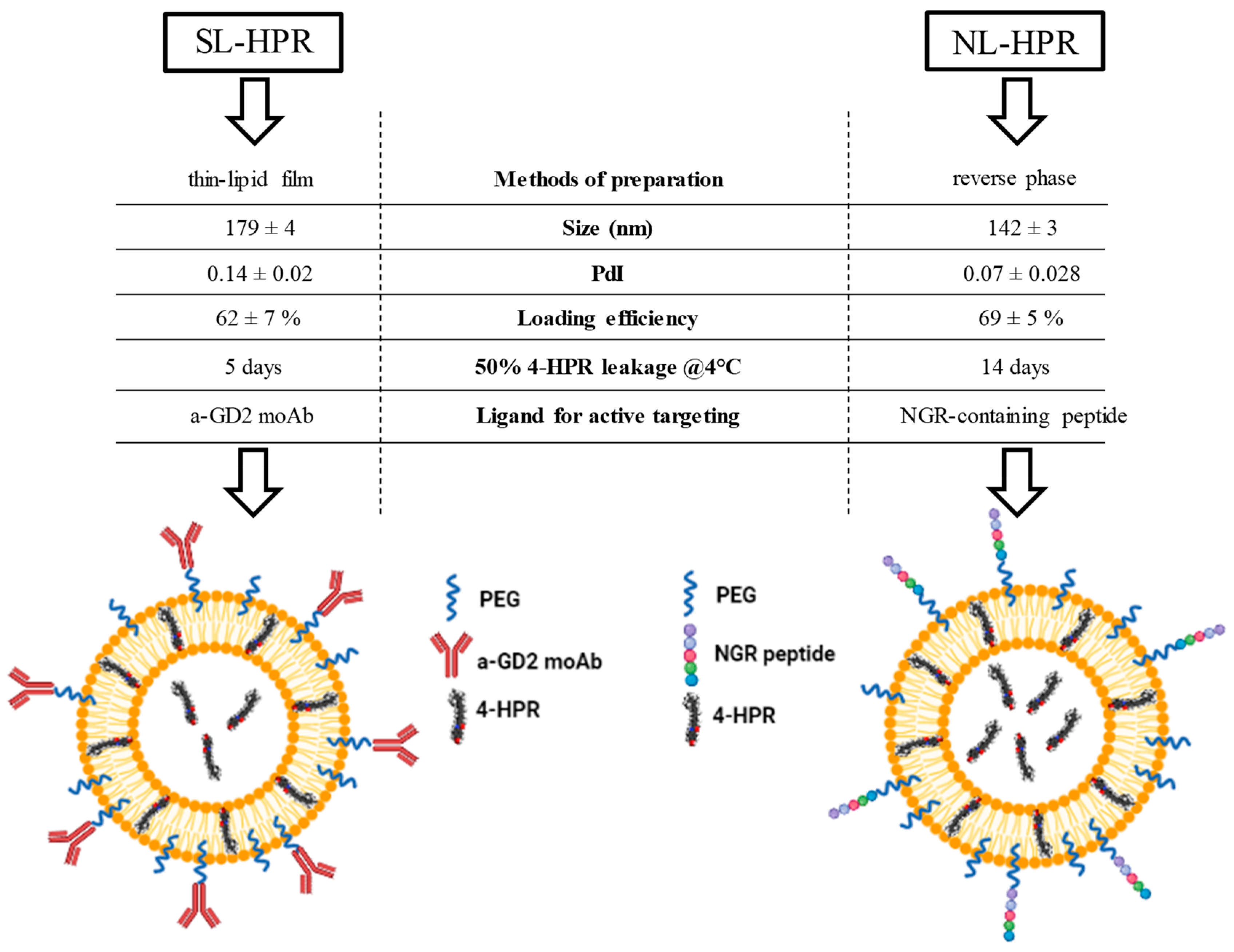
2.4.2. Development, Characterization, and Functionality of Fenretinide-Loaded Liposomes
2.4.3. Fenretinide-loaded Liposomes for Tumor Targeting
2.4.4. Fenretinide-Loaded Liposomes for Tumor Vasculature Targeting
3. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 4-HPR | N-4-hydroxyphenyl retinamide (fenretinide) |
| NB | neuroblastoma |
| SL | Stealth Liposomes |
| SIL | Stealth ImmunoLiposomes |
References
- Blomhoff, R.; Blomhoff, H.K. Overview of retinoid metabolism and function. J. Neurobiol. 2006, 66, 606–630. [Google Scholar] [CrossRef] [PubMed]
- Campo-Paysaa, F.; Marlétaz, F.; Laudet, V.; Schubert, M. Retinoic acid signaling in development: Tissue-specific functions and evolutionary origins. Genesis 2008, 46, 640–656. [Google Scholar] [CrossRef] [PubMed]
- Niederreither, K.; Subbarayan, V.; Dollé, P.; Chambon, P. Embryonic retinoic acid synthesis is essential for early mouse post-implantation development. Nat. Genet. 1999, 21, 444–448. [Google Scholar] [CrossRef]
- Sporn, M.B.; Dunlop, N.M.; Newton, D.L.; Henderson, W.R. Relationships between structure and activity of retinoids. Nature 1976, 263, 110–113. [Google Scholar] [CrossRef] [PubMed]
- Kiser, P.D.; Golczak, M.; Palczewski, K. Chemistry of the retinoid (visual) cycle. Chem. Rev. 2014, 114, 194–232. [Google Scholar] [CrossRef] [PubMed]
- Beckenbach, L.; Baron, J.M.; Merk, H.F.; Loffler, H.; Amann, P.M. Retinoid treatment of skin diseases. Eur. J. Dermatol. 2015, 25, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Dobrotkova, V.; Chlapek, P.; Mazanek, P.; Sterba, J.; Veselska, R. Traffic lights for retinoids in oncology: Molecular markers of retinoid resistance and sensitivity and their use in the management of cancer differentiation therapy. BMC Cancer 2018, 18, 1059. [Google Scholar] [CrossRef]
- Bollag, W. Retinoids and cancer. Cancer Chemother. Pharmacol. 1979, 3, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Zusi, F.C.; Lorenzi, M.V.; Vivat-Hannah, V. Selective retinoids and rexinoids in cancer therapy and chemoprevention. Drug Discov. Today 2002, 7, 1165–1174. [Google Scholar] [CrossRef]
- Ponzoni, M.; Bocca, P.; Chiesa, V.; Decensi, A.; Pistoia, V.; Raffaghello, L.; Rozzo, C.; Montaldo, P.G. Differential effects of N-(4-hydroxyphenyl)retinamide and retinoic acid on neuroblastoma cells: Apoptosis versus differentiation. Cancer Res. 1995, 55, 853–861. [Google Scholar]
- Montaldo, P.G.; Pagnan, G.; Pastorino, F.; Chiesa, V.; Raffaghello, L.; Kirchmeier, M.; Allen, T.M.; Ponzoni, M. N-(4-hydroxyphenyl) retinamide is cytotoxic to melanoma cells in vitro through induction of programmed cell death. Int. J. Cancer 1999, 81, 262–267. [Google Scholar] [CrossRef]
- Ferrara, F.F.; Fazi, F.; Bianchini, A.; Padula, F.; Gelmetti, V.; Minucci, S.; Mancini, M.; Pelicci, P.G.; Lo Coco, F.; Nervi, C. Histone deacetylase-targeted treatment restores retinoic acid signaling and differentiation in acute myeloid leukemia. Cancer Res. 2001, 61, 2–7. [Google Scholar] [PubMed]
- Liu, M.; Iavarone, A.; Freedman, L.P. Transcriptional activation of the human p21(WAF1/CIP1) gene by retinoic acid receptor. Correlation with retinoid induction of U937 cell differentiation. J. Biol. Chem. 1996, 271, 31723–31728. [Google Scholar] [CrossRef] [Green Version]
- Johnson, K.J. The retinoids: Biology, chemistry and medicine (second edition) Edited by M B Sporn, A B Roberts and the late D S Goodman. pp 311. Raven Press, NY. 1993. $250.00 ISBN 0-7817-0082-5. Biochem. Educ. 1994, 22, 56. [Google Scholar] [CrossRef]
- Hu, J.; Liu, Y.F.; Wu, C.F.; Xu, F.; Shen, Z.X.; Zhu, Y.M.; Li, J.M.; Tang, W.; Zhao, W.L.; Wu, W.; et al. Long-term efficacy and safety of all-trans retinoic acid/arsenic trioxide-based therapy in newly diagnosed acute promyelocytic leukemia. Proc. Natl. Acad. Sci. USA 2009, 106, 3342–3347. [Google Scholar] [CrossRef] [Green Version]
- Villablanca, J.G.; Khan, A.A.; Avramis, V.I.; Seeger, R.C.; Matthay, K.K.; Ramsay, N.K.; Reynolds, C.P. Phase I trial of 13-cis-retinoic acid in children with neuroblastoma following bone marrow transplantation. J. Clin. Oncol. 1995, 13, 894–901. [Google Scholar] [CrossRef]
- Reynolds, C.P.; Matthay, K.K.; Villablanca, J.G.; Maurer, B.J. Retinoid therapy of high-risk neuroblastoma. Cancer Lett. 2003, 197, 185–192. [Google Scholar] [CrossRef]
- Ferreira, R.; Napoli, J.; Enver, T.; Bernardino, L.; Ferreira, L. Advances and challenges in retinoid delivery systems in regenerative and therapeutic medicine. Nat. Commun. 2020, 11, 4265. [Google Scholar] [CrossRef]
- David, M.; Hodak, E.; Lowe, N.J. Adverse effects of retinoids. Med. Toxicol. Advers. Drug Exp. 1988, 3, 273–288. [Google Scholar] [CrossRef]
- LiverTox: Clinical and Research Information on Drug-Induced Liver Injury; National Institute of Diabetes and Digestive and Kidney Diseases: Bethesda, MD, USA, 2012.
- Khalil, S.; Bardawil, T.; Stephan, C.; Darwiche, N.; Abbas, O.; Kibbi, A.G.; Nemer, G.; Kurban, M. Retinoids: A journey from the molecular structures and mechanisms of action to clinical uses in dermatology and adverse effects. J. Dermatol. Treat. 2017, 28, 684–696. [Google Scholar] [CrossRef]
- Goncalves, A.; Estevinho, B.N.; Rocha, F. Formulation approaches for improved retinoids delivery in the treatment of several pathologies. Eur. J. Pharm. Biopharm. 2019, 143, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.J. Trifarotene: First Approval. Drugs 2019, 79, 1905–1909. [Google Scholar] [CrossRef] [PubMed]
- Torrisi, R.; Decensi, A. Fenretinide and cancer prevention. Curr. Oncol. Rep. 2000, 2, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Kelloff, G.J.; Crowell, J.A.; Boone, C.W.; Steele, V.E.; Lubet, R.A.; Greenwald, P.; Alberts, D.S.; Covey, J.M.; Doody, L.A.; Knapp, G.G.; et al. Strategy and planning for chemopreventive drug development: Clinical development plans. Chemoprevention Branch and Agent Development Committee. National Cancer Institute. J. Cell Biochem. Suppl. 1994, 20, 55–62. [Google Scholar] [CrossRef]
- Mehta, R.G.; Moon, R.C.; Hawthorne, M.; Formelli, F.; Costa, A. Distribution of fenretinide in the mammary gland of breast cancer patients. Eur. J. Cancer 1991, 27, 138–141. [Google Scholar] [CrossRef]
- Cooper, J.P.; Reynolds, C.P.; Cho, H.; Kang, M.H. Clinical development of fenretinide as an antineoplastic drug: Pharmacology perspectives. Exp. Biol. Med. (Maywood) 2017, 242, 1178–1184. [Google Scholar] [CrossRef] [Green Version]
- Formelli, F.; Barua, A.B.; Olson, J.A. Bioactivities of N-(4-hydroxyphenyl) retinamide and retinoyl beta-glucuronide. FASEB J. 1996, 10, 1014–1024. [Google Scholar] [CrossRef]
- Trapasso, E.; Cosco, D.; Celia, C.; Fresta, M.; Paolino, D. Retinoids: New use by innovative drug-delivery systems. Expert Opin. Drug Deliv. 2009, 6, 465–483. [Google Scholar] [CrossRef]
- Torchilin, V. Tumor delivery of macromolecular drugs based on the EPR effect. Adv. Drug Deliv. Rev. 2011, 63, 131–135. [Google Scholar] [CrossRef]
- Attia, M.F.; Anton, N.; Wallyn, J.; Omran, Z.; Vandamme, T.F. An overview of active and passive targeting strategies to improve the nanocarriers efficiency to tumour sites. J. Pharm. Pharmacol. 2019, 71, 1185–1198. [Google Scholar] [CrossRef] [Green Version]
- Pastorino, F.; Marimpietri, D.; Brignole, C.; Di Paolo, D.; Pagnan, G.; Daga, A.; Piccardi, F.; Cilli, M.; Allen, T.M.; Ponzoni, M. Ligand-targeted liposomal therapies of neuroblastoma. Curr. Med. Chem. 2007, 14, 3070–3078. [Google Scholar] [CrossRef] [PubMed]
- Corti, A.; Pastorino, F.; Curnis, F.; Arap, W.; Ponzoni, M.; Pasqualini, R. Targeted Drug Delivery and Penetration Into Solid Tumors. Med. Res. Rev. 2012, 32, 1078–1091. [Google Scholar] [CrossRef] [PubMed]
- Pastorino, F.; Brignole, C.; Di Paolo, D.; Perri, P.; Curnis, F.; Corti, A.; Ponzoni, M. Overcoming Biological Barriers in Neuroblastoma Therapy: The Vascular Targeting Approach with Liposomal Drug Nanocarriers. Small 2019, 15, e1804591. [Google Scholar] [CrossRef]
- Redmond, K.A.; Nguyen, T.S.; Ryan, R.O. All-trans-retinoic acid nanodisks. Int. J. Pharm. 2007, 339, 246–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, B.N.; Pfeffer, C.M.; Singh, A.T.K. Update on Nanotechnology-based Drug Delivery Systems in Cancer Treatment. Anticancer Res. 2017, 37, 5975–5981. [Google Scholar] [CrossRef] [Green Version]
- Ryan, R.O. Nanodisks: Hydrophobic drug delivery vehicles. Expert Opin. Drug Deliv. 2008, 5, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.T.; Evens, A.M.; Anderson, R.J.; Beckstead, J.A.; Sankar, N.; Sassano, A.; Bhalla, S.; Yang, S.; Platanias, L.C.; Forte, T.M.; et al. All trans retinoic acid nanodisks enhance retinoic acid receptor mediated apoptosis and cell cycle arrest in mantle cell lymphoma. Br. J. Haematol. 2010, 150, 158–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stauffer, R.G.; Mohammad, M.; Singh, A.T. Novel Nanoscale Delivery Particles Encapsulated with Anticancer Drugs, All-trans Retinoic Acid or Curcumin, Enhance Apoptosis in Lymphoma Cells Predominantly Expressing CD20 Antigen. Anticancer Res. 2015, 35, 6425–6429. [Google Scholar]
- Buehler, D.C.; Toso, D.B.; Kickhoefer, V.A.; Zhou, Z.H.; Rome, L.H. Vaults engineered for hydrophobic drug delivery. Small 2011, 7, 1432–1439. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, B.; Biswas, S. Polymeric micelles in cancer therapy: State of the art. J. Control. Release 2021, 332, 127–147. [Google Scholar] [CrossRef]
- Valerii, M.C.; Benaglia, M.; Caggiano, C.; Papi, A.; Strillacci, A.; Lazzarini, G.; Campieri, M.; Gionchetti, P.; Rizzello, F.; Spisni, E. Drug delivery by polymeric micelles: An in vitro and in vivo study to deliver lipophilic substances to colonocytes and selectively target inflamed colon. Nanomedicine 2013, 9, 675–685. [Google Scholar] [CrossRef]
- Orienti, I.; Salvati, V.; Sette, G.; Zucchetti, M.; Bongiorno-Borbone, L.; Peschiaroli, A.; Zolla, L.; Francescangeli, F.; Ferrari, M.; Matteo, C.; et al. A novel oral micellar fenretinide formulation with enhanced bioavailability and antitumour activity against multiple tumours from cancer stem cells. J. Exp. Clin. Cancer Res. 2019, 38, 373. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.C.; MacKay, J.A.; Fréchet, J.M.; Szoka, F.C. Designing dendrimers for biological applications. Nat. Biotechnol. 2005, 23, 1517–1526. [Google Scholar] [CrossRef]
- Palmerston Mendes, L.; Pan, J.; Torchilin, V.P. Dendrimers as Nanocarriers for Nucleic Acid and Drug Delivery in Cancer Therapy. Molecules 2017, 22, 1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tekade, R.K.; Dutta, T.; Tyagi, A.; Bharti, A.C.; Das, B.C.; Jain, N.K. Surface-engineered dendrimers for dual drug delivery: A receptor up-regulation and enhanced cancer targeting strategy. J. Drug Target. 2008, 16, 758–772. [Google Scholar] [CrossRef]
- Pan, J.; Attia, S.A.; Filipczak, N.; Torchilin, V.P. 10—Dendrimers for drug delivery purposes. In Nanoengineered Biomaterials for Advanced Drug Delivery; Mozafari, M., Ed.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 201–242. [Google Scholar]
- Wang, Y.; Wang, H.; Lv, X.; Liu, C.; Qi, L.; Song, X.; Yu, A. Enhancement of all-trans retinoic acid-induced differentiation by pH-sensitive nanoparticles for solid tumor cells. Macromol. Biosci. 2014, 14, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Yalcin, S.; Erkan, M.; Unsoy, G.; Parsian, M.; Kleeff, J.; Gunduz, U. Effect of gemcitabine and retinoic acid loaded PAMAM dendrimer-coated magnetic nanoparticles on pancreatic cancer and stellate cell lines. Biomed. Pharmacother. 2014, 68, 737–743. [Google Scholar] [CrossRef]
- Allam, A.; Thomsen, A.R.; Gothwal, M.; Saha, D.; Maurer, J.; Brunner, T.B. Pancreatic stellate cells in pancreatic cancer: In focus. Pancreatology 2017, 17, 514–522. [Google Scholar] [CrossRef] [PubMed]
- Akbarzadeh, A.; Rezaei-Sadabady, R.; Davaran, S.; Joo, S.W.; Zarghami, N.; Hanifehpour, Y.; Samiei, M.; Kouhi, M.; Nejati-Koshki, K. Liposome: Classification, preparation, and applications. Nanoscale Res. Lett. 2013, 8, 102. [Google Scholar] [CrossRef] [Green Version]
- Kopeckova, K.; Eckschlager, T.; Sirc, J.; Hobzova, R.; Plch, J.; Hrabeta, J.; Michalek, J. Nanodrugs used in cancer therapy. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc. Czech. Repub. 2019, 163, 122–131. [Google Scholar] [CrossRef] [Green Version]
- Alavi, M.; Hamidi, M. Passive and active targeting in cancer therapy by liposomes and lipid nanoparticles. Drug Metab. Pers. Ther. 2019, 34. [Google Scholar] [CrossRef] [PubMed]
- Barenholz, Y. Doxil (R)—The first FDA-approved nano-drug: Lessons learned. J. Control. Release 2012, 160, 117–134. [Google Scholar] [CrossRef] [PubMed]
- Gabizon, A.A.; Patil, Y.; La-Beck, N.M. New insights and evolving role of pegylated liposomal doxorubicin in cancer therapy. Drug Resist. Updates 2016, 29, 90–106. [Google Scholar] [CrossRef]
- Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal Formulations in Clinical Use: An Updated Review. Pharmaceutics 2017, 9, 12. [Google Scholar] [CrossRef]
- Viswanathan, S.; Berlin Grace, V.M.; Danisha, J.P. Enhancement of tumor suppressor RAR-β protein expression by cationic liposomal-ATRA treatment in benzo(a)pyrene-induced lung cancer mice model. Naunyn Schmiedebergs Arch. Pharmacol. 2019, 392, 415–426. [Google Scholar] [CrossRef]
- Cristiano, M.C.; Cosco, D.; Celia, C.; Tudose, A.; Mare, R.; Paolino, D.; Fresta, M. Anticancer activity of all-trans retinoic acid-loaded liposomes on human thyroid carcinoma cells. Colloids Surf. B Biointerfaces 2017, 150, 408–416. [Google Scholar] [CrossRef]
- Kawakami, S.; Suzuki, S.; Yamashita, F.; Hashida, M. Induction of apoptosis in A549 human lung cancer cells by all-trans retinoic acid incorporated in DOTAP/cholesterol liposomes. J. Control. Release 2006, 110, 514–521. [Google Scholar] [CrossRef]
- Simile, M.M.; Pagnan, G.; Pastorino, F.; Brignole, C.; De Miglio, M.R.; Muroni, M.R.; Asara, G.; Frau, M.; Seddaiu, M.A.; Calvisi, D.F.; et al. Chemopreventive N-(4-hydroxyphenyl)retinamide (fenretinide) targets deregulated NF-{kappa}B and Mat1A genes in the early stages of rat liver carcinogenesis. Carcinogenesis 2005, 26, 417–427. [Google Scholar] [CrossRef]
- Pagnan, G.; Montaldo, P.G.; Pastorino, F.; Raffaghello, L.; Kirchmeier, M.; Allen, T.M.; Ponzoni, M. GD2-mediated melanoma cell targeting and cytotoxicity of liposome-entrapped fenretinide. Int. J. Cancer 1999, 81, 268–274. [Google Scholar] [CrossRef]
- Raffaghello, L.; Pagnan, G.; Pastorino, F.; Cosimo, E.; Brignole, C.; Marimpietri, D.; Montaldo, P.G.; Gambini, C.; Allen, T.M.; Bogenmann, E.; et al. In vitro and in vivo antitumor activity of liposomal Fenretinide targeted to human neuroblastoma. Int. J. Cancer 2003, 104, 559–567. [Google Scholar] [CrossRef]
- Technau, U.; Scholz, C.B. Origin and evolution of endoderm and mesoderm. Int. J. Dev. Biol. 2003, 47, 531–539. [Google Scholar] [PubMed]
- Solnica-Krezel, L.; Sepich, D.S. Gastrulation: Making and shaping germ layers. Annu. Rev. Cell Dev. Biol. 2012, 28, 687–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferretti, E.; Hadjantonakis, A.K. Mesoderm specification and diversification: From single cells to emergent tissues. Curr. Opin. Cell Biol. 2019, 61, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Zorn, A.M.; Wells, J.M. Vertebrate endoderm development and organ formation. Annu. Rev. Cell Dev. Biol. 2009, 25, 221–251. [Google Scholar] [CrossRef] [Green Version]
- Elshazzly, M.; Lopez, M.J.; Reddy, V.; Caban, O. Embryology, Central Nervous System. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Darnell, D.; Gilbert, S.F. Neuroembryology. Wiley Interdiscip Rev. Dev. Biol. 2017, 6. [Google Scholar] [CrossRef] [Green Version]
- Pla, P.; Monsoro-Burq, A.H. The neural border: Induction, specification and maturation of the territory that generates neural crest cells. Dev. Biol. 2018, 444 Suppl. 1, S36–S46. [Google Scholar] [CrossRef]
- Krispin, S.; Nitzan, E.; Kalcheim, C. The dorsal neural tube: A dynamic setting for cell fate decisions. Dev. Neurobiol. 2010, 70, 796–812. [Google Scholar] [CrossRef]
- Maguire, L.H.; Thomas, A.R.; Goldstein, A.M. Tumors of the neural crest: Common themes in development and cancer. Dev. Dyn. 2015, 244, 311–322. [Google Scholar] [CrossRef]
- Erdei, E.; Torres, S.M. A new understanding in the epidemiology of melanoma. Expert Rev. Anticancer Ther. 2010, 10, 1811–1823. [Google Scholar] [CrossRef] [Green Version]
- Postow, M.A.; Callahan, M.K.; Wolchok, J.D. Immune Checkpoint Blockade in Cancer Therapy. J. Clin. Oncol. 2015, 33, 1974–1982. [Google Scholar] [CrossRef] [Green Version]
- Hamid, O.; Robert, C.; Daud, A.; Hodi, F.S.; Hwu, W.J.; Kefford, R.; Wolchok, J.D.; Hersey, P.; Joseph, R.; Weber, J.S.; et al. Five-year survival outcomes for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. Ann. Oncol. 2019, 30, 582–588. [Google Scholar] [CrossRef]
- Brodeur, G.M. Neuroblastoma: Biological insights into a clinical enigma. Nat. Rev. Cancer 2003, 3, 203–216. [Google Scholar] [CrossRef]
- Maris, J.M. Recent advances in neuroblastoma. N. Engl. J. Med. 2010, 362, 2202–2211. [Google Scholar] [CrossRef] [Green Version]
- MacFarland, S.; Bagatell, R. Advances in neuroblastoma therapy. Curr. Opin. Pediatr. 2019, 31, 14–20. [Google Scholar] [CrossRef]
- Papahadjopoulos, D.; Allen, T.M.; Gabizon, A.; Mayhew, E.; Matthay, K.; Huang, S.K.; Lee, K.D.; Woodle, M.C.; Lasic, D.D.; Redemann, C.; et al. Sterically stabilized liposomes: Improvements in pharmacokinetics and antitumor therapeutic efficacy. Proc. Natl. Acad. Sci. USA 1991, 88, 11460–11464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, T.M.; Cullis, P.R. Liposomal drug delivery systems: From concept to clinical applications. Adv. Drug Deliv. Rev. 2013, 65, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Di Paolo, D.; Pastorino, F.; Zuccari, G.; Caffa, I.; Loi, M.; Marimpietri, D.; Brignole, C.; Perri, P.; Cilli, M.; Nico, B.; et al. Enhanced anti-tumor and anti-angiogenic efficacy of a novel liposomal fenretinide on human neuroblastoma. J. Control. Release 2013, 170, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Formelli, F.; Clerici, M.; Campa, T.; Di Mauro, M.G.; Magni, A.; Mascotti, G.; Moglia, D.; De Palo, G.; Costa, A.; Veronesi, U. Five-year administration of fenretinide: Pharmacokinetics and effects on plasma retinol concentrations. J. Clin. Oncol. 1993, 11, 2036–2042. [Google Scholar] [CrossRef] [PubMed]
- Formelli, F.; De Palo, G.; Costa, A.; Veronesi, U. Human transplacental passage of the retinoid fenretinide (4HPR). Eur. J. Cancer 1998, 34, 428–429. [Google Scholar] [CrossRef]
- Zhang, H. Thin-Film Hydration Followed by Extrusion Method for Liposome Preparation. Methods Mol. Biol. 2017, 1522, 17–22. [Google Scholar] [CrossRef]
- Allen, T.M.; Hansen, C. Pharmacokinetics of stealth versus conventional liposomes: Effect of dose. Biochim. Biophys. Acta 1991, 1068, 133–141. [Google Scholar] [CrossRef]
- Mujoo, K.; Cheresh, D.A.; Yang, H.M.; Reisfeld, R.A. Disialoganglioside-Gd2 on Human Neuroblastoma-Cells—Target Antigen for Monoclonal Antibody-Mediated Cytolysis and Suppression of Tumor-Growth. Cancer Res. 1987, 47, 1098–1104. [Google Scholar] [PubMed]
- Lammie, G.; Cheung, N.; Gerald, W.; Rosenblum, M.; Cordoncardo, C. Ganglioside gd(2) expression in the human nervous-system and in neuroblastomas—An immunohistochemical study. Int. J. Oncol. 1993, 3, 909–915. [Google Scholar] [CrossRef] [PubMed]
- Pastorino, F.; Brignole, C.; Marimpietri, D.; Sapra, P.; Moase, E.H.; Allen, T.M.; Ponzoni, M. Doxorubicin-loaded Fab’ fragments of anti-disialoganglioside immunoliposomes selectively inhibit the growth and dissemination of human neuroblastoma in nude mice. Cancer Res. 2003, 63, 86–92. [Google Scholar] [PubMed]
- Brignole, C.; Bensa, V.; Fonseca, N.A.; Del Zotto, G.; Bruno, S.; Cruz, A.F.; Malaguti, F.; Carlini, B.; Morandi, F.; Calarco, E.; et al. Cell surface Nucleolin represents a novel cellular target for neuroblastoma therapy. J. Exp. Clin. Cancer Res. 2021, 40, 180. [Google Scholar] [CrossRef] [PubMed]
- Corti, A.; Ponzoni, M. Tumor vascular targeting with tumor necrosis factor alpha and chemotherapeutic drugs. Signal. Transduct. Commun. Cancer Cells 2004, 1028, 104–112. [Google Scholar] [CrossRef]
- Di Matteo, P.; Arrigoni, G.L.; Alberici, L.; Corti, A.; Gallo-Stampino, C.; Traversari, C.; Doglioni, C.; Rizzardi, G.P. Enhanced expression of CD13 in vessels of inflammatory and neoplastic tissues. J. Histochem. Cytochem. 2011, 59, 47–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eggert, A.; Ikegaki, N.; Kwiatkowski, J.; Zhao, H.; Brodeur, G.M.; Himelstein, B.P. High-level expression of angiogenic factors is associated with advanced tumor stage in human neuroblastomas. Clin. Cancer Res. 2000, 6, 1900–1908. [Google Scholar]
- Ribatti, D.; Ponzoni, M. Antiangiogenic strategies in neuroblastoma. Cancer Treat. Rev. 2005, 31, 27–34. [Google Scholar] [CrossRef]
- Ruoslahti, E.; Duza, T.; Zhang, L. Vascular homing peptides with cell-penetrating properties. Curr. Pharm. Des. 2005, 11, 3655–3660. [Google Scholar] [CrossRef]
- Szoka, F., Jr.; Papahadjopoulos, D. Procedure for preparation of liposomes with large internal aqueous space and high capture by reverse-phase evaporation. Proc. Natl. Acad. Sci. USA 1978, 75, 4194–4198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasqualini, R.; Koivunen, E.; Kain, R.; Lahdenranta, J.; Sakamoto, M.; Stryhn, A.; Ashmun, R.A.; Shapiro, L.H.; Arap, W.; Ruoslahti, E. Aminopeptidase N is a receptor for tumor-homing peptides and a target for inhibiting angiogenesis. Cancer Res. 2000, 60, 722–727. [Google Scholar]
- Pastorino, F.; Brignole, C.; Marimpietri, D.; Cilli, M.; Gambini, C.; Ribatti, D.; Longhi, R.; Allen, T.M.; Corti, A.; Ponzoni, M. Vascular damage and anti-angiogenic effects of tumor vessel-targeted liposomal chemotherapy. Cancer Res. 2003, 63, 7400–7409. [Google Scholar] [PubMed]
- Loi, M.; Di Paolo, D.; Becherini, P.; Zorzoli, A.; Perri, P.; Carosio, R.; Cilli, M.; Ribatti, D.; Brignole, C.; Pagnan, G.; et al. The use of the orthotopic model to validate antivascular therapies for cancer. Int. J. Dev. Biol. 2011, 55, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.M.; DiPietrantonio, A.M.; Hsieh, T.C. Mechanism of fenretinide (4-HPR)-induced cell death. Apoptosis 2001, 6, 377–388. [Google Scholar] [CrossRef]
- Pastorino, F.; Brignole, C.; Marimpietri, D.; Pagnan, G.; Morando, A.; Ribatti, D.; Semple, S.C.; Gambini, C.; Allen, T.M.; Ponzoni, M. Targeted liposomal c-myc antisense oligodeoxynucleotides induce apoptosis and inhibit tumor growth and metastases in human melanoma models. Clin. Cancer Res. 2003, 9, 4595–4605. [Google Scholar] [PubMed]
- Pastorino, F.; Brignole, C.; Di Paolo, D.; Nico, B.; Pezzolo, A.; Marimpietri, D.; Pagnan, G.; Piccardi, F.; Cilli, M.; Longhi, R.; et al. Targeting liposomal chemotherapy via both tumor cell-specific and tumor vasculature-specific ligands potentiates therapeutic efficacy. Cancer Res. 2006, 66, 10073–10082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loi, M.; Marchio, S.; Becherini, P.; Di Paolo, D.; Soster, M.; Curnis, F.; Brignole, C.; Pagnan, G.; Perri, P.; Caffa, I.; et al. Combined targeting of perivascular and endothelial tumor cells enhances anti-tumor efficacy of liposomal chemotherapy in neuroblastoma. J. Control. Release 2010, 145, 66–73. [Google Scholar] [CrossRef]
- Di Paolo, D.; Brignole, C.; Pastorino, F.; Carosio, R.; Zorzoli, A.; Rossi, M.; Loi, M.; Pagnan, G.; Emionite, L.; Cilli, M.; et al. Neuroblastoma-targeted nanoparticles entrapping siRNA specifically knockdown ALK. Mol. Ther. 2011, 19, 1131–1140. [Google Scholar] [CrossRef] [Green Version]
- Loi, M.; Di Paolo, D.; Soster, M.; Brignole, C.; Bartolini, A.; Emionite, L.; Sun, J.; Becherini, P.; Curnis, F.; Petretto, A.; et al. Novel phage display-derived neuroblastoma-targeting peptides potentiate the effect of drug nanocarriers in preclinical settings. J. Control. Release 2013, 170, 233–241. [Google Scholar] [CrossRef] [Green Version]
- Di Paolo, D.; Pastorino, F.; Brignole, C.; Corrias, M.V.; Emionite, L.; Cilli, M.; Tamma, R.; Priddy, L.; Amaro, A.; Ferrari, D.; et al. Combined Replenishment of miR-34a and let-7b by Targeted Nanoparticles Inhibits Tumor Growth in Neuroblastoma Preclinical Models. Small 2020, 16, e1906426. [Google Scholar] [CrossRef] [PubMed]
- Cossu, I.; Bottoni, G.; Loi, M.; Emionite, L.; Bartolini, A.; Di Paolo, D.; Brignole, C.; Piaggio, F.; Perri, P.; Sacchi, A.; et al. Neuroblastoma-targeted nanocarriers improve drug delivery and penetration, delay tumor growth and abrogate metastatic diffusion. Biomaterials 2015, 68, 89–99. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| First Generation | Second Generation | Third Generation | Fourth Generation | |
|---|---|---|---|---|
| Features |
|
|
|
|
| Examples |  |  |  |  |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bensa, V.; Calarco, E.; Giusto, E.; Perri, P.; Corrias, M.V.; Ponzoni, M.; Brignole, C.; Pastorino, F. Retinoids Delivery Systems in Cancer: Liposomal Fenretinide for Neuroectodermal-Derived Tumors. Pharmaceuticals 2021, 14, 854. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14090854
Bensa V, Calarco E, Giusto E, Perri P, Corrias MV, Ponzoni M, Brignole C, Pastorino F. Retinoids Delivery Systems in Cancer: Liposomal Fenretinide for Neuroectodermal-Derived Tumors. Pharmaceuticals. 2021; 14(9):854. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14090854
Chicago/Turabian StyleBensa, Veronica, Enzo Calarco, Elena Giusto, Patrizia Perri, Maria Valeria Corrias, Mirco Ponzoni, Chiara Brignole, and Fabio Pastorino. 2021. "Retinoids Delivery Systems in Cancer: Liposomal Fenretinide for Neuroectodermal-Derived Tumors" Pharmaceuticals 14, no. 9: 854. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14090854