
Semaphorin 3A—Glycosaminoglycans Interaction as Therapeutic Target for Axonal Regeneration

 , and
, and
Abstract
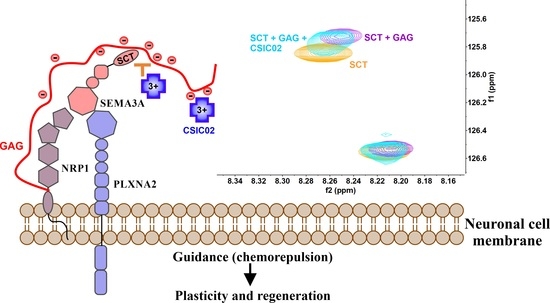
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
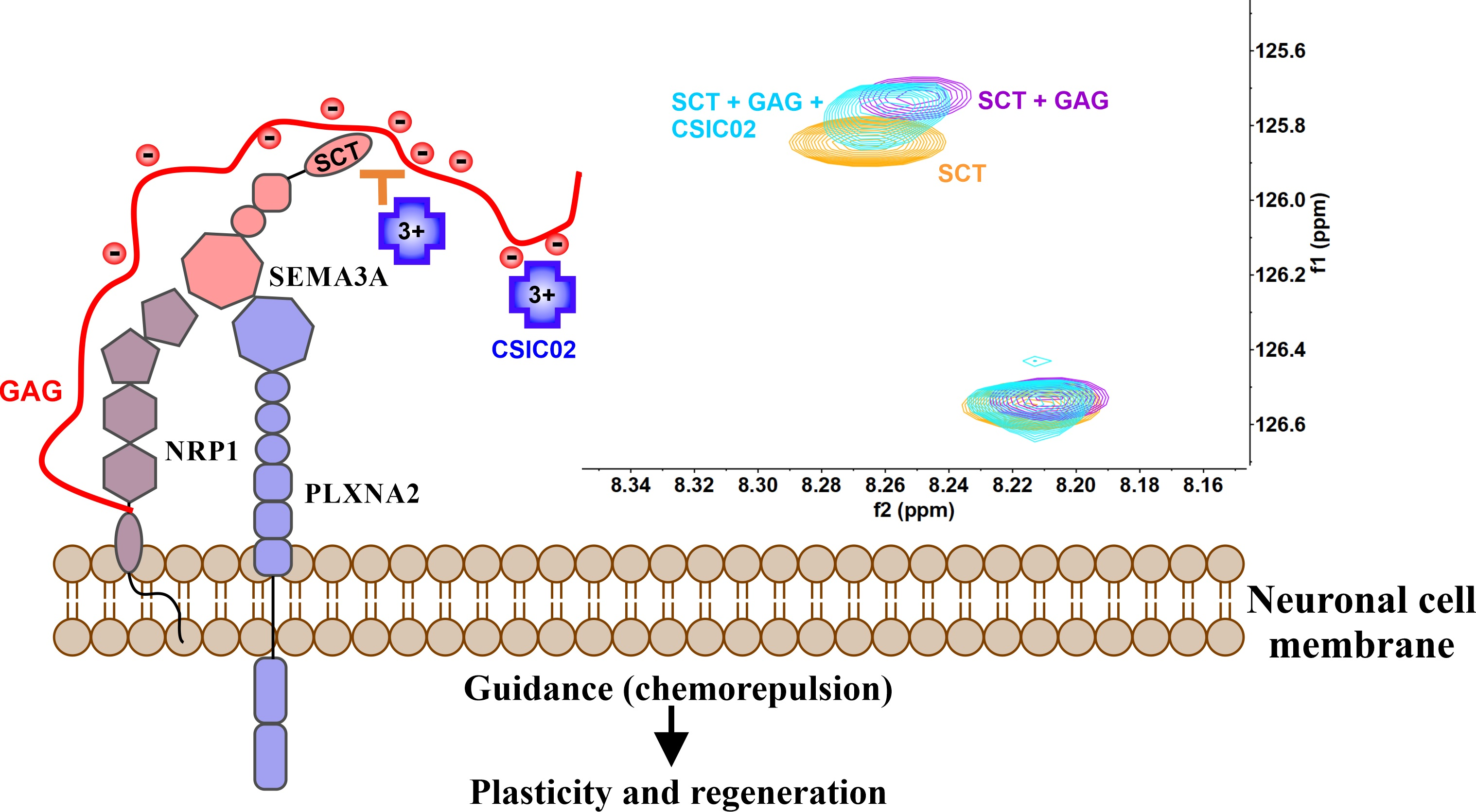
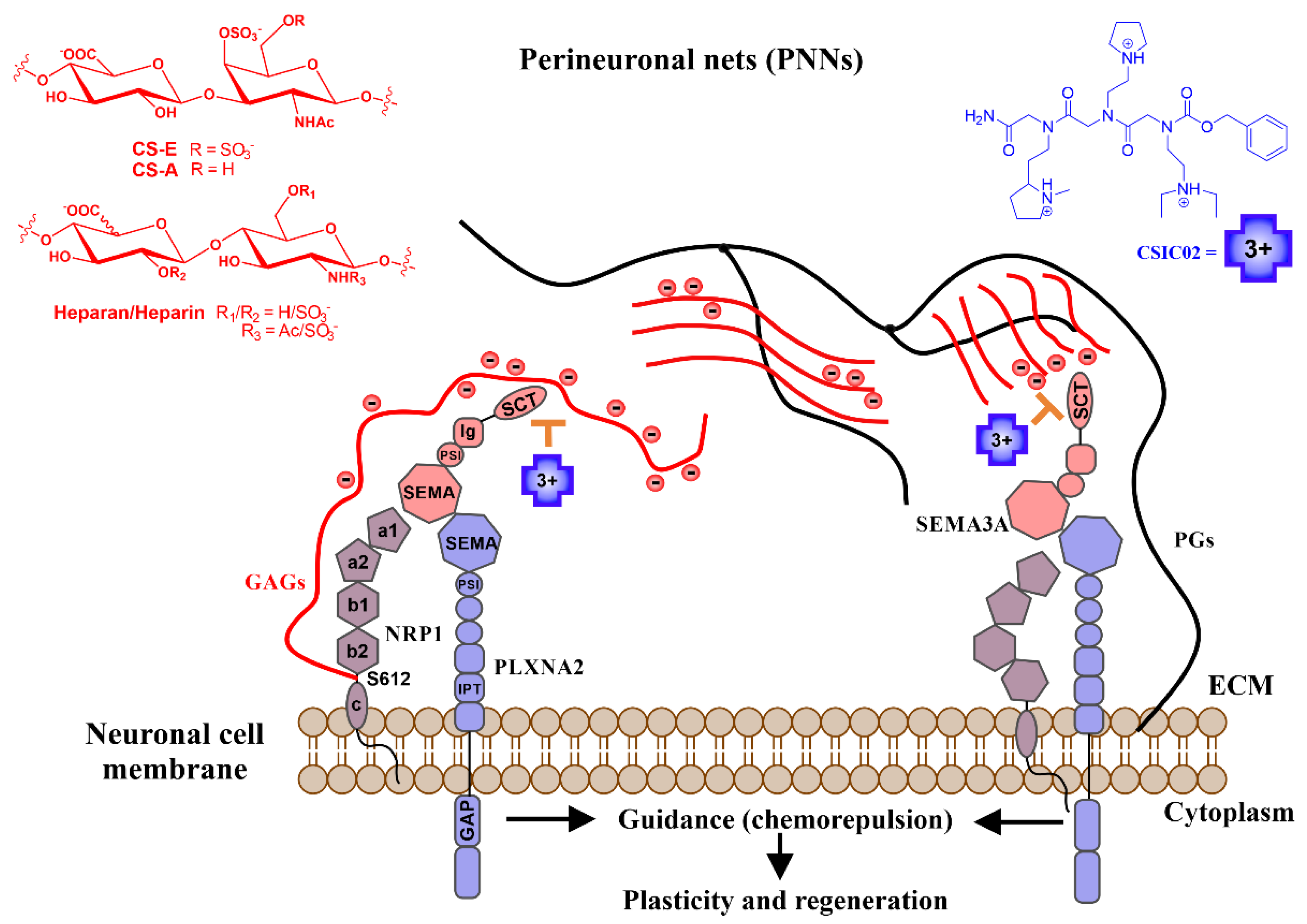
2.1. Sema3A C-Terminal Domain Interaction with Glycosaminoglycans (GAGs)
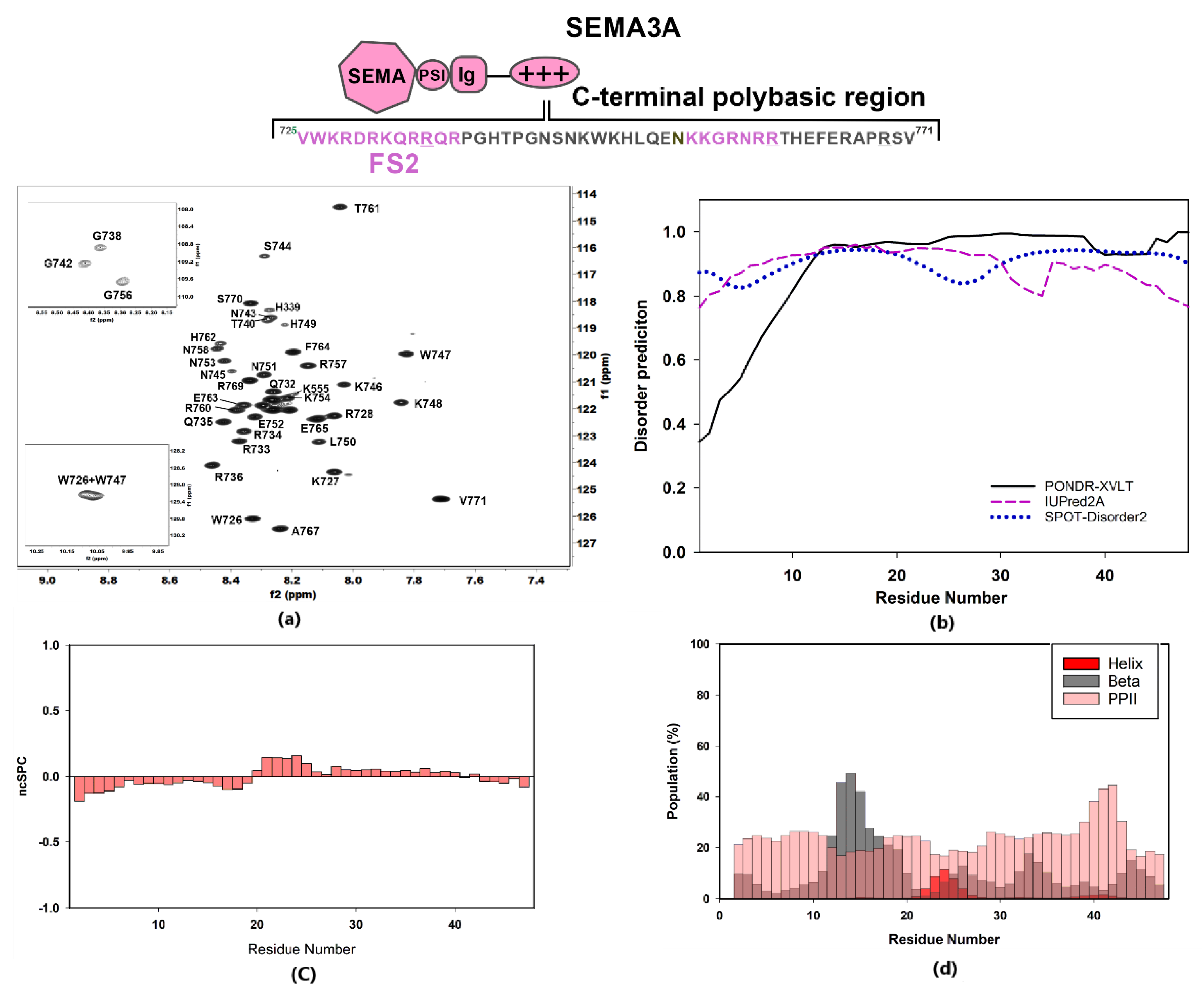
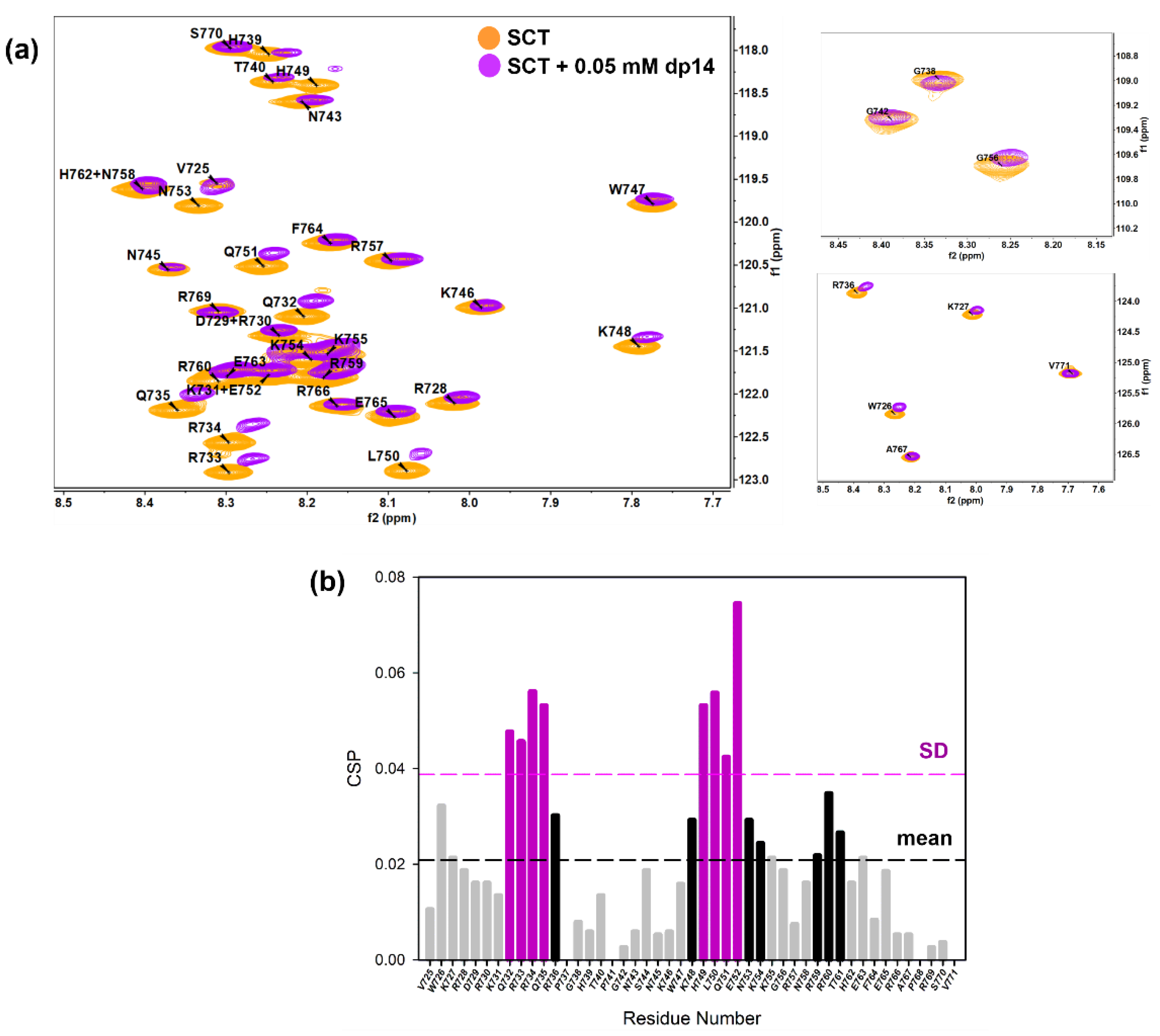
2.1.1. NMR Sequence Specific Assignment and Structural Features
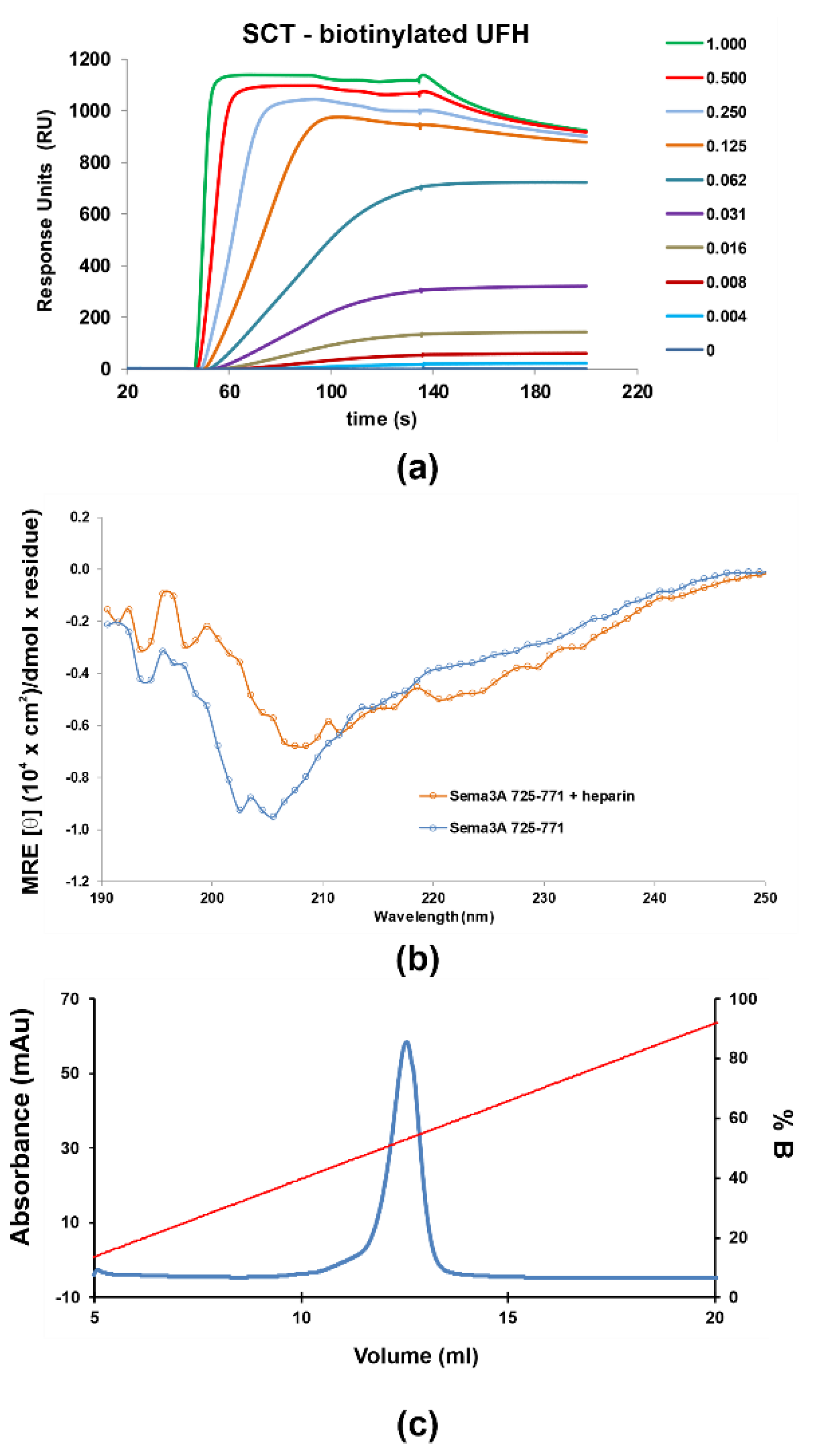
2.1.2. Characterization of the Interaction between GAGs and Sema3A Basic Tail
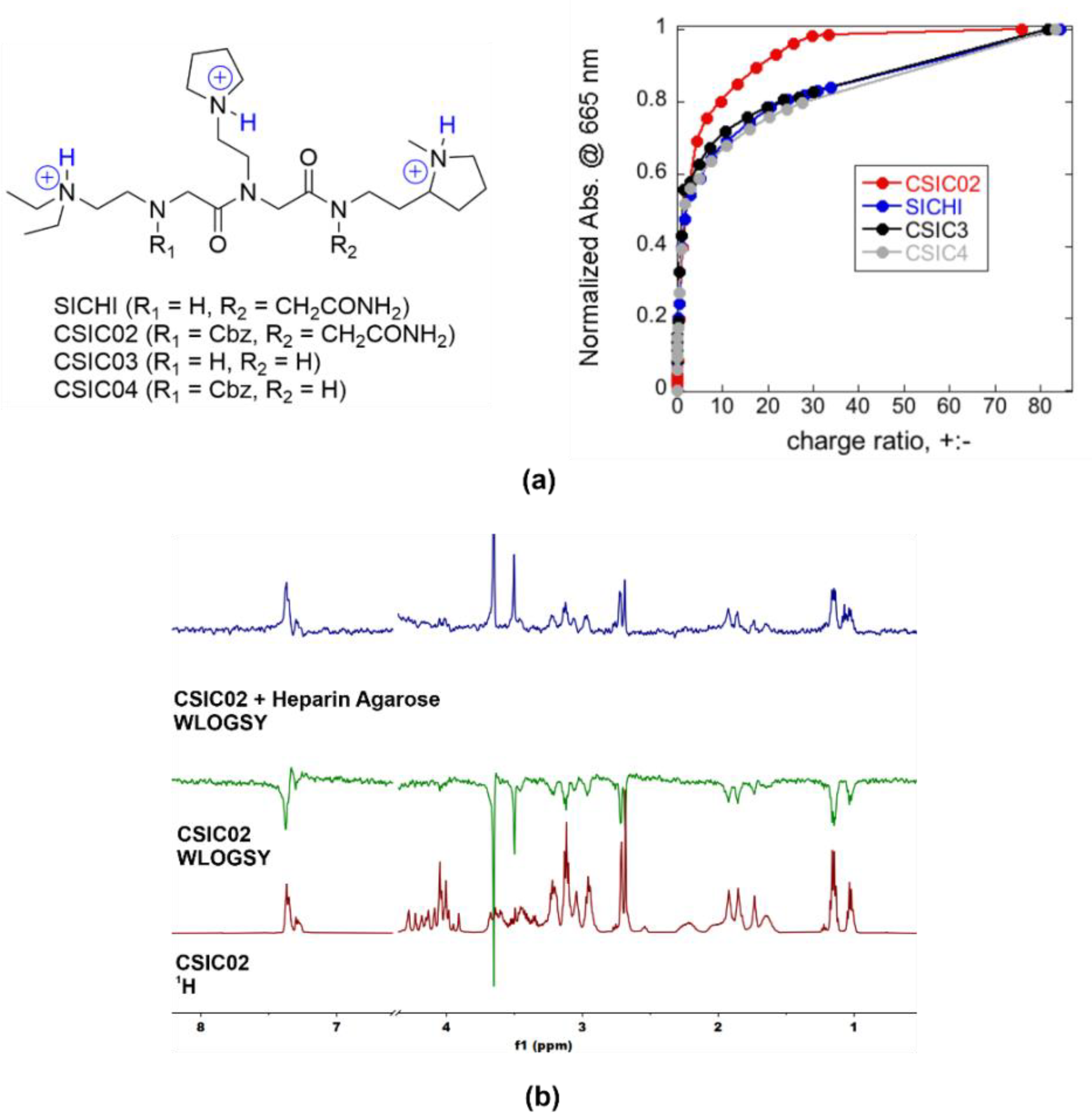
2.2. Analysis of Peptoids Interaction with GAGs
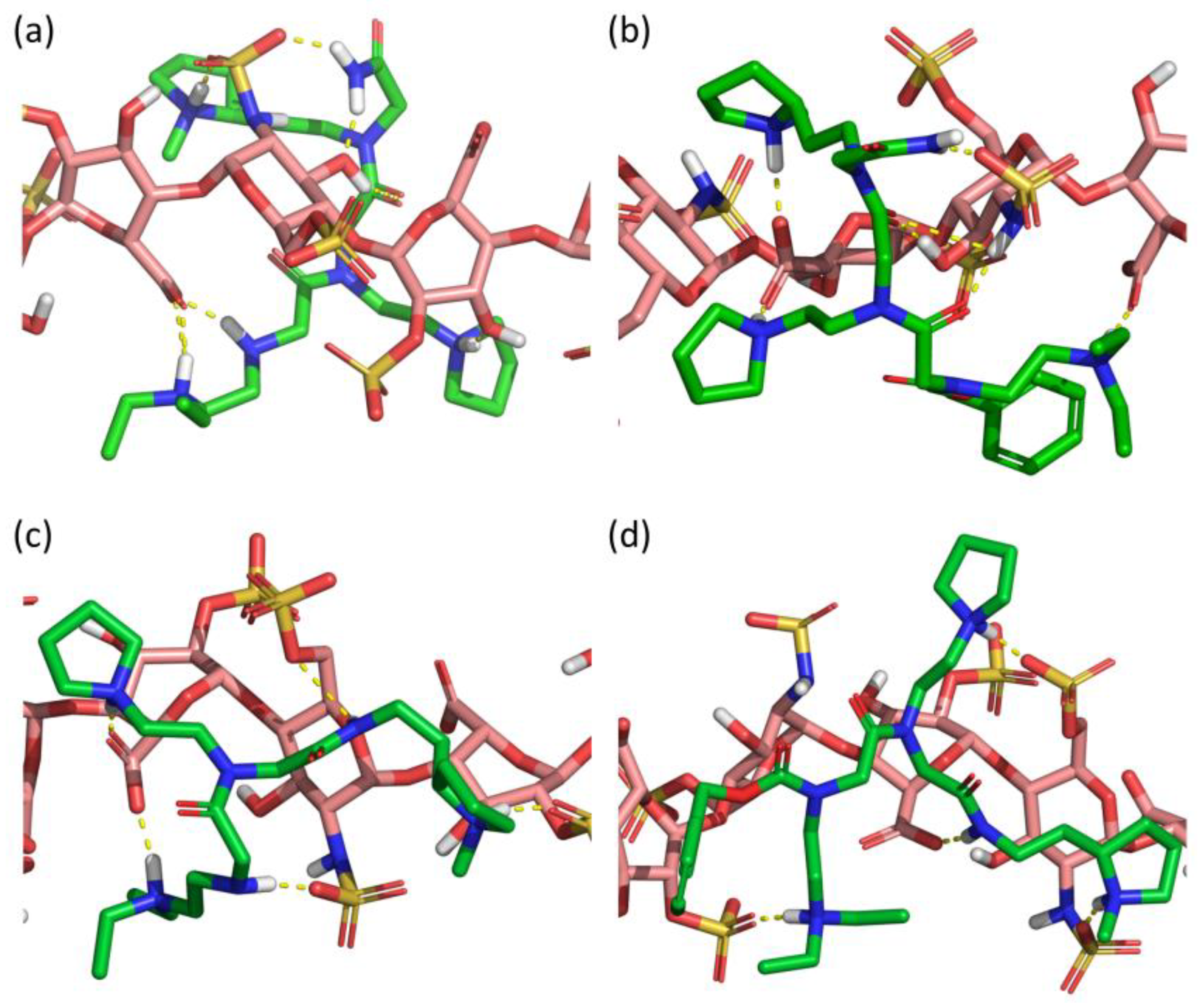
2.3. CSIC02 Targets and Modulates the Interaction between Sema3A Basic Tail and Heparin
3. Discussion
4. Materials and Methods
4.1. Glycosaminoglycans, Peptoids and Peptides
4.2. Recombinant Protein Expression and Purification
4.3. Methylene Blue Assay and pKa Determination by Potentiometry
4.4. NMR Experiments
4.5. Surface Plasmon Resonance
4.6. Circular Dichroism
4.7. Heparin Agarose Affinity
4.8. Bioinformatics Tools
4.9. Computational Methods
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Soleman, S.; Filippov, M.A.; Dityatev, A.; Fawcett, J.W. Targeting the neural extracellular matrix in neurological disorders. Neuroscience 2013, 253, 194–213. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, N.S.; Mancera, R.L. The structure of glycosaminoglycans and their interactions with proteins. Chem. Biol. Drug. Des. 2008, 72, 455–482. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Esko, J.D. Demystifying Heparan Sulfate–Protein Interactions. Annu. Rev. Biochem. 2014, 83, 129–157. [Google Scholar] [CrossRef] [PubMed]
- Townley, R.A.; Bülow, H.E. Deciphering functional glycosaminoglycan motifs in development. Cur. Opin. Struct. Biol. 2018, 50, 144–154. [Google Scholar] [CrossRef]
- Vallet, S.D.; Clerc, O.; Ricard-blum, S. Glycosaminoglycan–Protein Interactions: The First Draft of the Glycosaminoglycan Interactome. J. Histochem. Cytochem. 2021, 69, 93–104. [Google Scholar] [CrossRef]
- Afratis, N.; Gialeli, C.; Nikitovic, D.; Tsegenidis, T.; Karousou, E.; Theocharis, A.D.; Pavão, M.S.; Tzanakakis, G.N.; Karamanos, N.K. Glycosaminoglycans: Key players in cancer cell biology and treatment. FEBS J. 2012, 279, 1177–1197. [Google Scholar] [CrossRef]
- Shi, D.; Sheng, A.; Chi, L. Glycosaminoglycan-Protein Interactions and Their Roles in Human Disease. Front. Mol. Biosci. 2021, 8, 639666. [Google Scholar] [CrossRef]
- De Wit, J.; De Winter, F.; Klooster, J.; Verhaagen, J. Semaphorin 3A displays a punctate distribution on the surface of neuronal cells and interacts with proteoglycans in the extracellular matrix. Mol. Cell. Neurosci. 2005, 29, 40–55. [Google Scholar] [CrossRef]
- Zimmer, G.; Schanuel, S.M.; Burger, S.; Weth, F.; Steinecke, A.; Bolz, J.; Lent, R. Chondroitin sulfate acts in concert with semaphorin 3A to guide tangential migration of cortical interneurons in the ventral telencephalon. Cereb. Cortex 2010, 20, 2411–2422. [Google Scholar] [CrossRef] [Green Version]
- Dick, G.; Tan, C.L.; Alves, J.N.; Ehlert, E.M.; Miller, G.M.; Hsieh-Wilson, L.C.; Sugahara, K.; Oosterhof, A.; van Kuppevelt, T.H.; Verhaagen, J.; et al. Semaphorin 3A binds to the perineuronal nets via chondroitin sulfate type E motifs in rodent brains. J. Biol. Chem. 2013, 288, 27384–27395. [Google Scholar] [CrossRef] [Green Version]
- Vo, T.; Carulli, D.; Ehlert, E.; Kwok, J.; Dick, G.; Mecollari, V.; Moloney, E.; Neufeld, G.; de Winter, F.; Fawcett, J.; et al. The chemorepulsive axon guidance protein semaphorin3A is a constituent of perineuronal nets in the adult rodent brain. Mol. Cell. Neurosci. 2013, 56, 186–200. [Google Scholar] [CrossRef] [PubMed]
- Carulli, D.; Verhaagen, J. An extracellular perspective on cns maturation: Perineuronal nets and the control of plasticity. Int. J. Mol. Sci. 2021, 22, 2434. [Google Scholar] [CrossRef]
- de Winter, F.; Kwok, J.C.; Fawcett, J.W.; Vo, T.T.; Carulli, D.; Verhaagen, J. The Chemorepulsive Protein Semaphorin 3A and Perineuronal Net-Mediated Plasticity. Neural Plast. 2016, 3679545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semaphorin Nomenclature Committee. Unified Nomenclature for the Semaphorins/Collapsins. Cell 1999, 97, 551–552. [Google Scholar] [CrossRef] [Green Version]
- Masu, M. Proteoglycans and axon guidance: A new relationship between old partners. J. Neurochem. 2016, 139, 58–75. [Google Scholar] [CrossRef]
- Ahammad, I. A comprehensive review of tumor proliferative and suppressive role of semaphorins and therapeutic approaches. Biophys. Rev. 2020, 12, 1233–1247. [Google Scholar] [CrossRef]
- Jiao, B.; Liu, S.; Tan, X.; Lu, P.; Wang, D.; Xu, H. Class-3 semaphorins: Potent multifunctional modulators for angiogenesis-associated diseases. Biomed. Pharmacother. 2021, 137, 111329. [Google Scholar] [CrossRef]
- Takamatsu, H.; Kumanogoh, A. Diverse roles for semaphorin-plexin signaling in the immune system. Trends. Immunol. 2012, 33, 127–135. [Google Scholar] [CrossRef]
- Iragavarapu-Charyulu, V.; Wojcikiewicz, E.; Urdaneta, A. Semaphorins in Angiogenesis and Autoimmune Diseases: Therapeutic Targets? Front. Immunol. 2020, 11, 346. [Google Scholar] [CrossRef]
- Worzfeld, T.; Offermanns, S. Semaphorins and plexins as therapeutic targets. Nat. Rev. Drug Discov. 2014, 13, 603–621. [Google Scholar] [CrossRef]
- Zhang, X.; Shao, S.; Li, L. Characterization of class-3 semaphorin receptors, neuropilins and plexins, as therapeutic targets in a pan-cancer study. Cancers 2020, 12, 1816. [Google Scholar] [CrossRef]
- Testa, D.; Prochiantz, A.; Di Nardo, A.A. Perineuronal nets in brain physiology and disease. Semin. Cell Dev. Biol. 2019, 89, 125–135. [Google Scholar] [CrossRef]
- Mecollari, V.; Nieuwenhuis, B.; Verhaagen, J. A perspective on the role of class III semaphorin signaling in central nervous system trauma. Front. Cell. Neurosci. 2014, 8, 328. [Google Scholar] [CrossRef] [Green Version]
- De Winter, F.; Cui, Q.; Symons, N.; Verhaagen, J.; Harvey, A.R. Expression of class-3 semaphorins and their receptors in the neonatal and adult rat retina. Investig. Ophthalmol. Vis. Sci. 2004, 45, 4554–4562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harvey, A.R.; Ooi, J.W.W.; Rodger, J. Neurotrophic factors and the regeneration of adult retinal ganglion cell axons. In International Review of Neurobiology, 1st ed.; Goldberg, J.L., Trakhtenberg, E.F., Eds.; Academic Press: Cambridge, MA, USA, 2012; Volume 106, pp. 1–33. ISBN 9780124071780. [Google Scholar] [CrossRef]
- Tropea, D.; Caleo, M.; Maffei, L. Synergistic Effects of Brain-Derived Neurotrophic Factor and Chondroitinase ABC on Retinal Fiber Sprouting after Denervation of the Superior Colliculus in Adult Rats. J. Neurosci. 2003, 23, 7034–7044. Available online: http://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/pubmed/12904464 (accessed on 3 September 2021). [CrossRef]
- Dekeyster, E.; Aerts, J.; Valiente-Soriano, F.J.; De Groef, L.; Vreysen, S.; Salinas-Navarro, M.; Vidal-Sanz, M.; Arckens, L.; Moons, L. Ocular Hypertension Results in Retinotopic Alterations in the Visual Cortex of Adult Mice. Curr. Eye Res. 2015, 40, 1269–1283. [Google Scholar] [CrossRef]
- Boggio, E.M.; Ehlert, E.M.; Lupori, L.; Moloney, E.B.; De Winter, F.; Vander Kooi, C.W.; Baroncelli, L.; Mecollari, V.; Blits, B.; Fawcett, J.W.; et al. Inhibition of Semaphorin3A Promotes Ocular Dominance Plasticity in the Adult Rat Visual Cortex. Mol. Neurobiol. 2019, 56, 5987–5997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boia, R.; Ruzafa, N.; Aires, I.D.; Pereiro, X.; Ambrósio, A.F.; Vecino, E.; Santiago, A.R. Neuroprotective strategies for retinal ganglion cell degeneration: Current status and challenges ahead. Int. J. Mol. Sci. 2020, 21, 2262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeon, K.I.; Nehrke, K.; Huxlin, K.R. Semaphorin 3A potentiates the profibrotic effects of transforming growth factor-β1 in the cornea. Biochem. Biophys. Res. Commun. 2020, 521, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Tribble, J.R.; Williams, P.A.; Caterson, B.; Sengpiel, F.; Morgan, J.E. Digestion of the glycosaminoglycan extracellular matrix by chondroitinase ABC supports retinal ganglion cell dendritic preservation in a rodent model of experimental glaucoma. Mol. Brain 2018, 11, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Alto, L.T.; Terman, J.R. Semaphorins and their signaling mechanisms. In Semaphorin Signaling. Methods in Molecular Biology; Terman, J.R., Ed.; Humana Press: New York, NY, USA, 2017; Volume 1493, pp. 1–25. ISBN 978-1-4939-6448-2. [Google Scholar] [CrossRef] [Green Version]
- Meyer, L.A.T.; Fritz, J.; Pierdant-Mancera, M.; Bagnard, D. Current drug design to target the Semaphorin/Neuropilin/Plexin complexes. Cell Adhes. Migr. 2016, 10, 700–708. [Google Scholar] [CrossRef] [Green Version]
- Kikuchi, K.; Kishino, A.; Konishi, O.; Kumagai, K.; Hosotani, N.; Saji, I.; Nakayama, C.; Kimura, T. In vitro and in vivo characterization of a novel semaphorin 3A inhibitor, SM-216289 or xanthofulvin. J. Biol. Chem. 2003, 278, 42985–42991. [Google Scholar] [CrossRef] [Green Version]
- Montolio, M.; Messeguer, J.; Masip, I.; Guijarro, P.; Gavin, R.; Antonio Del Rio, J.; Messeguer, A.; Soriano, E. A semaphorin 3A inhibitor blocks axonal chemorepulsion and enhances axon regeneration. Chem. Biol. 2009, 16, 691–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirvan, A.; Kimron, M.; Holdengreber, V.; Ziv, I.; Ben-Shaul, Y.; Melamed, S.; Melamed, E.; Barzilai, A.; Solomon, A.S. Anti-semaphorin 3A antibodies rescue retinal ganglion cells from cell death following optic nerve axotomy. J. Biol. Chem. 2002, 277, 49799–49807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamashita, N.; Jitsuki-Takahashi, A.; Ogawara, M.; Ohkubo, W.; Araki, T.; Hotta, C.; Tamura, T.; Hashimoto, S.I.; Yabuki, T.; Tsuji, T.; et al. Anti-Semaphorin3A Neutralization Monoclonal Antibody Prevents Sepsis Development in Lipopolysaccharide-treated Mice. Int. Immunol. 2015, 27, 459–466. [Google Scholar] [CrossRef]
- Lindahl, U. Heparan sulfate-protein interactions-A concept for drug design? J. Thromb. Haemost. 2007, 98, 109–115. [Google Scholar] [CrossRef]
- Luo, Y.; Raible, D.; Raper, J.A. Collapsin: A Protein in Brain That Induces the Collapse and Paralysis of Neuronal Growth Cones. Cell 1993, 75, 217–227. [Google Scholar] [CrossRef]
- Corredor, M.; Bonet, R.; Moure, A.; Domingo, C.; Bujons, J.; Alfonso, I.; Perez, Y.; Messeguer, A. Cationic Peptides and Peptidomimetics Bind Glycosaminoglycans as Potential Sema3A Pathway Inhibitors. Biophys. J. 2016, 110, 1291–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, J.; Wang, Y.; Yao, K. Protection of retinal ganglion cells in glaucoma: Current status and future. Exp. Eye Res. 2021, 205, 108506. [Google Scholar] [CrossRef]
- Wareham, L.K.; Risner, M.L.; Calkins, D.J. Protect, Repair, and Regenerate: Towards Restoring Vision in Glaucoma. Curr. Ophthalmol. Rep. 2020, 8, 301–310. [Google Scholar] [CrossRef]
- Janssen, B.J.; Malinauskas, T.; Weir, G.A.; Cader, M.Z.; Siebold, C.; Jones, E.Y. Neuropilins lock secreted semaphorins onto plexins in a ternary signaling complex. Nat. Struct. Mol. Biol. 2012, 19, 1293–1299. [Google Scholar] [CrossRef] [Green Version]
- Schubert, M.; Labudde, D.; Oschkinat, H.; Schmieder, P. A software tool for the prediction of Xaa-Pro peptide bond conformations in proteins based on 13C chemical shift statistics. J. Biomol. NMR 2002, 24, 149–154. [Google Scholar] [CrossRef]
- Borcherds, W.M.; Daughdrill, G.W. Using NMR Chemical Shifts to Determine Residue-Specific Secondary Structure Populations for Intrinsically Disordered Proteins. In Methods in Enzymology; Rhoades, E., Ed.; Academic Press: Cambridge, MA, USA, 2018; Volume 611, pp. 101–136. ISBN 9780128156490. [Google Scholar] [CrossRef]
- Sormanni, P.; Camilloni, C.; Fariselli, P.; Vendruscolo, M. The s2D method: Simultaneous sequence-based prediction of the statistical populations of ordered and disordered regions in proteins. J. Mol. Biol. 2015, 427, 982–996. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 41, 819. [Google Scholar] [CrossRef]
- Guo, H.F.; Li, X.; Parker, M.W.; Waltenberger, J.; Becker, P.M.; Vander Kooi, C.W. Mechanistic basis for the potent anti-angiogenic activity of semaphorin 3F. Biochemistry 2013, 52, 7551–7558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rapper, J.A.; Koppel, A.M. Collapsin-1 Covalently Dimerizes, and Dimerization Is Necessary for Collapsing Activity. J. Biol. Chem. 1998, 273, 15708–15713. [Google Scholar] [CrossRef] [Green Version]
- Klostermann, A.; Lohrum, M.; Adams, R.H.; Puschel, A.W. The chemorepulsive activity of the axonal guidance signal semaphorin D requires dimerization. J. Biol. Chem. 1998, 273, 7326–7331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Moniz, H.A.; Walcott, B.; Moremen, K.W.; Linhardt, R.J.; Wang, L. Characterization of the interaction between Robo1 and heparin and other glycosaminoglycans. Biochimie 2013, 95, 2345–2353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Y.; Zhang, F.; Linhardt, R.J. Analysis of the Glycosaminoglycan Chains of Proteoglycans. J. Histochem. Cytochem. 2020, 69, 121–135. [Google Scholar] [CrossRef]
- Olson, S.T.; Frances-Chmura, A.M.; Swanson, R.; Björk, I.; Zettlmeissl, G. Effect of individual carbohydrate chains of recombinant antithrombin on heparin affinity and on the generation of glycoforms differing in heparin affinity. Arch. Biochem. Biophys. 1997, 341, 212–221. [Google Scholar] [CrossRef]
- Esko, J.D.; Prestegard, J.; Linhardt, R.J. Proteins That Bind Sulfated Glycosaminoglycans. In Essentials of Glycobiology [Internet], 3rd ed.; Varki, A., Cummings, R.D., Esko, J.D., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2015–2017; Chapter 38. [CrossRef]
- Herndon, M.E.; Stipp, C.S.; Lander, A.D. Interactions of neural glycosaminoglycans and proteoglycans with protein ligands: Assessment of selectivity, heterogeneity and the participation of core proteins in binding. Glycobiology 1999, 9, 143–155. [Google Scholar] [CrossRef] [Green Version]
- Bu, C.; Jin, L. NMR Characterization of the Interactions Between Glycosaminoglycans and Proteins. Front. Mol. Biosci. 2021, 8, 646808. [Google Scholar] [CrossRef]
- Williamson, M.P. Using chemical shift perturbation to characterize ligand binding. Prog. Nucl. Magn. Reson. Spectrosc. 2013, 73, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Fromm, J.R.; Hileman, R.E.; Caldwell, E.E.; Weiler, J.M.; Linhardt, R.J. Pattern and spacing of basic amino acids in heparin binding sites. Arch. Biochem. Biophys. 1997, 343, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Hileman, R.E.; Fromm, J.R.; Weiler, J.M.; Linhardt, R.J. Glycosaminoglycan-protein interactions: Definition of consensus sites in glycosaminoglycan binding proteins. Bioessays 1998, 20, 156–167. [Google Scholar] [CrossRef]
- Hanchate, N.K.; Giacobini, P.; Lhuillier, P.; Parkash, J.; Espy, C.; Fouveaut, C.; Leroy, C.; Baron, S.; Campagne, C.; Vanacker, C.; et al. SEMA3A, a Gene Involved in Axonal Pathfinding, Is Mutated in Patients with Kallmann Syndrome. PLoS Genet. 2012, 8, e1002896. [Google Scholar] [CrossRef]
- Nadanaka, S.; Miyata, S.; Yaqiang, B.; Tamura, J.I.; Habuchi, O.; Kitagawa, H. Reconsideration of the semaphorin-3a binding motif found in chondroitin sulfate using galnac4s-6st-knockout mice. Biomolecules 2020, 10, 1499. [Google Scholar] [CrossRef]
- Jiao, Q.C.; Liu, Q.; Sun, C.; He, H. Investigation on the binding site in heparin by spectrophotometry. Talanta 1999, 48, 1095–1101. [Google Scholar] [CrossRef]
- LaPlante, S.R.; Aubry, N.; Bolger, G.; Bonneau, P.; Carson, R.; Coulombe, R.; Sturino, C.; Beaulieu, P.L. Monitoring drug self-aggregation and potential for promiscuity in off-target in vitro pharmacology screens by a practical NMR strategy. J. Med. Chem. 2013, 56, 7073–7083. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, A.; Seelig, J. Binding and clustering of glycosaminoglycans: A common property of mono- and multivalent cell-penetrating compounds. Biophys. J. 2008, 94, 2142–2149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kantor, D.B.; Chivatakarn, O.; Peer, K.L.; Oster, S.F.; Inatani, M.; Hansen, M.J.; Flanagan, J.G.; Yamaguchi, Y.; Sretavan, D.W.; Giger, R.J.; et al. Semaphorin 5A is a bifunctional axon guidance cue regulated by heparan and chondroitin sulfate proteoglycans. Neuron 2004, 44, 961–975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conrad, A.H.; Zhang, Y.; Tasheva, E.S.; Conrad, G.W. Proteomic analysis of potential keratan sulfate, chondroitin sulfate A, and hyaluronic acid molecular interactions. Investig. Ophthalmol. Vis. Sci. 2010, 51, 4500–4515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, R.J.; Gordts, P.; Le, D.; Xu, D.; Esko, J.D.; Tor, Y. Small molecule antagonists of cell-surface heparan sulfate and heparin–protein interactions. Chem. Sci. 2015, 6, 5984–5993. [Google Scholar] [CrossRef] [Green Version]
- Weiss, R.J.; Esko, J.D.; Tor, Y. Targeting heparin and heparan sulfate protein interactions. Org. Biomol. Chem. 2017, 15, 5656–5668. [Google Scholar] [CrossRef] [PubMed]
- Corredor, M.; Carbajo, D.; Domingo, C.; Pérez, Y.; Bujons, J.; Messeguer, A.; Alfonso, I. Dynamic Covalent Identification of an Efficient Heparin Ligand. Angew. Chem. Int. Ed. Engl. 2018, 130, 12149–12153. [Google Scholar] [CrossRef]
- Schuksz, M.; Fuster, M.M.; Brown, J.R.; Crawford, B.E.; Ditto, D.P.; Lawrence, R.; Glass, C.A.; Wang, L.; Tor, Y.; Esko, J.D. Surfen, a small molecule antagonist of heparan sulfate. Proc. Natl. Acad. Sci. USA 2008, 105, 13075–13080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, R.H.; Lohrum, M.; Klostermann, A.; Betz, H.; Puschel, A.W. The chemorepulsive activity of secreted semaphorins is regulated by furin-dependent proteolytic processing. EMBO J. 1997, 16, 6077–6086. [Google Scholar] [CrossRef] [Green Version]
- Cardin, A.D.; Weintraub, H.J. Molecular Modeling of Protein-Glycosaminoglycan Interactions. Arteriosclerosis 1989, 9, 21–32. Available online: http://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/pubmed/2463827 (accessed on 3 September 2021). [CrossRef] [PubMed]
- Habchi, J.; Tompa, P.; Longhi, S.; Uversky, V.N. Introducing protein intrinsic disorder. Chem. Rev. 2014, 114, 6561–6588. [Google Scholar] [CrossRef] [Green Version]
- Xie, H.; Vucetic, S.; Iakoucheva, L.M.; Oldfield, C.J.; Dunker, A.K.; Obradovic, Z.; Uversky, V.N. Functional anthology of intrinsic disorder. 3. Ligands, post-translational modifications, and diseases associated with intrinsically disordered proteins. J. Proteome Res. 2007, 6, 1917–1932. [Google Scholar] [CrossRef] [Green Version]
- Peysselon, F.; Ricard-Blum, S. Heparin-protein interactions: From affinity and kinetics to biological roles. Application to an interaction network regulating angiogenesis. Matrix Biol. 2013, 35, 73–81. [Google Scholar] [CrossRef]
- Bae, J.; Desai, U.R.; Pervin, A.; Caldwell, E.E.O.; Weiler, J.M.; Linhardt, R.J. Interaction of heparin with synthetic antithrombin III peptide analogues. Biochem. J. 1994, 301, 121–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz, E.M.; Linhardt, R.J. Heparin-binding domains in vascular biology. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1549–1557. [Google Scholar] [CrossRef] [Green Version]
- Menezes, R.; Hashemi, S.; Vincent, R.; Collins, G.; Meyer, J.; Foston, M.; Arinzeh, T.L. Investigation of Glycosaminoglycan Mimetic Scaffolds for Neurite Growth. Acta Biomater. 2019, 90, 169–178. [Google Scholar] [CrossRef]
- Mizumoto, S.; Murakoshi, S.; Kalayanamitra, K.; Deepa, S.S.; Fukui, S.; Kongtawelert, P.; Yamada, S.; Sugahara, K. Highly sulfated hexasaccharide sequences isolated from chondroitin sulfate of shark fin cartilage: Insights into the sugar sequences with bioactivities. Glycobiology 2013, 23, 155–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markley, J.L.; Bax, A.; Arata, Y.; Hilbers, C.W.; Kaptein, R.; Sykes, B.D.; Wright, P.E.; Wüthrich, K. Recommendations for the presentation of NMR structures of proteins and nucleic acids. IUPAC-IUBMB-IUPAB inter-union task group on the standardization of data bases of protein and nucleic acid structures determined by NMR spectroscopy. Eur. J. Biochem. 1998, 256, 1–15. [Google Scholar] [CrossRef]
- Vranken, W.F.; Boucher, W.; Stevens, T.J.; Fogh, R.H.; Pajon, A.; Llinas, M.; Ulrich, E.L.; Markley, J.L.; Ionides, J.; Laue, E.D. The CCPN data model for NMR spectroscopy: Development of a software pipeline. Proteins 2005, 59, 687–696. [Google Scholar] [CrossRef]
- Cain, S.A.; Baldock, C.; Gallagher, J.; Morgan, A.; Bax, D.V.; Weiss, A.S.; Shuttleworth, C.A.; Kielty, C.M. Fibrillin-1 interactions with heparin. Implications for microfibril and elastic fiber assembly. J. Biol. Chem. 2005, 280, 30526–30537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitmore, L.; Wallace, B.A. Protein secondary structure analyses from circular dichroism spectroscopy: Methods and reference databases. Biopolymers 2008, 89, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Romero, P.; Obradovic, Z.; Li, X.; Garner, E.C.; Brown, C.J.; Dunker, A.K. Sequence complexity of disordered protein. Proteins 2001, 42, 38–48. [Google Scholar] [CrossRef]
- Erdős, G.; Dosztányi, Z. Analyzing Protein Disorder with IUPred2A. Curr. Prot. Bioinform. 2020, 70, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanson, J.; Paliwal, K.K.; Litfin, T.; Zhou, Y. SPOT-Disorder2: Improved Protein Intrinsic Disorder Prediction by Ensembled Deep Learning. Genom. Proteom. Bioinform. 2019, 17, 645–656. [Google Scholar] [CrossRef]
- Camilloni, C.; De Simone, A.; Vranken, W.F.; Vendruscolo, M. Determination of secondary structure populations in disordered states of proteins using nuclear magnetic resonance chemical shifts. Biochemistry 2012, 51, 2224–2231. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger. Release 2021-2; LLC: New York, NY, USA, 2021. [Google Scholar]
- Schrödinger. Release 2021-2: Maestro; LLC: New York, NY, USA, 2021. [Google Scholar]
- Schrödinger. Release 2021-2: Macromodel; LLC: New York, NY, USA, 2021. [Google Scholar]
- Lu, C.; Wu, C.; Ghoreishi, D.; Chen, W.; Wang, L.; Damm, W.; Ross, G.A.; Dahlgren, M.K.; Russell, E.; Von Bargen, C.D.; et al. OPLS4: Improving Force Field Accuracy on Challenging Regimes of Chemical Space. J. Chem. Theory Comp. 2021. [Google Scholar] [CrossRef]
- Still, W.C.; Tempczyk, A.; Hawley, R.C.; Hendrickson, T. Semianalytical treatment of solvation for molecular mechanics and dynamics. J. Am. Chem. Soc. 1990, 112, 6127–6129. [Google Scholar] [CrossRef]
- Schrödinger. Release 2021-2: Glide; LLC: New York, NY, USA, 2021. [Google Scholar]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [Green Version]
- Schrödinger. Release 2021-2: Desmond Molecular Dynamics System; D.E. Shaw Research: New York, NY, USA, 2021. [Google Scholar]
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossváry, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the ACM/IEEE Conference on Supercomputing (SC06), Tampa, FL, USA, 11–17 November 2006. [Google Scholar]
- Schrödinger. Release 2021-2: Maestro-Desmond Interoperability Tools; D.E. Shaw Research: New York, NY, USA, 2021. [Google Scholar]
- Evans, D.J.; Holian, B.L. The Nose–Hoover thermostat. J. Chem. Phys. 1985, 83, 4069–4074. [Google Scholar] [CrossRef]
- Martyna, G.J.; Klein, M.L.; Tuckerman, M. Nosé–Hoover chains: The canonical ensemble via continuous dynamics. J. Chem. Phys. 1992, 97, 2635–2643. [Google Scholar] [CrossRef]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Tuckerman, M.; Berne, B.J.; Martyna, G.J. Reversible multiple time scale molecular dynamics. J. Chem. Phys. 1992, 97, 1990–2001. [Google Scholar] [CrossRef] [Green Version]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Kräutler, V.; van Gunsteren, W.F.; Hünenberger, P.H. A fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J. Comp. Chem. 2001, 22, 501–508. [Google Scholar] [CrossRef]
- Schrödinger. The PyMOL Molecular Graphics System, 2.4 ed.; LLC: New York, NY, USA, 2021. [Google Scholar]
- Khan, S.; Gor, J.; Mulloy, B.; Perkins, S.J. Semi-rigid solution structures of heparin by constrained X-ray scattering modelling: New insight into heparin-protein complexes. J. Mol. Biol. 2010, 395, 504–521. [Google Scholar] [CrossRef] [PubMed]
- Berma, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winter, W.T.; Arnott, S.; Isaac, D.H.; Atkins, E.D. Chondroitin 4-sulfate: The structure of a sulfated glycosaminoglycan. J. Mol. Biol. 1978, 125, 1–19. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pérez, Y.; Bonet, R.; Corredor, M.; Domingo, C.; Moure, A.; Messeguer, À.; Bujons, J.; Alfonso, I. Semaphorin 3A—Glycosaminoglycans Interaction as Therapeutic Target for Axonal Regeneration. Pharmaceuticals 2021, 14, 906. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14090906
Pérez Y, Bonet R, Corredor M, Domingo C, Moure A, Messeguer À, Bujons J, Alfonso I. Semaphorin 3A—Glycosaminoglycans Interaction as Therapeutic Target for Axonal Regeneration. Pharmaceuticals. 2021; 14(9):906. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14090906
Chicago/Turabian StylePérez, Yolanda, Roman Bonet, Miriam Corredor, Cecilia Domingo, Alejandra Moure, Àngel Messeguer, Jordi Bujons, and Ignacio Alfonso. 2021. "Semaphorin 3A—Glycosaminoglycans Interaction as Therapeutic Target for Axonal Regeneration" Pharmaceuticals 14, no. 9: 906. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14090906