
Pilot Sub-Study of the Effect of Hepatitis C Cure by Glecaprevir/Pibrentasvir on the Gut Microbiome of Patients with Chronic Hepatitis C Genotypes 1 to 6 in the Mythen Study

 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. Population and Baseline Characteristics
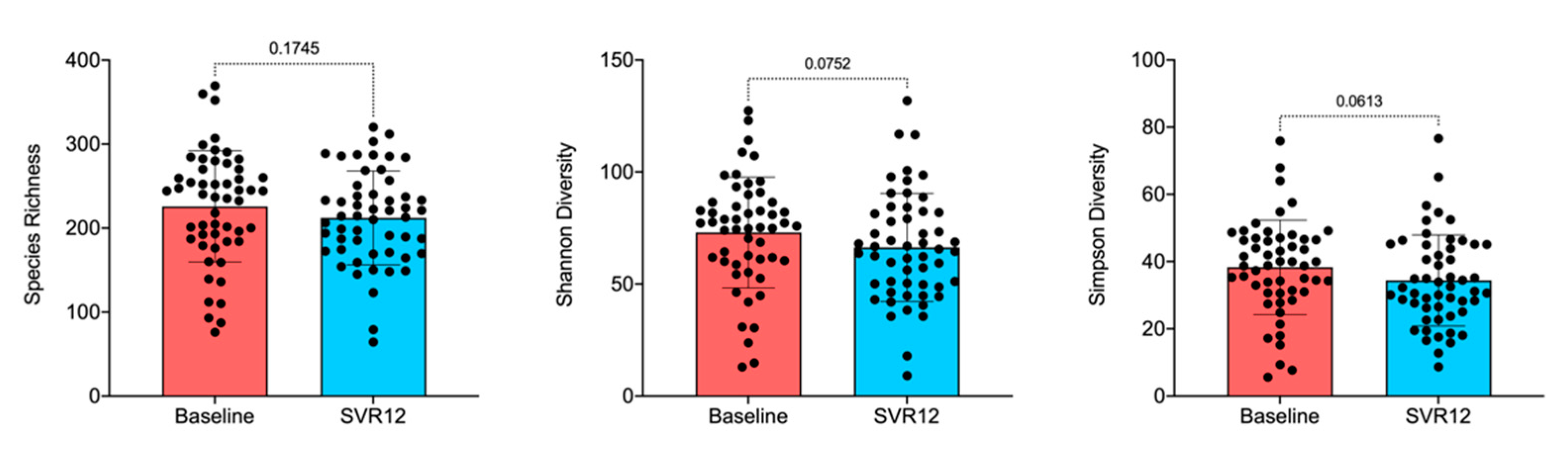
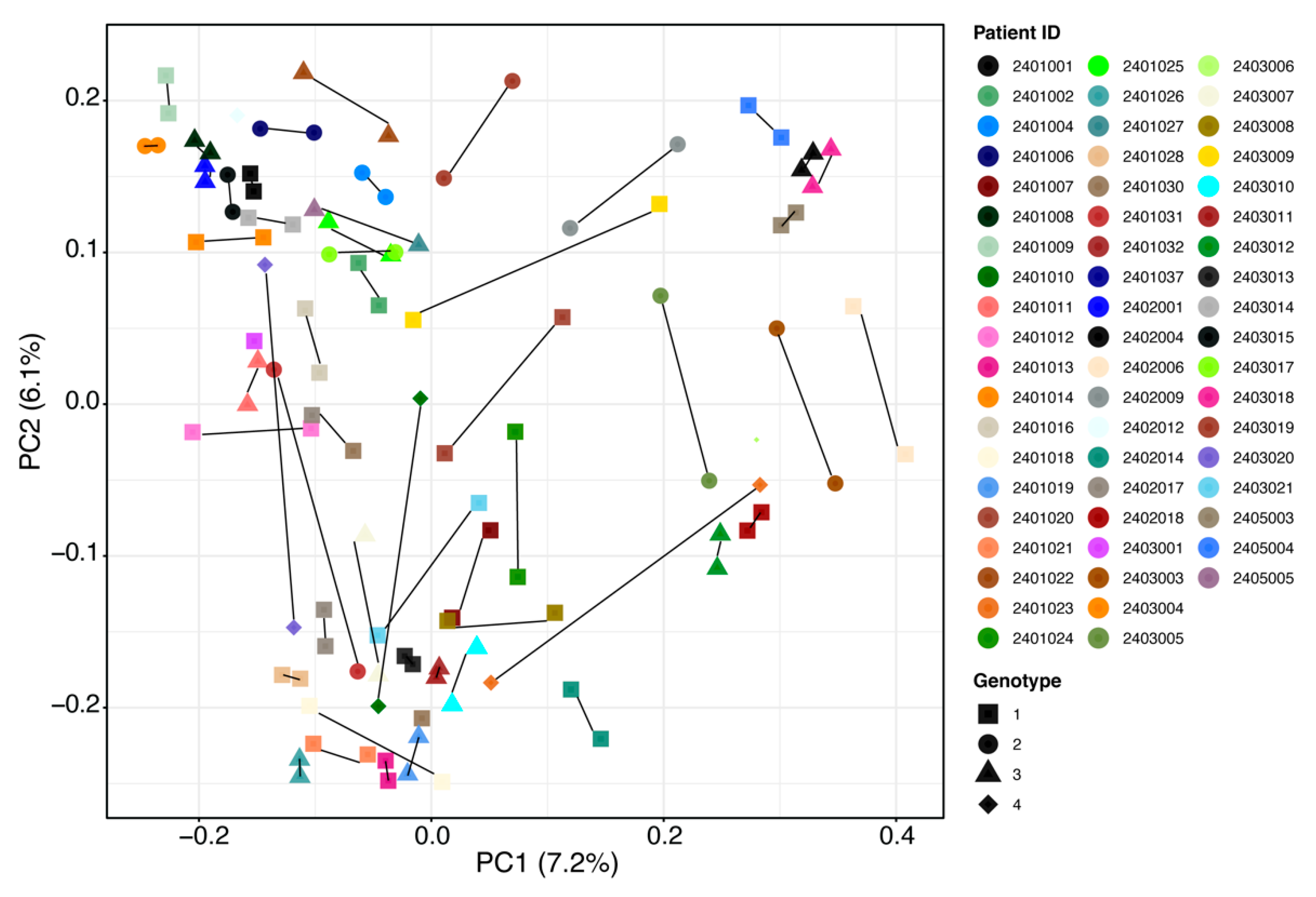
2.2. Gut Microbiota Analysis
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Heidrich, B.; Vital, M.; Plumeier, I.; Doscher, N.; Kahl, S.; Kirschner, J.; Ziegert, S.; Solbach, P.; Lenzen, H.; Potthoff, A.; et al. Intestinal microbiota in patients with chronic hepatitis C with and without cirrhosis compared with healthy controls. Liver Int. 2018, 38, 50–58. [Google Scholar] [CrossRef]
- Hooper, L.V.; Midtvedt, T.; Gordon, J.I. How host-microbial interactions shape the nutrient environment of the mammalian intestine. Annu. Rev. Nutr. 2002, 22, 283–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hooper, L.V.; Macpherson, A.J. Immune adaptations that maintain homeostasis with the intestinal microbiota. Nat. Rev. Immunol. 2010, 10, 159–169. [Google Scholar] [CrossRef]
- Soares, M.P.; Yilmaz, B. Microbiota Control of Malaria Transmission. Trends Parasitol. 2016, 32, 120–130. [Google Scholar] [CrossRef] [Green Version]
- Stecher, B.; Macpherson, A.J.; Hapfelmeier, S.; Kremer, M.; Stallmach, T.; Hardt, W.D. Comparison of Salmonella enterica serovar Typhimurium colitis in germfree mice and mice pretreated with streptomycin. Infect. Immun. 2005, 73, 3228–3241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yilmaz, B.; Schibli, S.; Macpherson, A.J.; Sokollik, C. D-lactic Acidosis: Successful Suppression of D-lactate-Producing Lactobacillus by Probiotics. Pediatrics 2018, 142, e20180337. [Google Scholar] [CrossRef] [Green Version]
- Yilmaz, B.; Portugal, S.; Tran, T.M.; Gozzelino, R.; Ramos, S.; Gomes, J.; Regalado, A.; Cowan, P.J.; d’Apice, A.J.; Chong, A.S.; et al. Gut microbiota elicits a protective immune response against malaria transmission. Cell 2014, 159, 1277–1289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, K.; McCoy, K.D.; Macpherson, A.J. Use of axenic animals in studying the adaptation of mammals to their commensal intestinal microbiota. Semin. Immunol. 2007, 19, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Cahenzli, J.; Balmer, M.L.; McCoy, K.D. Microbial-immune cross-talk and regulation of the immune system. Immunology 2013, 138, 12–22. [Google Scholar] [CrossRef]
- Ganal, S.C.; Sanos, S.L.; Kallfass, C.; Oberle, K.; Johner, C.; Kirschning, C.; Lienenklaus, S.; Weiss, S.; Staeheli, P.; Aichele, P.; et al. Priming of Natural Killer Cells by Nonmucosal Mononuclear Phagocytes Requires Instructive Signals from Commensal Microbiota. Immunity 2012, 37, 171–186. [Google Scholar] [CrossRef] [Green Version]
- Macpherson, A.J.; Yilmaz, B.; Limenitakis, J.P.; Ganal-Vonarburg, S.C. IgA Function in Relation to the Intestinal Microbiota. Annu. Rev. Immunol. 2018, 36, 359–381. [Google Scholar] [CrossRef]
- Lynch, J.B.; Hsiao, E.Y. Microbiomes as sources of emergent host phenotypes. Science 2019, 365, 1405–1409. [Google Scholar] [CrossRef]
- Uchimura, Y.; Fuhrer, T.; Li, H.; Lawson, M.A.; Zimmermann, M.; Yilmaz, B.; Zindel, J.; Ronchi, F.; Sorribas, M.; Hapfelmeier, S.; et al. Antibodies Set Boundaries Limiting Microbial Metabolite Penetration and the Resultant Mammalian Host Response. Immunity 2018, 49, 545–559.e5. [Google Scholar] [CrossRef] [Green Version]
- Clayton, T.A.; Lindon, J.C.; Cloarec, O.; Antti, H.; Charuel, C.; Hanton, G.; Provost, J.P.; Le Net, J.L.; Baker, D.; Walley, R.J.; et al. Pharmaco-metabonomic phenotyping and personalized drug treatment. Nature 2006, 440, 1073–1077. [Google Scholar] [CrossRef]
- de Aguiar Vallim, T.Q.; Tarling, E.J.; Edwards, P.A. Pleiotropic roles of bile acids in metabolism. Cell Metab. 2013, 17, 657–669. [Google Scholar] [CrossRef] [Green Version]
- Grundmann, O. The Gut Microbiome and Pre-systemic Metabolism: Current State and Evolving Research. J. Drug Metab. Toxicol. 2010, 1, 1000104. [Google Scholar] [CrossRef]
- Holmes, E.; Li, J.V.; Athanasiou, T.; Ashrafian, H.; Nicholson, J.K. Understanding the role of gut microbiome-host metabolic signal disruption in health and disease. Trends Microbiol. 2011, 19, 349–359. [Google Scholar] [CrossRef]
- Nieuwdorp, M.; Gilijamse, P.W.; Pai, N.; Kaplan, L.M. Role of the microbiome in energy regulation and metabolism. Gastroenterology 2014, 146, 1525–1533. [Google Scholar] [CrossRef] [PubMed]
- Sekirov, I.; Russell, S.L.; Antunes, L.C.M.; Finlay, B.B. Gut Microbiota in Health and Disease. Physiol. Rev. 2010, 90, 859–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sender, R.; Fuchs, S.; Milo, R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol. 2016, 14, e1002533. [Google Scholar] [CrossRef] [Green Version]
- Manichanh, C.; Borruel, N.; Casellas, F.; Guarner, F. The gut microbiota in IBD. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Marasco, G.; Di Biase, A.R.; Schiumerini, R.; Eusebi, L.H.; Iughetti, L.; Ravaioli, F.; Scaioli, E.; Colecchia, A.; Festi, D. Gut Microbiota and Celiac Disease. Dig. Dis. Sci. 2016, 61, 1461–1472. [Google Scholar] [CrossRef]
- Wong, S.H.; Yu, J. Gut microbiota in colorectal cancer: Mechanisms of action and clinical applications. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 690–704. [Google Scholar] [CrossRef]
- Kim, D.; Zeng, M.Y.; Nunez, G. The interplay between host immune cells and gut microbiota in chronic inflammatory diseases. Exp. Mol. Med. 2017, 49, e339. [Google Scholar] [CrossRef] [Green Version]
- Arora, T.; Backhed, F. The gut microbiota and metabolic disease: Current understanding and future perspectives. J. Intern. Med. 2016, 280, 339–349. [Google Scholar] [CrossRef]
- Webster, D.P.; Klenerman, P.; Dusheiko, G.M. Hepatitis C. Lancet 2015, 385, 1124–1135. [Google Scholar] [CrossRef] [Green Version]
- Messina, J.P.; Humphreys, I.; Flaxman, A.; Brown, A.; Cooke, G.S.; Pybus, O.G.; Barnes, E. Global distribution and prevalence of hepatitis C virus genotypes. Hepatology 2015, 61, 77–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- European Association for the Study of the Liver. Electronic address, eee, Clinical Practice Guidelines Panel C; representative, EGB.; Panel m. EASL recommendations on treatment of hepatitis C: Final update of the series. J. Hepatol. 2020, 73, 1170–1218. [Google Scholar] [CrossRef] [PubMed]
- Summary of Product Characteristics. AbbVie Deutschland GmbH & Co. KG: Ludwigshafen, Germany. Available online: https://www.ema.europa.eu/en/documents/product-information/maviret-epar-product-information_en.pdf (accessed on 16 April 2021).
- Mullhaupt, B.; Semela, D.; Ruckstuhl, L.; Magenta, L.; Clerc, O.; Torgler, R.; Negro, F.; Semmo, N. Real-world effectiveness and safety of glecaprevir/pibrentasvir therapy in patients with chronic hepatitis C virus infection in Switzerland. Swiss Med. Wkly. 2021, 151, w20399. [Google Scholar] [CrossRef]
- Morgan, X.C.; Tickle, T.L.; Sokol, H.; Gevers, D.; Devaney, K.L.; Ward, D.V.; Reyes, J.A.; Shah, S.A.; LeLeiko, N.; Snapper, S.B.; et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 2012, 13, R79. [Google Scholar] [CrossRef]
- Yilmaz, B.; Juillerat, P.; Oyas, O.; Ramon, C.; Bravo, F.D.; Franc, Y.; Fournier, N.; Michetti, P.; Mueller, C.; Geuking, M.; et al. Microbial network disturbances in relapsing refractory Crohn’s disease. Nat. Med. 2019, 25, 323–336. [Google Scholar] [CrossRef] [PubMed]
- Elvers, K.T.; Wilson, V.J.; Hammond, A.; Duncan, L.; Huntley, A.L.; Hay, A.D.; van der Werf, E.T. Antibiotic-induced changes in the human gut microbiota for the most commonly prescribed antibiotics in primary care in the UK: A systematic review. BMJ Open 2020, 10, e035677. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, A.; Metzler-Zebeli, B.U.; Karembe, H.; Sperling, D.; Koger, S.; Joachim, A. Shifts in the Fecal Microbial Community of Cystoisospora suis Infected Piglets in Response to Toltrazuril. Front. Microbiol. 2020, 11, 983. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Andrews, S.C. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/2015 (accessed on 16 April 2021).
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Callahan, B.J.; Sankaran, K.; Fukuyama, J.A.; McMurdie, P.J.; Holmes, S.P. Bioconductor Workflow for Microbiome Data Analysis: From raw reads to community analyses. F1000Res 2016, 5, 1492. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [Green Version]
- Reitmeier, S.; Hitch, T.C.A.; Treichel, N.; Fikas, N.; Hausmann, B.; Ramer-Tait, A.E.; Neuhaus, K.; Berry, D.; Haller, D.; Lagkouvardos, I.; et al. Handling of spurious sequences affects the outcome of high-throughput 16S rRNA gene amplicon profiling. ISME Commun. 2021, 1, 31. [Google Scholar] [CrossRef]
- Jost, L. Partitioning diversity into independent alpha and beta components. Ecology 2007, 88, 2427–2439. [Google Scholar] [CrossRef] [Green Version]
- Chao, A.; Gotelli, N.J.; Hsieh, T.C.; Sander, E.L.; Ma, K.H.; Colwell, R.K.; Ellison, A.M. Rarefaction and extrapolation with Hill numbers: A framework for sampling and estimation in species diversity studies. Ecol. Monogr. 2014, 84, 45–67. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, T.C.; Ma, K.H.; Chao, A. iNEXT: An R package for rarefaction and extrapolation of species diversity (Hill numbers). Methods Ecol. Evol. 2016, 7, 1451–1456. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Characteristics | Gut Microbiota Sub-Group N = 58 |
|---|---|
| Mean ± SD age, years | 54.3 ± 11.8 |
| ≥ 65, n | 7 |
| Mean BMI, kg/m2 | 24.0 |
| ≥ 30.0, clinically obese, n | 4 |
| Sex, n | |
| Men | 33 |
| Women | 25 |
| HCV genotype, n | |
| 1 a | 27 |
| 2 | 10 |
| 3 | 16 |
| 4 | 5 |
| Cirrhosis, n | 5 |
| Pre-treatment for HCV, n | 6 |
| Illicit drug use, n | 33 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yilmaz, B.; Ruckstuhl, L.; Müllhaupt, B.; Magenta, L.; Kuster, M.H.; Clerc, O.; Torgler, R.; Semmo, N. Pilot Sub-Study of the Effect of Hepatitis C Cure by Glecaprevir/Pibrentasvir on the Gut Microbiome of Patients with Chronic Hepatitis C Genotypes 1 to 6 in the Mythen Study. Pharmaceuticals 2021, 14, 931. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14090931
Yilmaz B, Ruckstuhl L, Müllhaupt B, Magenta L, Kuster MH, Clerc O, Torgler R, Semmo N. Pilot Sub-Study of the Effect of Hepatitis C Cure by Glecaprevir/Pibrentasvir on the Gut Microbiome of Patients with Chronic Hepatitis C Genotypes 1 to 6 in the Mythen Study. Pharmaceuticals. 2021; 14(9):931. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14090931
Chicago/Turabian StyleYilmaz, Bahtiyar, Lisa Ruckstuhl, Beat Müllhaupt, Lorenzo Magenta, Melanie Harrer Kuster, Olivier Clerc, Ralph Torgler, and Nasser Semmo. 2021. "Pilot Sub-Study of the Effect of Hepatitis C Cure by Glecaprevir/Pibrentasvir on the Gut Microbiome of Patients with Chronic Hepatitis C Genotypes 1 to 6 in the Mythen Study" Pharmaceuticals 14, no. 9: 931. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14090931