
The Metabolomic Approach Reveals the Alteration in Human Serum and Cerebrospinal Fluid Composition in Parkinson’s Disease Patients

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Results
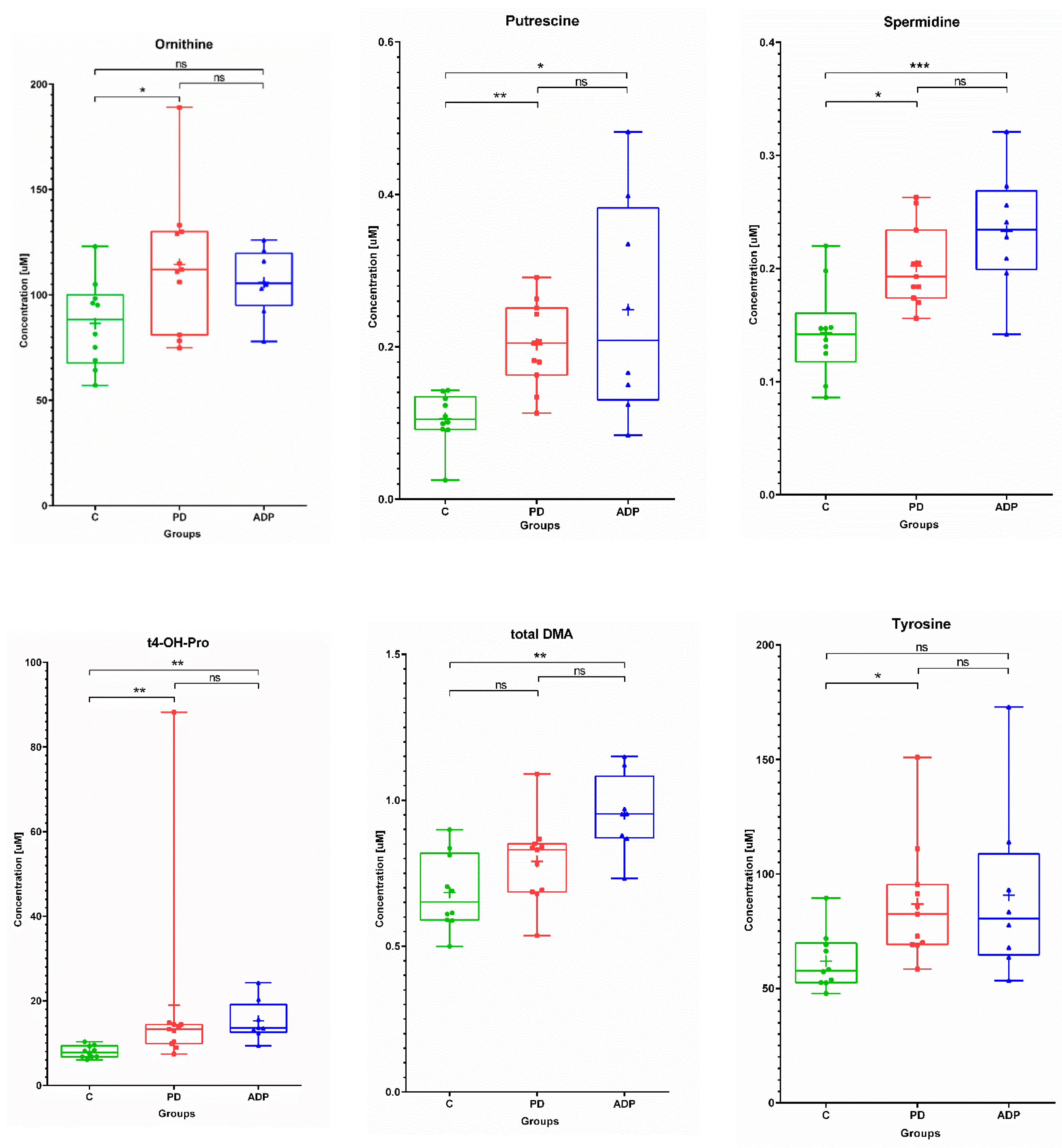
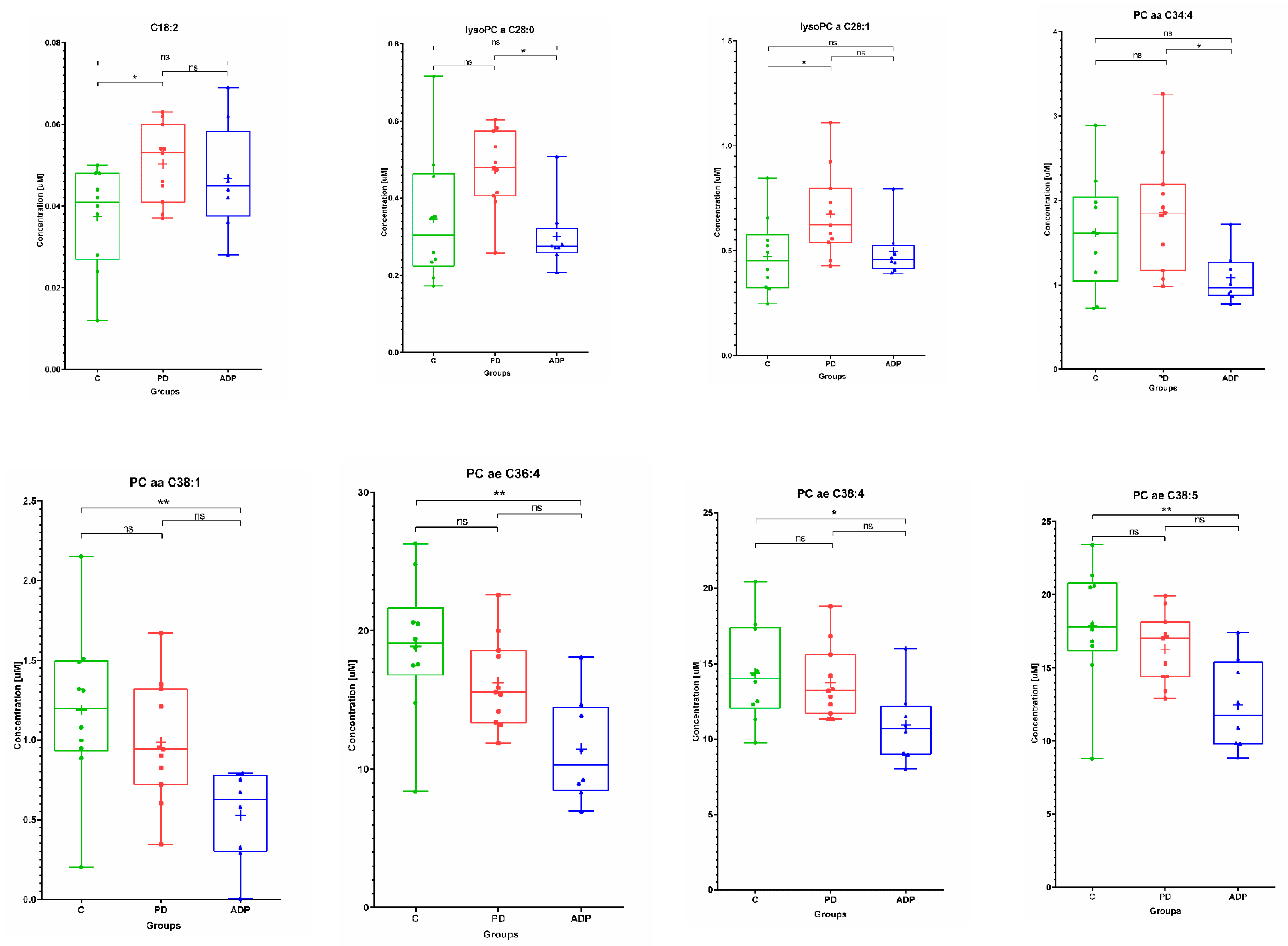
2.1. Serum Metabolome
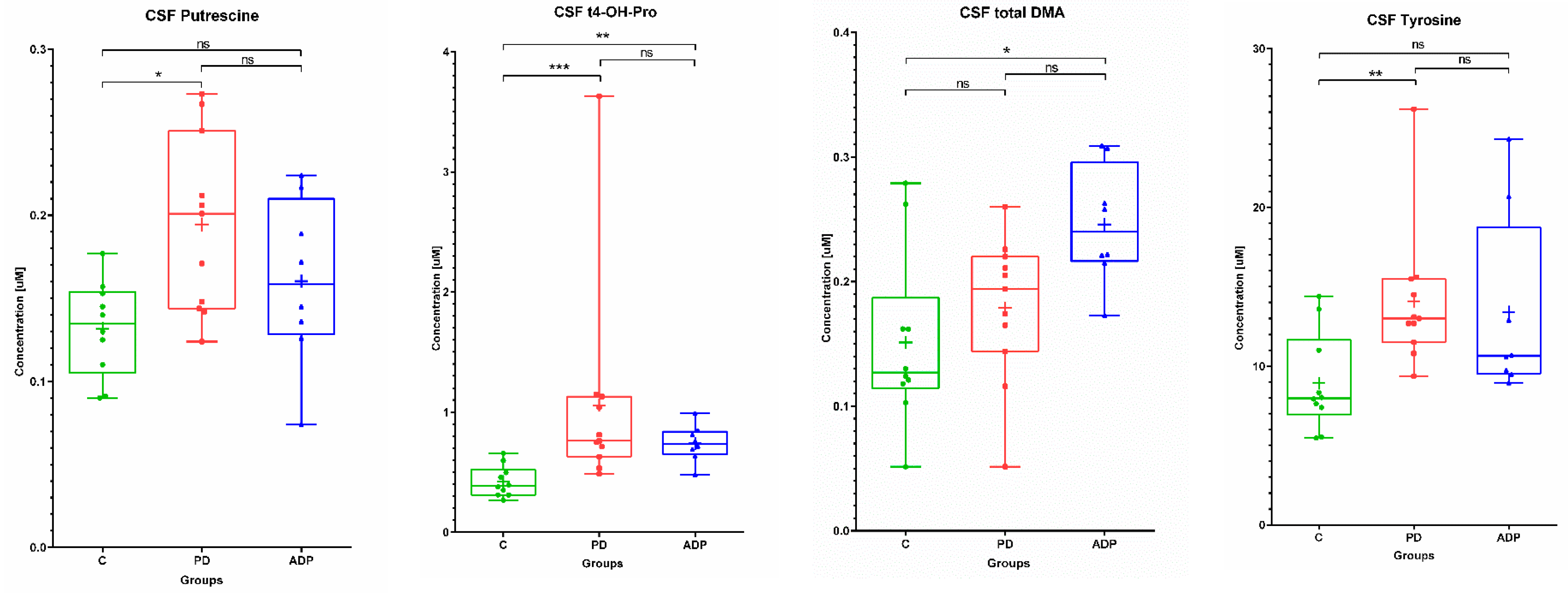
2.2. CSF Metabolome
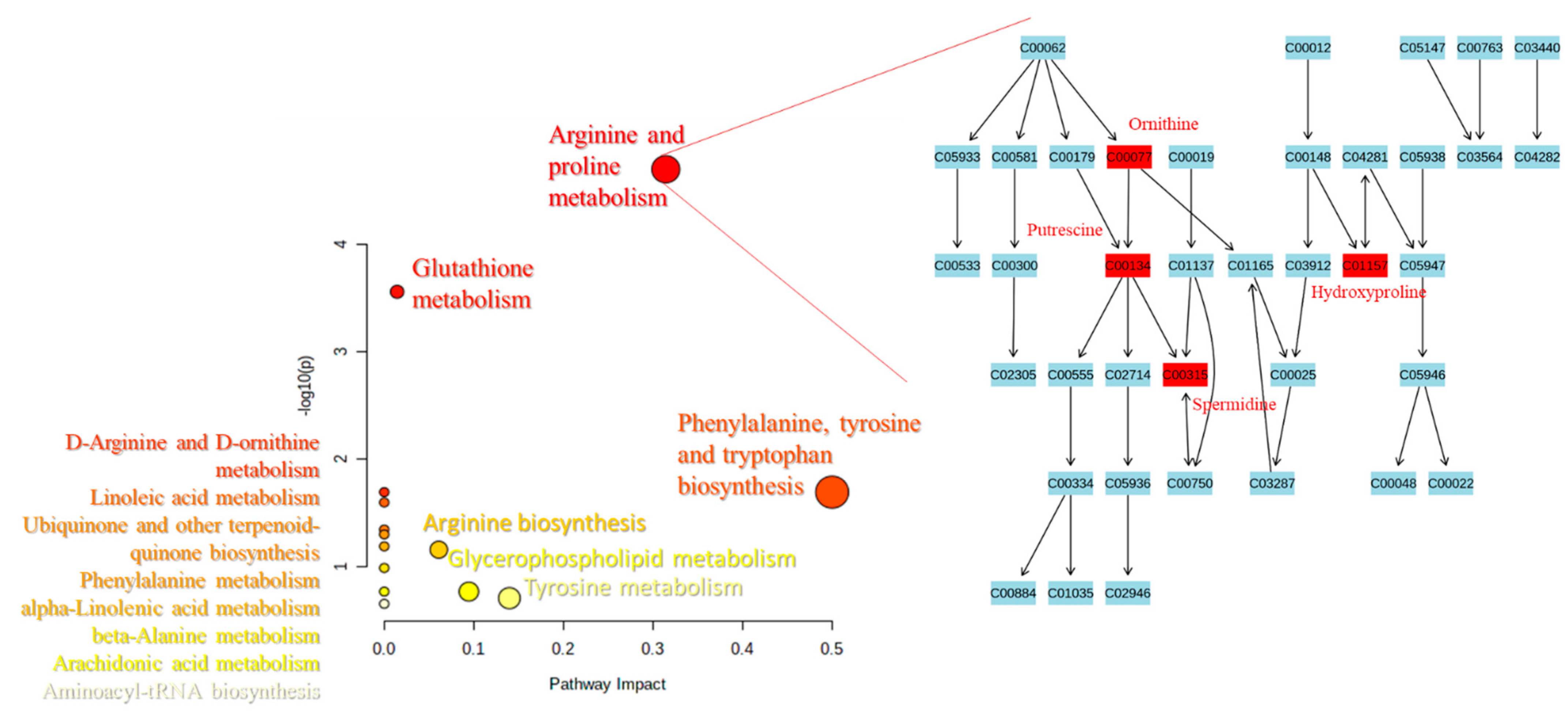
2.3. Pathways Analysis
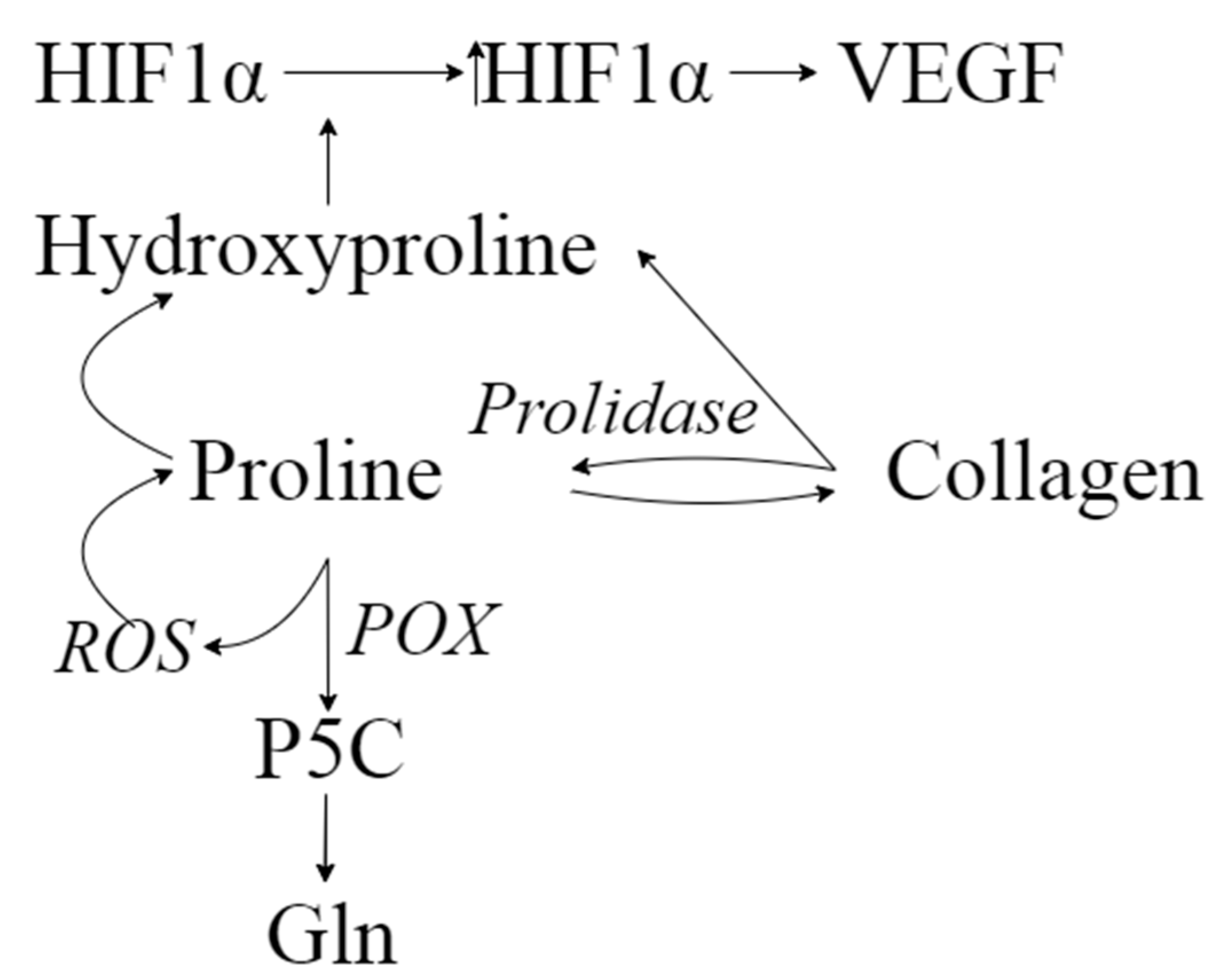
3. Discussion
4. Materials and Methods
4.1. Patient Recruitment and Diagnosis
4.2. Sample Collection
4.3. Sample Preparation
4.4. Instrumentation and Metabolite Assays
4.5. Data Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- De Rijk, M.C.; Tzourio, C.; Breteler, M.M.; Dartigues, J.F.; Amaducci, L.; Lopez-Pousa, S.; Manubens-Bertran, J.M.; Alpérovitch, A.; Rocca, W.A. Prevalence of parkinsonism and Parkinson’s disease in Europe: The EUROPARKINSON Collaborative Study. European Community Concerted Action on the Epidemiology of Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 1997, 62, 10–15. [Google Scholar] [CrossRef] [Green Version]
- Miller, D.B.; O’Callaghan, J.P. Biomarkers of Parkinson’s disease: Present and future. Metabolism 2015, 64, S40–S46. [Google Scholar] [CrossRef] [Green Version]
- LeWitt, P.A.; Li, J.; Lu, M.; Guo, L.; Auinger, P. Parkinson Study Group–DATATOP Investigators Metabolomic biomarkers as strong correlates of Parkinson disease progression. Neurology 2017, 88, 862–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burté, F.; Houghton, D.; Lowes, H.; Pyle, A.; Nesbitt, S.; Yarnall, A.; Yu-Wai-Man, P.; Burn, D.J.; Santibanez-Koref, M.; Hudson, G. Metabolic profiling of Parkinson’s disease and mild cognitive impairment. Mov. Disord. 2017, 32, 927–932. [Google Scholar] [CrossRef]
- Emamzadeh, F.N.; Surguchov, A. Parkinson’s disease: Biomarkers, treatment, and risk factors. Front. Neurosci. 2018, 12, 612. [Google Scholar] [CrossRef]
- Moretti, D.V. Available and future treatments for atypical parkinsonism. A systematic review. CNS Neurosci. Ther. 2019, 25, 159–174. [Google Scholar] [CrossRef]
- Wuolikainen, A.; Jonsson, P.; Ahnlund, M.; Antti, H.; Marklund, S.L.; Moritz, T.; Forsgren, L.; Andersen, P.M.; Trupp, M. Multi-platform mass spectrometry analysis of the CSF and plasma metabolomes of rigorously matched amyotrophic lateral sclerosis, Parkinson’s disease and control subjects. Mol. Biosyst. 2016, 12, 1287–1298. [Google Scholar] [CrossRef]
- Andersen, A.D.; Binzer, M.; Stenager, E.; Gramsbergen, J.B. Cerebrospinal fluid biomarkers for Parkinson’s disease—A systematic review. Acta Neurol. Scand. 2017, 135, 34–56. [Google Scholar] [CrossRef]
- Wang, J.; Hoekstra, J.G.; Zuo, C.; Cook, T.J.; Zhang, J. Biomarkers of Parkinson’s disease: Current status and future perspectives. Drug Discov. Today 2013, 18, 155–162. [Google Scholar] [CrossRef] [Green Version]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Hasegawa, M.; Goedert, M. alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc. Natl. Acad. Sci. USA 1998, 95, 6469–6473. [Google Scholar] [CrossRef] [Green Version]
- Lei, P.; Ayton, S.; Finkelstein, D.I.; Adlard, P.A.; Masters, C.L.; Bush, A.I. Tau protein: Relevance to Parkinson’s disease. Int. J. Biochem. Cell Biol. 2010, 42, 1775–1778. [Google Scholar] [CrossRef]
- Gomperts, S.N.; Rentz, D.M.; Moran, E.; Becker, J.A.; Locascio, J.J.; Klunk, W.E.; Mathis, C.A.; Elmaleh, D.R.; Shoup, T.; Fischman, A.J.; et al. Imaging amyloid deposition in Lewy body diseases. Neurology 2008, 71, 903–910. [Google Scholar] [CrossRef]
- Schapira, A.H.V. Mitochondria in the aetiology and pathogenesis of Parkinson’s disease. Lancet Neurol. 2008, 7, 97–109. [Google Scholar] [CrossRef]
- Blesa, J.; Trigo-Damas, I.; Quiroga-Varela, A.; Jackson-Lewis, V.R. Oxidative stress and Parkinson’s disease. Front. Neuroanat. 2015, 9, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivero-Ríos, P.; Gómez-Suaga, P.; Fdez, E.; Hilfiker, S. Upstream deregulation of calcium signaling in Parkinson’s disease. Front. Mol. Neurosci. 2014, 7, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamberts, J.T.; Hildebrandt, E.N.; Brundin, P. Spreading of α-synuclein in the face of axonal transport deficits in Parkinson’s disease: A speculative synthesis. Neurobiol. Dis. 2015, 77, 276–283. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, Y.; Zhou, J. Neuroinflammation in Parkinson’s disease and its potential as therapeutic target. Transl. Neurodegener. 2015, 4, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogdanov, M.; Matson, W.R.; Wang, L.; Matson, T.; Saunders-Pullman, R.; Bressman, S.S.; Flint Beal, M. Metabolomic profiling to develop blood biomarkers for Parkinson’s disease. Brain 2008, 131, 389–396. [Google Scholar] [CrossRef] [Green Version]
- Lewitt, P.A.; Li, J.; Lu, M.; Beach, T.G.; Adler, C.H.; Guo, L. Arizona Parkinson’s Disease Consortium 3-hydroxykynurenine and other Parkinson’s disease biomarkers discovered by metabolomic analysis. Mov. Disord. 2013, 28, 1653–1660. [Google Scholar] [CrossRef]
- Trupp, M.; Jonsson, P.; Ohrfelt, A.; Zetterberg, H.; Obudulu, O.; Malm, L.; Wuolikainen, A.; Linder, J.; Moritz, T.; Blennow, K.; et al. Metabolite and peptide levels in plasma and CSF differentiating healthy controls from patients with newly diagnosed Parkinson’s disease. J. Parkinsons. Dis. 2014, 4, 549–560. [Google Scholar] [CrossRef]
- García-Sanz, P.; Orgaz, L.; Bueno-Gil, G.; Espadas, I.; Rodríguez-Traver, E.; Kulisevsky, J.; Gutierrez, A.; Dávila, J.C.; González-Polo, R.A.; Fuentes, J.M.; et al. N370S-GBA1 mutation causes lysosomal cholesterol accumulation in Parkinson’s disease. Mov. Disord. 2017, 32, 1409–1422. [Google Scholar] [CrossRef] [PubMed]
- Johansen, K.K.; Wang, L.; Aasly, J.O.; White, L.R.; Matson, W.R.; Henchcliffe, C.; Beal, M.F.; Bogdanov, M. Metabolomic Profiling in LRRK2-Related Parkinson’s Disease. PLoS ONE 2009, 4, e7551. [Google Scholar] [CrossRef] [Green Version]
- Luan, H.; Liu, L.-F.; Meng, N.; Tang, Z.; Chua, K.-K.; Chen, L.-L.; Song, J.-X.; Mok, V.C.T.; Xie, L.-X.; Li, M.; et al. LC–MS-Based Urinary Metabolite Signatures in Idiopathic Parkinson’s Disease. J. Proteome Res. 2015, 14, 467–478. [Google Scholar] [CrossRef]
- Luan, H.; Liu, L.-F.; Tang, Z.; Zhang, M.; Chua, K.-K.; Song, J.-X.; Mok, V.C.T.; Li, M.; Cai, Z. Comprehensive urinary metabolomic profiling and identification of potential noninvasive marker for idiopathic Parkinson’s disease. Sci. Rep. 2015, 5, 13888. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, S.S.; Santosh, W.; Kumar, S.; Christlet, H.T.T. Metabolic profiling of Parkinson’s disease: Evidence of biomarker from gene expression analysis and rapid neural network detection. J. Biomed. Sci. 2009, 16, 63. [Google Scholar] [CrossRef] [Green Version]
- Öhman, A.; Forsgren, L. NMR metabonomics of cerebrospinal fluid distinguishes between Parkinson’s disease and controls. Neurosci. Lett. 2015, 594, 36–39. [Google Scholar] [CrossRef]
- Roede, J.R.; Uppal, K.; Park, Y.; Lee, K.; Tran, V.; Walker, D.; Strobel, F.H.; Rhodes, S.L.; Ritz, B.; Jones, D.P. Serum Metabolomics of Slow vs. Rapid Motor Progression Parkinson’s Disease: A Pilot Study. PLoS ONE 2013, 8, e77629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatano, T.; Saiki, S.; Okuzumi, A.; Mohney, R.P.; Hattori, N. Identification of novel biomarkers for Parkinson’s disease by metabolomic technologies. J. Neurol. Neurosurg. Psychiatry 2016, 87, 295–301. [Google Scholar] [CrossRef]
- Havelund, J.F.; Andersen, A.D.; Binzer, M.; Blaabjerg, M.; Heegaard, N.H.; Stenager, E.; Faergeman, N.J.; Gramsbergen, J.B. Changes in kynurenine pathway metabolism in Parkinson patients with L-DOPA-induced dyskinesia. J. Neurochem. 2017, 142, 756–766. [Google Scholar] [CrossRef]
- Saiki, S.; Hatano, T.; Fujimaki, M.; Ishikawa, K.-I.; Mori, A.; Oji, Y.; Okuzumi, A.; Fukuhara, T.; Koinuma, T.; Imamichi, Y.; et al. Decreased long-chain acylcarnitines from insufficient β-oxidation as potential early diagnostic markers for Parkinson’s disease. Sci. Rep. 2017, 7, 7328. [Google Scholar] [CrossRef]
- Trezzi, J.-P.; Galozzi, S.; Jaeger, C.; Barkovits, K.; Brockmann, K.; Maetzler, W.; Berg, D.; Marcus, K.; Betsou, F.; Hiller, K.; et al. Distinct metabolomic signature in cerebrospinal fluid in early parkinson’s disease. Mov. Disord. 2017, 32, 1401–1408. [Google Scholar] [CrossRef]
- Patti, G.J.; Yanes, O.; Shriver, L.P.; Courade, J.-P.; Tautenhahn, R.; Manchester, M.; Siuzdak, G. Metabolomics implicates altered sphingolipids in chronic pain of neuropathic origin. Nat. Chem. Biol. 2012, 8, 232–234. [Google Scholar] [CrossRef] [Green Version]
- Botas, A.; Campbell, H.M.; Han, X.; Maletic-Savatic, M. Metabolomics of neurodegenerative diseases. Int. Rev. Neurobiol. 2015, 122, 53–80. [Google Scholar] [CrossRef]
- Siskos, A.P.; Jain, P.; Römisch-Margl, W.; Bennett, M.; Achaintre, D.; Asad, Y.; Marney, L.; Richardson, L.; Koulman, A.; Griffin, J.L.; et al. Interlaboratory Reproducibility of a Targeted Metabolomics Platform for Analysis of Human Serum and Plasma. Anal. Chem. 2017, 89, 656–665. [Google Scholar] [CrossRef]
- Phang, J.M. Proline Metabolism in Cell Regulation and Cancer Biology: Recent Advances and Hypotheses. Antioxid. Redox Signal. 2019, 30, 635–649. [Google Scholar] [CrossRef] [Green Version]
- Phang, J.M.; Liu, W.; Hancock, C.N.; Fischer, J.W. Proline metabolism and cancer: Emerging links to glutamine and collagen. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 71–77. [Google Scholar] [CrossRef] [Green Version]
- Çelik, V.K.; Çiğdem, B.; Kapancik, S.; Kiliçgün, H.; Bolayir, E. The Importance of Increased Serum Ornithine Levels in the Pathogenesıs of Alzheımer and Parkınson’s Dıseases. Asian J. Res. Rep. Neurol. 2018, 1, 1–8. [Google Scholar]
- Jiménez-Jiménez, F.J.; Alonso-Navarro, H.; García-Martín, E.; Agúndez, J.A.G. Cerebrospinal and blood levels of amino acids as potential biomarkers for Parkinson’s disease: Review and meta-analysis. Eur. J. Neurol. 2020, 27, 2336–2347. [Google Scholar] [CrossRef]
- Palka, J.; Oscilowska, I.; Szoka, L. Collagen metabolism as a regulator of proline dehydrogenase/proline oxidase-dependent apoptosis/autophagy. Amino Acids 2021, 1–9. [Google Scholar] [CrossRef]
- Cooper, S.K.; Pandhare, J.; Donald, S.P.; Phang, J.M. A novel function for hydroxyproline oxidase in apoptosis through generation of reactive oxygen species. J. Biol. Chem. 2008, 283, 10485–10492. [Google Scholar] [CrossRef] [Green Version]
- Singh, D.; Srivastava, S.K.; Chaudhuri, T.K.; Upadhyay, G. Multifaceted role of matrix metalloproteinases (MMPs). Front. Mol. Biosci. 2015, 2, 19. [Google Scholar] [CrossRef] [PubMed]
- Henchcliffe, C.; Beal, M.F. Mitochondrial biology and oxidative stress in Parkinson disease pathogenesis. Nat. Clin. Pract. Neurol. 2008, 4, 600–609. [Google Scholar] [CrossRef]
- Li, X.; Cui, X.-X.; Chen, Y.-J.; Wu, T.-T.; Xu, H.; Yin, H.; Wu, Y.-C. Therapeutic Potential of a Prolyl Hydroxylase Inhibitor FG-4592 for Parkinson’s Diseases in Vitro and in Vivo: Regulation of Redox Biology and Mitochondrial Function. Front. Aging Neurosci. 2018, 10, 121. [Google Scholar] [CrossRef] [Green Version]
- Surazynski, A.; Donald, S.P.; Cooper, S.K.; Whiteside, M.A.; Salnikow, K.; Liu, Y.; Phang, J.M. Extracellular matrix and HIF-1 signaling: The role of prolidase. Int. J. Cancer 2008, 122, 1435–1440. [Google Scholar] [CrossRef]
- Janelidze, S.; Lindqvist, D.; Francardo, V.; Hall, S.; Zetterberg, H.; Blennow, K.; Adler, C.H.; Beach, T.G.; Serrano, G.E.; van Westen, D.; et al. Increased CSF biomarkers of angiogenesis in Parkinson disease. Neurology 2015, 85, 1834–1842. [Google Scholar] [CrossRef] [Green Version]
- Pandhare, J.; Dash, S.; Jones, B.; Villalta, F.; Dash, C. A Novel Role of Proline Oxidase in HIV-1 Envelope Glycoprotein-induced Neuronal Autophagy. J. Biol. Chem. 2015, 290, 25439–25451. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.; Zhang, L.; Natarajan, S.K.; Becker, D.F. Proline mechanisms of stress survival. Antioxid. Redox Signal. 2013, 19, 998–1011. [Google Scholar] [CrossRef] [Green Version]
- Raghunathan, R.; Hogan, J.D.; Labadorf, A.; Myers, R.H.; Zaia, J. A glycomics and proteomics study of aging and Parkinson’s disease in human brain. Sci. Rep. 2020, 10, 12804. [Google Scholar] [CrossRef]
- Qiu, B.; Wei, F.; Sun, X.; Wang, X.; Duan, B.; Shi, C.; Zhang, J.; Zhang, J.; Qiu, W.; Mu, W. Measurement of hydroxyproline in collagen with three different methods. Mol. Med. Rep. 2014, 10, 1157–1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hijioka, M.; Inden, M.; Yanagisawa, D.; Kitamura, Y. DJ-1/PARK7: A New Therapeutic Target for Neurodegenerative Disorders. Biol. Pharm. Bull. 2017, 40, 548–552. [Google Scholar] [CrossRef] [Green Version]
- Selvaraj, S.; Piramanayagam, S. Impact of gene mutation in the development of Parkinson’s disease. Genes Dis. 2019, 6, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, T.; Kaji, Y.; Agatsuma, T.; Niki, T.; Arisawa, M.; Shuto, S.; Ariga, H.; Iguchi-Ariga, S.M.M. DJ-1 cooperates with PYCR1 in cell protection against oxidative stress. Biochem. Biophys. Res. Commun. 2013, 436, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Doria, M.; Maugest, L.; Moreau, T.; Lizard, G.; Vejux, A. Contribution of cholesterol and oxysterols to the pathophysiology of Parkinson’s disease. Free Radic. Biol. Med. 2016, 101, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Tohgi, H.; Abe, T.; Takahashi, S.; Nozaki, Y.; Kikuchi, T. Concentrations of tyrosine, L-dihydroxyphenylalanine, dopamine, and 3-O-methyldopa in the cerebrospinal fluid of Parkinson’s disease. Neurosci. Lett. 1991, 127, 212–214. [Google Scholar] [CrossRef]
- Antony, T.; Hoyer, W.; Cherny, D.; Heim, G.; Jovin, T.M.; Subramaniam, V. Cellular polyamines promote the aggregation of alpha-synuclein. J. Biol. Chem. 2003, 278, 3235–3240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Büttner, S.; Broeskamp, F.; Sommer, C.; Markaki, M.; Habernig, L.; Alavian-Ghavanini, A.; Carmona-Gutierrez, D.; Eisenberg, T.; Michael, E.; Kroemer, G.; et al. Spermidine protects against α-synuclein neurotoxicity. Cell Cycle 2014, 13, 3903–3908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, W.; Sapkota, S.; Camicioli, R.; Dixon, R.A.; Li, L. Profiling novel metabolic biomarkers for Parkinson’s disease using in-depth metabolomic analysis. Mov. Disord. 2017, 32, 1720–1728. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, X.; Wang, L.; Yang, C. High Performance Liquid Chromatography-Mass Spectrometry (LC-MS) Based Quantitative Lipidomics Study of Ganglioside-NANA-3 Plasma to Establish Its Association with Parkinson’s Disease Patients. Med. Sci. Monit. 2017, 23, 5345–5353. [Google Scholar] [CrossRef]
- Wood, P.L.; Tippireddy, S.; Feriante, J.; Woltjer, R.L. Augmented frontal cortex diacylglycerol levels in Parkinson’s disease and Lewy Body Disease. PLoS ONE 2018, 13, e0191815. [Google Scholar] [CrossRef]
- Seyfried, T.N.; Choi, H.; Chevalier, A.; Hogan, D.; Akgoc, Z.; Schneider, J.S. Sex-Related Abnormalities in Substantia Nigra Lipids in Parkinson’s Disease. ASN Neuro 2018, 10, 1759091418781889. [Google Scholar] [CrossRef] [Green Version]
- Stoessel, D.; Schulte, C.; Teixeira Dos Santos, M.C.; Scheller, D.; Rebollo-Mesa, I.; Deuschle, C.; Walther, D.; Schauer, N.; Berg, D.; Nogueira da Costa, A.; et al. Promising Metabolite Profiles in the Plasma and CSF of Early Clinical Parkinson’s Disease. Front. Aging Neurosci. 2018, 10, 51. [Google Scholar] [CrossRef] [PubMed]
- Lobasso, S.; Tanzarella, P.; Vergara, D.; Maffia, M.; Cocco, T.; Corcelli, A. Lipid profiling of parkin-mutant human skin fibroblasts. J. Cell. Physiol. 2017, 232, 3540–3551. [Google Scholar] [CrossRef]
- Zhao, H.; Wang, C.; Zhao, N.; Li, W.; Yang, Z.; Liu, X.; Le, W.; Zhang, X. Potential biomarkers of Parkinson’s disease revealed by plasma metabolic profiling. J. Chromatogr. B 2018, 1081–1082, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.-S.Y.; Chen, H.; Charlton, C.G.; Soliman, K.F.A. The Role of Phospholipid Methylation in 1-Methyl-4-Phenyl-Pyridinium Ion (MPP+)-Induced Neurotoxicity in PC12 Cells. Neurotoxicology 2005, 26, 945–957. [Google Scholar] [CrossRef] [PubMed]
- Rebouche, C.J. Kinetics, pharmacokinetics, and regulation of L-carnitine and acetyl-L-carnitine metabolism. Ann. N. Y. Acad. Sci. 2004, 1033, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Hagen, T.M.; Liu, J.; Lykkesfeldt, J.; Wehr, C.M.; Ingersoll, R.T.; Vinarsky, V.; Bartholomew, J.C.; Ames, B.N. Feeding acetyl-L-carnitine and lipoic acid to old rats significantly improves metabolic function while decreasing oxidative stress. Proc. Natl. Acad. Sci. USA 2002, 99, 1870–1875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fritz, I.B.; Arrigoni-Martelli, E. Sites of action of carnitine and its derivatives on the cardiovascular system: Interactions with membranes. Trends Pharmacol. Sci. 1993, 14, 355–360. [Google Scholar] [CrossRef]
- Chang, K.-H.; Cheng, M.-L.; Tang, H.-Y.; Huang, C.-Y.; Wu, Y.-R.; Chen, C.-M. Alternations of Metabolic Profile and Kynurenine Metabolism in the Plasma of Parkinson’s Disease. Mol. Neurobiol. 2018, 55, 6319–6328. [Google Scholar] [CrossRef]
- Hauser, D.N.; Hastings, T.G. Mitochondrial dysfunction and oxidative stress in Parkinson’s disease and monogenic parkinsonism. Neurobiol. Dis. 2013, 51, 35–42. [Google Scholar] [CrossRef] [Green Version]
- Kwiecien, S.; Ptak-Belowska, A.; Krzysiek-Maczka, G.; Targosz, A.; Jasnos, K.; Magierowski, M.; Szczyrk, U.; Brzozowski, B.; Konturek, S.J.; Konturek, P.C.; et al. Asymmetric dimethylarginine, an endogenous inhibitor of nitric oxide synthase, interacts with gastric oxidative metabolism and enhances stress-induced gastric lesions. J. Physiol. Pharmacol. 2012, 63, 515–524. [Google Scholar] [PubMed]
- Sankowski, B.; Księżarczyk, K.; Raćkowska, E.; Szlufik, S.; Koziorowski, D.; Giebułtowicz, J. Higher cerebrospinal fluid to plasma ratio of p-cresol sulfate and indoxyl sulfate in patients with Parkinson’s disease. Clin. Chim. Acta. 2020, 501, 165–173. [Google Scholar] [CrossRef]
- Watanabe, H.; Miyamoto, Y.; Honda, D.; Tanaka, H.; Wu, Q.; Endo, M.; Noguchi, T.; Kadowaki, D.; Ishima, Y.; Kotani, S.; et al. p-Cresyl sulfate causes renal tubular cell damage by inducing oxidative stress by activation of NADPH oxidase. Kidney Int. 2013, 83, 582–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arlt, S.; Schwedhelm, E.; Kölsch, H.; Jahn, H.; Linnebank, M.; Smulders, Y.; Jessen, F.; Böger, R.H.; Popp, J. Dimethylarginines, homocysteine metabolism, and cerebrospinal fluid markers for Alzheimer’s disease. J. Alzheimers. Dis. 2012, 31, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Hughes, A.J.; Daniel, S.E.; Kilford, L.; Lees, A.J. Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: A clinico-pathological study of 100 cases. J. Neurol. Neurosurg. Psychiatry 1992, 55, 181–184. [Google Scholar] [CrossRef] [Green Version]
- Tomlinson, C.L.; Stowe, R.; Patel, S.; Rick, C.; Gray, R.; Clarke, C.E. Systematic review of levodopa dose equivalency reporting in Parkinson’s disease. Mov. Disord. 2010, 25, 2649–2653. [Google Scholar] [CrossRef]
- Plewa, S.; Dereziński, P.; Florczak-Wyspiańska, J.; Popławska-Domaszewicz, K.; Kozubski, W.; Sokół, B.; Jankowski, R.; Matysiak, J.; Kokot, Z.J. LC-MS/MS based targeted metabolomics method for analysis of serum and cerebrospinal fluid. J. Med. Sci. 2019, 88, 12–20. [Google Scholar] [CrossRef]
- Xia, J.; Wishart, D.S. Web-based inference of biological patterns, functions and pathways from metabolomic data using MetaboAnalyst. Nat. Protoc. 2011, 6, 743–760. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Serum | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| No. | Metabolite | Control (n = 10) | PD (n = 11) | APDs (n = 8) | ||||||
| Mean | SD | Median | Mean | SD | Median | Mean | SD | Median | ||
| 1 | C18:2 | 0.04 | 0.01 | 0.04 | 0.05 | 0.01 | 0.05 | 0.05 | 0.01 | 0.05 |
| 2 | Ornithine | 86.43 | 20.57 | 88.30 | 114.47 | 32.3 | 112.00 | 105.91 | 15.63 | 105.50 |
| 3 | Tyrosine | 61.88 | 12.51 | 57.80 | 86.95 | 25.96 | 82.5 | 90.75 | 38.16 | 80.55 |
| 4 | Putrescine | 0.11 | 0.03 | 0.11 | 0.20 | 0.06 | 0.21 | 0.25 | 0.14 | 0.21 |
| 5 | Spermidine | 0.14 | 0.04 | 0.14 | 0.20 | 0.04 | 0.19 | 0.23 | 0.05 | 0.23 |
| 6 | t4-OH-Proline | 7.96 | 1.41 | 7.80 | 18.95 | 23.11 | 13.30 | 15.29 | 4.78 | 13.55 |
| 7 | Total DMA | 0.68 | 0.13 | 0.65 | 0.79 | 0.14 | 0.83 | 0.95 | 0.14 | 0.95 |
| 8 | lysoPC a C26:0 | 0.31 | 0.14 | 0.28 | 0.48 | 0.19 | 0.38 | 0.30 | 0.08 | 0.31 |
| 9 | lysoPC a C28:0 | 0.35 | 0.17 | 0.30 | 0.47 | 0.1 | 0.48 | 0.30 | 0.09 | 0.27 |
| 10 | lysoPC a C28:1 | 0.47 | 0.18 | 0.45 | 0.67 | 0.21 | 0.62 | 0.50 | 0.13 | 0.456 |
| 11 | PC aa C34:4 | 1.62 | 0.67 | 1.62 | 1.85 | 0.68 | 1.85 | 1.08 | 0.31 | 0.97 |
| 12 | PC aa C38:1 | 1.19 | 0.51 | 1.20 | 0.99 | 0.38 | 0.94 | 0.53 | 0.29 | 0.63 |
| 13 | PC ae C36:4 | 18.87 | 5.01 | 19.10 | 16.27 | 3.25 | 15.60 | 11.45 | 3.81 | 10.34 |
| 14 | PC ae C38:4 | 14.38 | 3.23 | 14.05 | 13.75 | 2.41 | 13.20 | 10.91 | 2.52 | 10.70 |
| 15 | PC ae C38:5 | 17.87 | 4.07 | 17.80 | 16.29 | 2.37 | 17.00 | 12.46 | 3.13 | 11.75 |
| 16 | PC ae C40:1 | 1.31 | 0.30 | 1.31 | 1.33 | 0.27 | 1.25 | 1.03 | 0.18 | 1.06 |
| 17 | PC ae C42:1 | 0.31 | 0.088 | 0.31 | 0.37 | 0.06 | 0.37 | 0.29 | 0.07 | 0.28 |
| 18 | PC ae C42:4 | 0.68 | 0.27 | 0.77 | 0.65 | 0.25 | 0.67 | 0.39 | 0.22 | 0.39 |
| Serum | ||||
|---|---|---|---|---|
| No | Metabolite | Post Hoc Test p-Values | ||
| C vs. PD | C vs. APDs | PD vs. APDs | ||
| 1 | C18:2 a | 0.049849 | NS | NS |
| 2 | Ornithine a | 0.045345 | NS | NS |
| 3 | Tyrosine b | 0.015431 | NS | NS |
| 4 | Putrescine b | 0.004395 | 0.011878 | NS |
| 5 | Spermidine a | 0.013458 | 0.000915 | NS |
| 6 | t4-OH-Proline b | 0.006106 | 0.00141 | NS |
| 7 | Total DMA a | NS | 0.001545 | NS |
| 8 | lysoPC a C28:0 b | NS | NS | 0.041845 |
| 9 | lysoPC a C28:1 b | 0.038853 | NS | NS |
| 10 | PC aa C34:4 a | NS | NS | 0.042019 |
| 11 | PC aa C38:1 a | NS | 0.008727 | NS |
| 12 | PC ae C36:4 a | NS | 0.00344 | NS |
| 13 | PC ae C38:4 a | NS | 0.046699 | NS |
| 14 | PC ae C38:5 a | NS | 0.007192 | NS |
| CSF | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| No. | Metabolite | Control (n = 10) | PD (n = 11) | APDs (n = 8) | ||||||
| Mean | SD | Median | Mean | SD | Median | Mean | SD | Median | ||
| 1 | Tyrosine | 8.93 | 3.09 | 7.98 | 14.09 | 4.44 | 13.00 | 13.42 | 5.81 | 10.65 |
| 2 | Putrescine | 0.13 | 0.03 | 0.14 | 0.19 | 0.05 | 0.20 | 0.16 | 0.05 | 0.16 |
| 3 | t4-OH-Proline | 0.42 | 0.13 | 0.39 | 1.06 | 0.88 | 0.76 | 0.74 | 0.15 | 0.74 |
| 4 | total DMA | 0.15 | 0.07 | 0.13 | 0.18 | 0.06 | 0.19 | 0.25 | 0.05 | 0.24 |
| CSF | ||||
|---|---|---|---|---|
| No | Metabolite | Post-Hoc Test p-Values | ||
| C vs. PD | C vs. APDs | PD vs. APDs | ||
| 1 | Tyrosine b | 0.008478 | NS | NS |
| 2 | Putrescine a | 0.01245 | NS | NS |
| 3 | t4-OH-Proline b | 0.000673 | 0.00936 | NS |
| 4 | total DMA a | NS | 0.011032 | NS |
| Feature | PD (n = 10), Mean ± SD | Controls (n = 10), Mean ± SD | APDs (n = 8), Mean ± SD |
|---|---|---|---|
| Male/female (N) | 4/6 | 7/3 | 3/5 |
| Age (years) | 60 ± 52 | 52 ± 13 | 63 ± 12 |
| Disease duration (years) | 7 ± 5 | - | 7 ± 7.5 |
| LEDD | 804 ± 514 | - | 641 ± 446 |
| HY | 2 ± 1 | - | |
| UPDRS III | 19 ± 12 | - | |
| Dementia | 0 | 0 | 1 |
| Depression | 0 | 0 | 3 |
| BMI | 27 ± 5 | 24 ± 3 | 26 ± 4 |
| Other drugs | Levothyroxine, aspirin, quetiapine, and angiotensin-converting enzyme inhibitor | Levothyroxine, beta-blockers, statins, and angiotensin-converting enzyme inhibitor | Quetiapine, sertraline, beta- blockers, statins, angiotensin- converting enzyme inhibitor, and tiapride |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Plewa, S.; Poplawska-Domaszewicz, K.; Florczak-Wyspianska, J.; Klupczynska-Gabryszak, A.; Sokol, B.; Miltyk, W.; Jankowski, R.; Kozubski, W.; Kokot, Z.J.; Matysiak, J. The Metabolomic Approach Reveals the Alteration in Human Serum and Cerebrospinal Fluid Composition in Parkinson’s Disease Patients. Pharmaceuticals 2021, 14, 935. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14090935
Plewa S, Poplawska-Domaszewicz K, Florczak-Wyspianska J, Klupczynska-Gabryszak A, Sokol B, Miltyk W, Jankowski R, Kozubski W, Kokot ZJ, Matysiak J. The Metabolomic Approach Reveals the Alteration in Human Serum and Cerebrospinal Fluid Composition in Parkinson’s Disease Patients. Pharmaceuticals. 2021; 14(9):935. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14090935
Chicago/Turabian StylePlewa, Szymon, Karolina Poplawska-Domaszewicz, Jolanta Florczak-Wyspianska, Agnieszka Klupczynska-Gabryszak, Bartosz Sokol, Wojciech Miltyk, Roman Jankowski, Wojciech Kozubski, Zenon J. Kokot, and Jan Matysiak. 2021. "The Metabolomic Approach Reveals the Alteration in Human Serum and Cerebrospinal Fluid Composition in Parkinson’s Disease Patients" Pharmaceuticals 14, no. 9: 935. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14090935