
High-Throughput Screening and Molecular Dynamics Simulation of Natural Product-like Compounds against Alzheimer’s Disease through Multitarget Approach

 ,
,  ,
,  , ,
, ,  , , and
, , and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Virtual Screening Analysis
2.2. Prediction of Physicochemical, Pharmacokinetics Properties, Drug-Likeness, and Toxicity Potentials
2.3. Molecular Docking Analysis
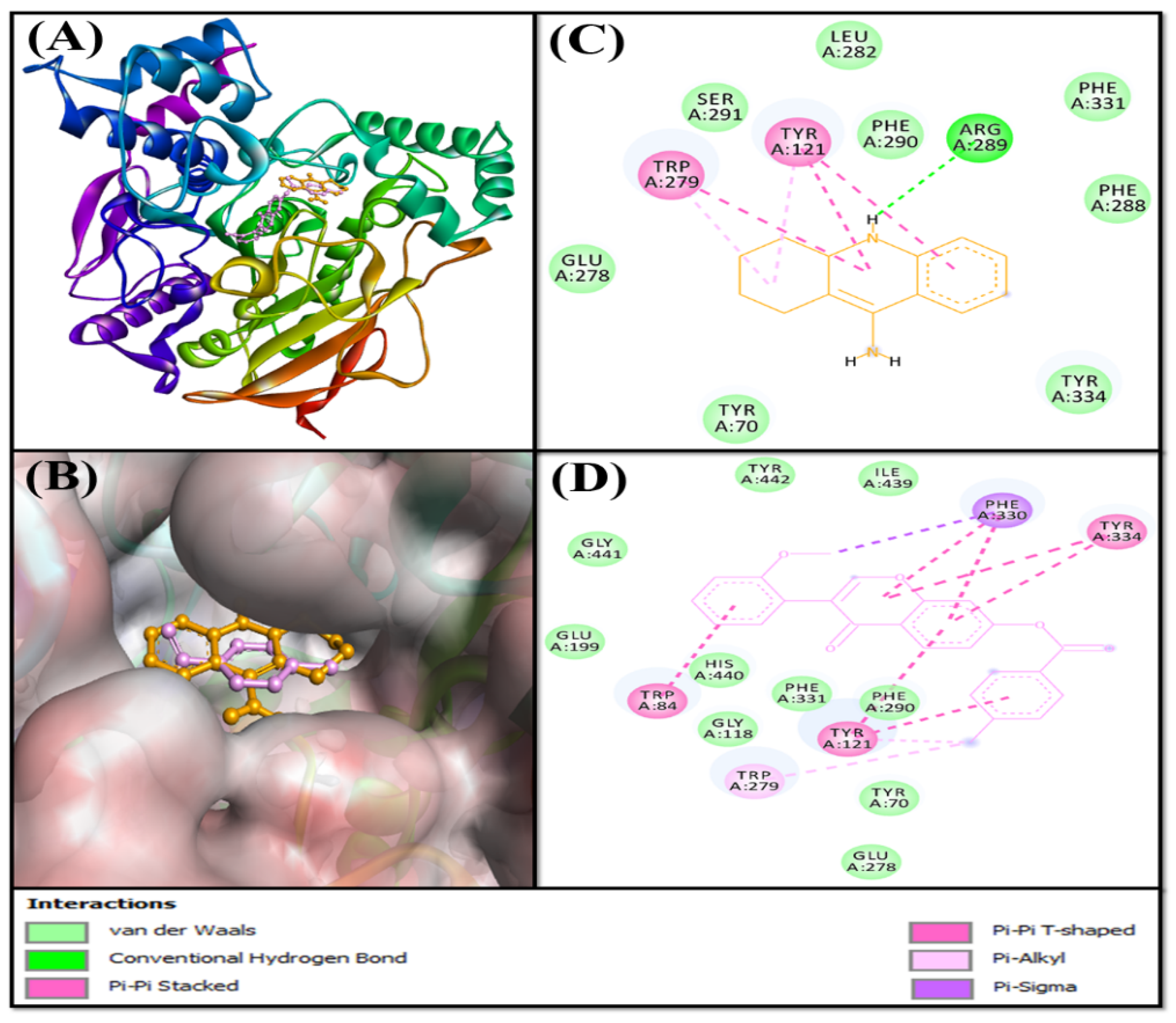
2.3.1. Analysis of the Interaction between AChE and F0850-4777
2.3.2. Analysis of the Interaction between BChE and F0850-4777
2.3.3. Analysis of the Interaction between Monoamine Oxidases and F0850-4777
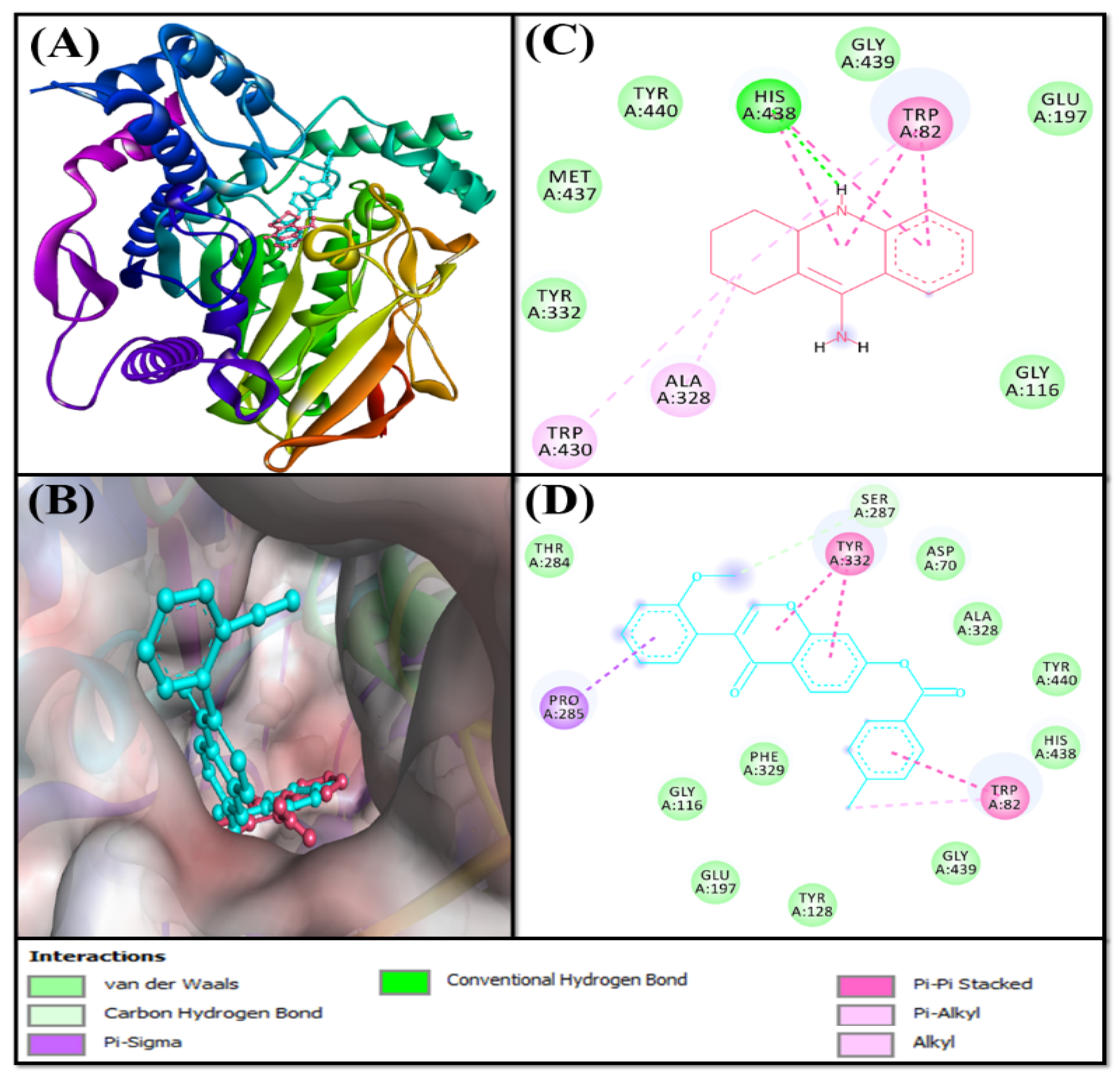
Analysis of the Interaction between MAO-A and F0850-4777
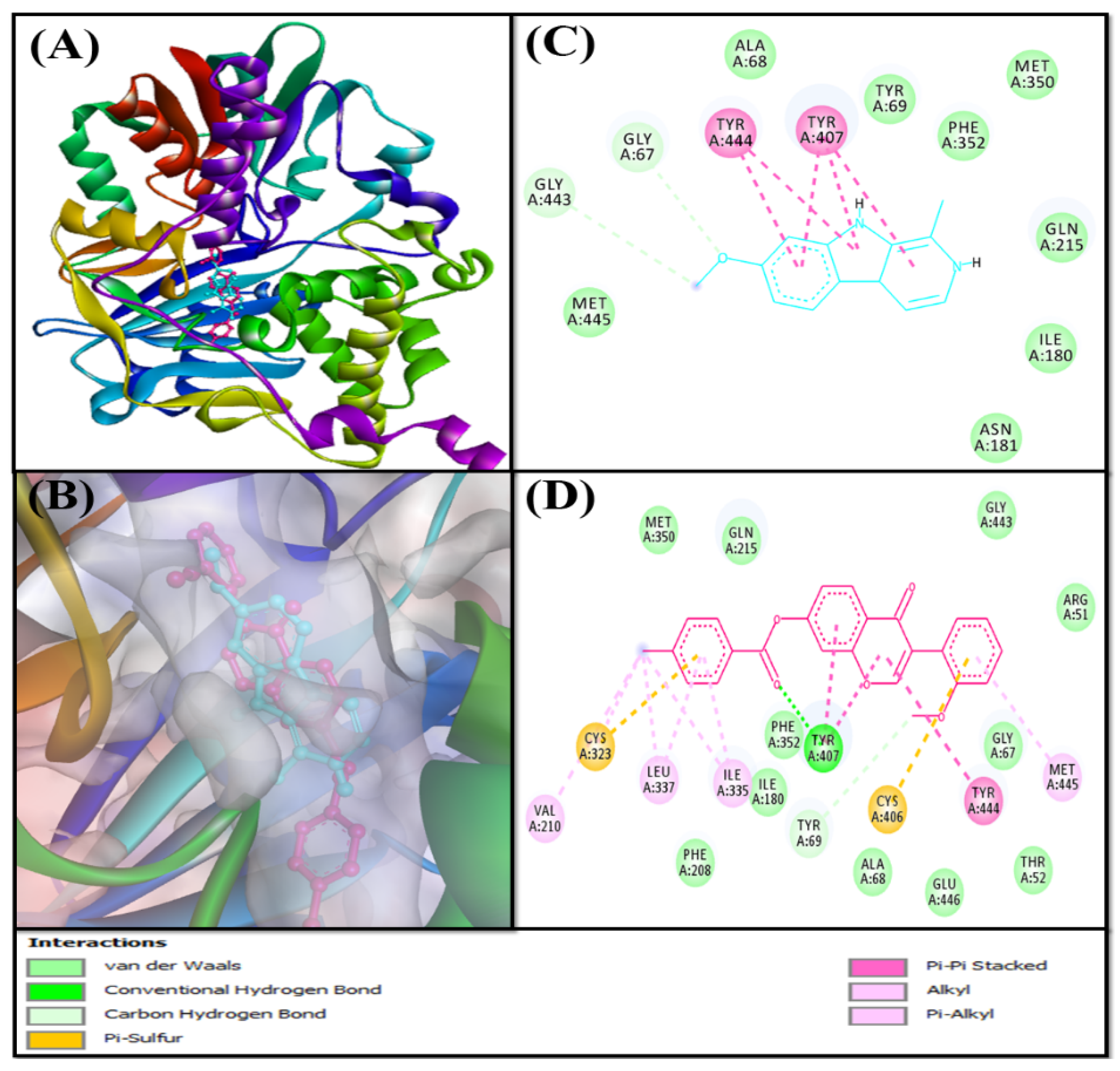
Analysis of the Interaction between MAO-B and F0850-4777
2.4. Analysis of Molecular Dynamics Simulation
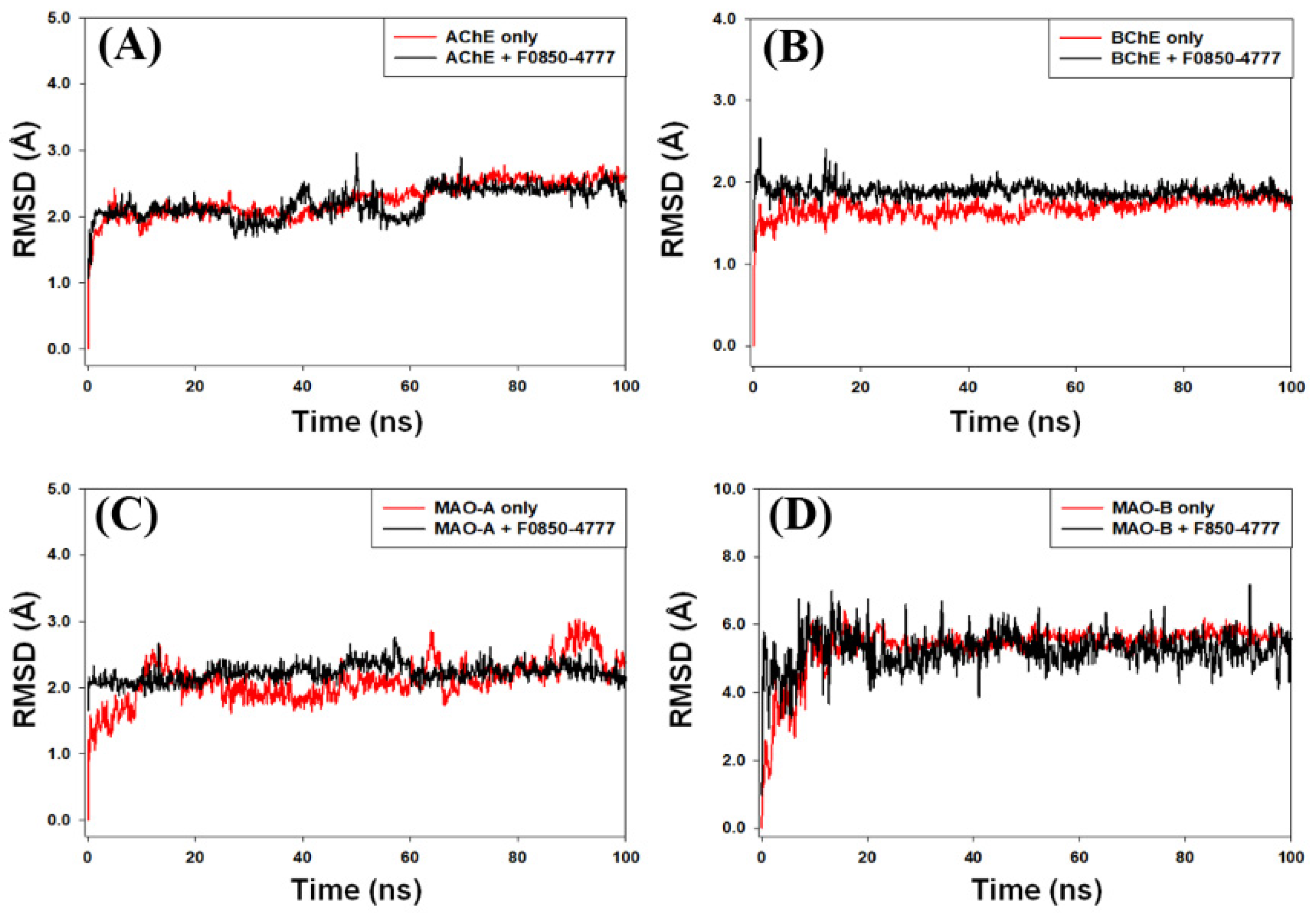
2.4.1. Root Mean Square Deviation (RMSD) Analysis
2.4.2. Root Mean Square Fluctuation (RMSF) Analysis
2.4.3. Analysis of Radius of Gyration (Rg) and Solvent Accessible Surface Area (SASA)
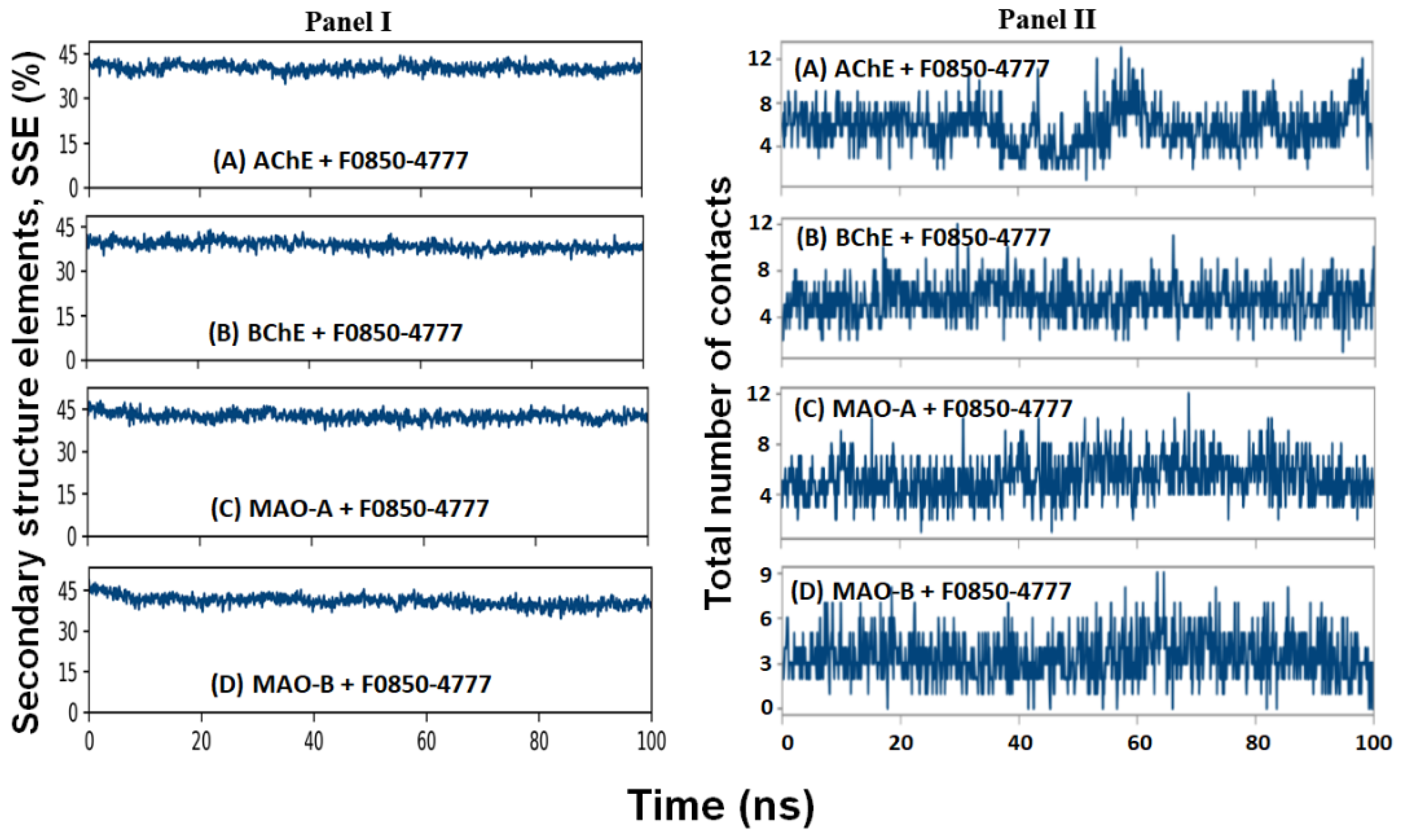
2.4.4. Secondary Structure Analysis
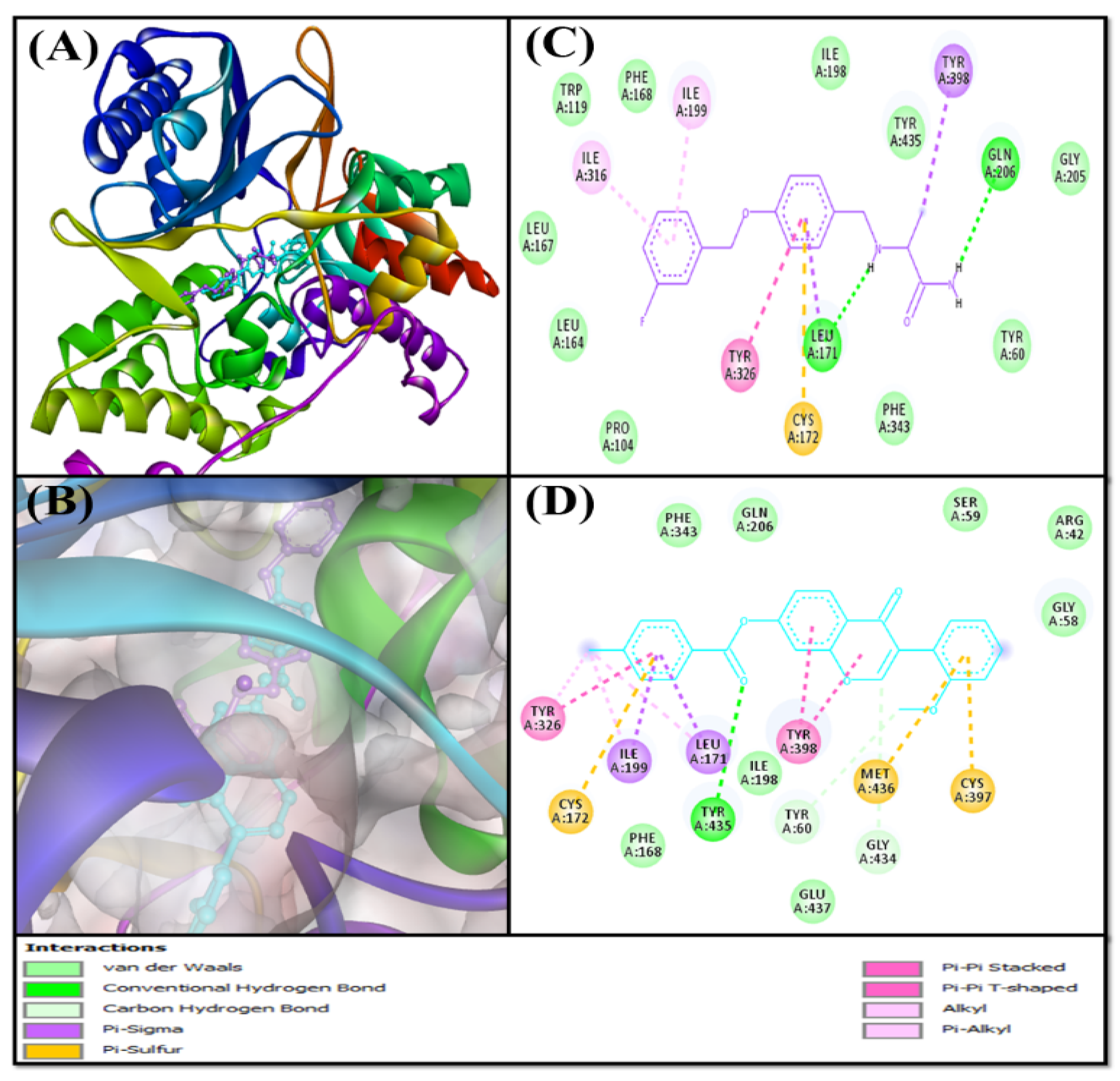
2.4.5. Contact between F0850-4777 and Target Proteins
2.4.6. Analysis of Free Energy (Prime-MM/GBSA) Calculations
3. Materials and Methods
3.1. Hardware and Software Used
3.2. Ligands Preparation
3.3. Protein Target Preparation
3.4. Molecular Docking
3.5. Prediction of Physicochemical, Pharmacokinetics Properties, Drug-Likeness, and Toxicity Potentials
3.6. Molecular Dynamics (MD) Simulation
3.7. Free Energy (Prime-MM/GBSA) Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kim, B.; Noh, G.O.; Kim, K. Behavioural and Psychological Symptoms of Dementia in Patients with Alzheimer’s Disease and Family Caregiver Burden: A Path Analysis. BMC Geriatr. 2021, 21, 160. [Google Scholar] [CrossRef]
- Dementia. Available online: https://www.who.int/news-room/fact-sheets/detail/dementia (accessed on 18 August 2021).
- The Top 10 Causes of Death. Available online: https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death (accessed on 18 August 2021).
- Health Days 2020—World Alzheimer’s Day. Available online: https://www.moh.gov.sa/en/HealthAwareness/healthDay/2020/Pages/HealthDay-2020-09-21.aspx (accessed on 18 August 2021).
- Abeysinghe, A.A.D.T.; Deshapriya, R.D.U.S.; Udawatte, C. Alzheimer’s Disease: A Review of the Pathophysiological Basis and Therapeutic Interventions. Life Sci. 2020, 256, 117996. [Google Scholar] [CrossRef]
- Sayeed Ahmad, S.; Akhtar, S.; Mohammad Sajid Jamal, Q.; Mohd Danish Rizvi, S.A.; Kamal, M.; Kalim, A.; Khan, M.; Haris Siddiqui, M. Multiple Targets for the Management of Alzheimer’s Disease. CNS Neurol. Disord.—Drug Targets—CNS Neurol. Disord. 2016, 15, 1279–1289. [Google Scholar] [CrossRef] [PubMed]
- Calabrò, M.; Rinaldi, C.; Santoro, G.; Crisafulli, C. The Biological Pathways of Alzheimer Disease: A Review. AIMS Neurosci. 2020, 8, 86–132. [Google Scholar] [CrossRef] [PubMed]
- Turner, P.R.; O’Connor, K.; Tate, W.P.; Abraham, W.C. Roles of Amyloid Precursor Protein and Its Fragments in Regulating Neural Activity, Plasticity and Memory. Prog. Neurobiol. 2003, 70, 1–32. [Google Scholar] [CrossRef]
- Reid, G.A.; Chilukuri, N.; Darvesh, S. Butyrylcholinesterase and the Cholinergic System. Neuroscience 2013, 234, 53–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, K.; Li, Y.; Zhang, H.; An, N.; Wei, Y.; Wang, L.; Tian, C.; Yuan, M.; Sun, Y.; Xing, Y.; et al. Oxidative Stress-Mediated Blood-Brain Barrier (BBB) Disruption in Neurological Diseases. Oxid. Med. Cell. Longev. 2020, 2020, e4356386. [Google Scholar] [CrossRef]
- Whitehouse, P.J.; Price, D.L.; Clark, A.W.; Coyle, J.T.; DeLong, M.R. Alzheimer Disease: Evidence for Selective Loss of Cholinergic Neurons in the Nucleus Basalis. Ann. Neurol. 1981, 10, 122–126. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.G.; Crowther, R.A. Tau Proteins and neurofibrillary Degeneration. Brain Pathol. Zur. Switz. 1991, 1, 279–286. [Google Scholar] [CrossRef]
- Strolin Benedetti, M.; Dostert, P. Monoamine oxidase, Brain Ageing and Degenerative Diseases. Biochem. Pharmacol. 1989, 38, 555–561. [Google Scholar] [CrossRef]
- Rosenberg, P.B.; Nowrangi, M.A.; Lyketsos, C.G. Neuropsychiatric Symptoms in Alzheimer’s Disease: What Might be Associated Brain Circuits? Mol. Asp. Med. 2015, 43–44, 25–37. [Google Scholar] [CrossRef] [Green Version]
- Dou, K.-X.; Tan, M.-S.; Tan, C.-C.; Cao, X.-P.; Hou, X.-H.; Guo, Q.-H.; Tan, L.; Mok, V.; Yu, J.-T. Comparative Safety and Effectiveness of Cholinesterase Inhibitors and Memantine for Alzheimer’s Disease: A Network Meta-Analysis of 41 Randomized Controlled Trials. Alzheimers Res. Ther. 2018, 10, 126. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.-K.; Chao, S.-P.; Hu, C.-J. Clinical Trials of New Drugs for Alzheimer Disease. J. Biomed. Sci. 2020, 27, 18. [Google Scholar] [CrossRef] [PubMed]
- Benek, O.; Korabecny, J.; Soukup, O. A Perspective on Multi-Target Drugs for Alzheimer’s Disease. Trends Pharmacol. Sci. 2020, 41, 434–445. [Google Scholar] [CrossRef]
- Grewal, A.S.; Singh, S.; Sharma, N.; Grover, R. In Silico Docking Studies of Some Flavonoids Against Multiple Targets of Alzheimer’s Disease. Plant Arch. 2020, 20, 3271–3278. [Google Scholar]
- Khatoon, A.; Khan, F.; Ahmad, N.; Shaikh, S.; Rizvi, S.M.D.; Shakil, S.; Al-Qahtani, M.H.; Abuzenadah, A.M.; Tabrez, S.; Ahmed, A.B.F.; et al. Silver Nanoparticles from Leaf Extract of Mentha piperita: Eco-Friendly Synthesis and Effect on Acetylcholinesterase Activity. Life Sci. 2018, 209, 430–434. [Google Scholar] [CrossRef]
- Maramai, S.; Benchekroun, M.; Gabr, M.T.; Yahiaoui, S. Multitarget Therapeutic Strategies for Alzheimer’s Disease: Review on Emerging Target Combinations. BioMed Res. Int. 2020, 2020, e5120230. [Google Scholar] [CrossRef]
- Shamsi, A.; Anwar, S.; Mohammad, T.; Alajmi, M.F.; Hussain, A.; Rehman, M.d.T.; Hasan, G.M.; Islam, A.; Hassan, M.d.I. MARK4 Inhibited by AChE Inhibitors, Donepezil and Rivastigmine Tartrate: Insights into Alzheimer’s Disease Therapy. Biomolecules 2020, 10, 789. [Google Scholar] [CrossRef]
- Ahmad, S.S.; Khan, H.; Danish Rizvi, S.M.; Ansari, S.A.; Ullah, R.; Rastrelli, L.; Mahmood, H.M.; Siddiqui, M.H. Computational Study of Natural Compounds for the Clearance of Amyloid-Βeta: A Potential Therapeutic Management Strategy for Alzheimer’s Disease. Molecules 2019, 24, 3233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvi, S.S.; Iqbal, D.; Ahmad, S.; Khan, M.S. Molecular Rationale Delineating the Role of Lycopene as a Potent HMG-CoA Reductase Inhibitor: In Vitro and In Silico Study. Nat. Prod. Res. 2016, 30, 2111–2114. [Google Scholar] [CrossRef]
- Iqbal, D.; Khan, M.S.; Khan, M.S.; Ahmad, S.; Srivastava, A.K. An In Vitro and Molecular Informatics Study to Evaluate the Antioxidative and β-hydroxy-β-methylglutaryl-CoA Reductase Inhibitory Property of Ficus Virens Ait. Phytother. Res. PTR 2014, 28, 899–908. [Google Scholar] [CrossRef]
- Jabir, N.R.; Shakil, S.; Tabrez, S.; Khan, M.S.; Rehman, M.T.; Ahmed, B.A. In Silico Screening of Glycogen Synthase kinase-3β Targeted Ligands against Acetylcholinesterase and Its Probable Relevance to Alzheimer’s Disease. J. Biomol. Struct. Dyn. 2021, 39, 5083–5092. [Google Scholar] [CrossRef]
- Rehman, M.T.; AlAjmi, M.F.; Hussain, A.; Rather, G.M.; Khan, M.A. High-Throughput Virtual Screening, Molecular Dynamics Simulation, and Enzyme Kinetics Identified ZINC84525623 as a Potential Inhibitor of NDM-1. Int. J. Mol. Sci. 2019, 20, 819. [Google Scholar] [CrossRef] [Green Version]
- Shamsi, A.; Mohammad, T.; Khan, M.S.; Shahwan, M.; Husain, F.M.; Rehman, M.T.; Hassan, M.I.; Ahmad, F.; Islam, A. Unraveling Binding Mechanism of Alzheimer’s Drug Rivastigmine Tartrate with Human Transferrin: Molecular Docking and Multi-Spectroscopic Approach towards Neurodegenerative Diseases. Biomolecules 2019, 9, 495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; Supuran, C.T. Natural Products in Drug Discovery: Advances and Opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216. [Google Scholar] [CrossRef]
- Ramsay, R.R.; Popovic-Nikolic, M.R.; Nikolic, K.; Uliassi, E.; Bolognesi, M.L. A perspective on multi-target drug discovery and design for complex diseases. Clin. Transl. Med. 2018, 7, 3. [Google Scholar] [CrossRef] [Green Version]
- Talevi, A. Multi-target pharmacology: Possibilities and limitations of the “skeleton key approach” from a medicinal chemist perspective. Front. Pharmacol. 2015, 6, 205. [Google Scholar] [CrossRef] [Green Version]
- DiMasi, J.A.; Hansen, R.W.; Grabowski, H.G. The price of innovation: New estimates of drug development costs. J. Health Econ. 2003, 22, 151–185. [Google Scholar] [CrossRef] [Green Version]
- Walters, W.P.; Stahl, M.T.; Murcko, M.A. Virtual screening—An overview. Drug Discov. Today 1998, 4, 160–178. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Muñoz-Muriedas, J.; Lopez, J.M.; Orozco, M.; Luque, F.J. Molecular modelling approaches to the design of acetylcholinesterase inhibitors: New challenges for the treatment of Alzheimer’s disease. Curr. Pharm. Des. 2004, 10, 3131–3140. [Google Scholar] [CrossRef]
- Crismon, M.L. Tacrine: First drug approved for Alzheimer’s disease. Ann. Pharmacother. 1994, 28, 744–751. [Google Scholar] [CrossRef]
- Sussman, J.L.; Harel, M.; Frolow, F.; Oefner, C.; Goldman, A.; Toker, L.; Silman, I. Atomic structure of acetylcholinesterase from Torpedo californica: A prototypic acetylcholine-binding protein. Science 1991, 253, 872–879. [Google Scholar] [CrossRef] [PubMed]
- Barak, D.; Kronman, C.; Ordentlich, A.; Ariel, N.; Bromberg, A.; Marcus, D.; Lazar, A.; Velan, B.; Shafferman, A. Acetylcholinesterase peripheral anionic site degeneracy conferred by amino acid arrays sharing a common core. J. Biol. Chem. 1994, 269, 6296–6305. [Google Scholar] [CrossRef]
- Inestrosa, N.C.; Alvarez, A.; Pérez, C.A.; Moreno, R.D.; Vicente, M.; Linker, C.; Casanueva, O.I.; Soto, C.; Garrido, J. Acetylcholinesterase accelerates assembly of amyloid-beta-peptides into Alzheimer’s fibrils: Possible role of the peripheral site of the enzyme. Neuron 1996, 16, 881–891. [Google Scholar] [CrossRef] [Green Version]
- Inestrosa, N.C.; Dinamarca, M.C.; Alvarez, A. Amyloid-cholinesterase interactions. Implications for Alzheimer’s disease. FEBS J. 2008, 275, 625–632. [Google Scholar] [CrossRef]
- Alles, G.A.; Hawes, R.C. Cholinesterases in the Blood of Man. J. Biol. Chem. 1940, 133, 375–390. [Google Scholar] [CrossRef]
- Mendel, B.; Rudney, H. On the Type of Cholinesterase Present in Brain Tissue. Science 1943, 98, 201–202. [Google Scholar] [CrossRef]
- Darvesh, S.; Grantham, D.L.; Hopkins, D.A. Distribution of butyrylcholinesterase in the human amygdala and hippocampal formation. J. Comp. Neurol. 1998, 393, 374–390. [Google Scholar] [CrossRef]
- Tago, H.; Maeda, T.; McGeer, P.L.; Kimura, H. Butyrylcholinesterase-rich neurons in rat brain demonstrated by a sensitive histochemical method. J. Comp. Neurol. 1992, 325, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Mesulam, M.-M.; Guillozet, A.; Shaw, P.; Levey, A.; Duysen, E.G.; Lockridge, O. Acetylcholinesterase knockouts establish central cholinergic pathways and can use butyrylcholinesterase to hydrolyze acetylcholine. Neuroscience 2002, 110, 627–639. [Google Scholar] [CrossRef]
- Layer, P.G. Cholinesterases during development of the avian nervous system. Cell. Mol. Neurobiol. 1991, 11, 7–33. [Google Scholar] [CrossRef]
- Dubovy, P.; Haninec, P. Non-specific cholinesterase activity of the developing peripheral nerves and its possible function in cells in intimate contact with growing axons of chick embryo. Int. J. Dev. Neurosci. Off. J. Int. Soc. Dev. Neurosci. 1990, 8, 589–602. [Google Scholar] [CrossRef]
- Geula, C.; Mesulam, M. Special properties of cholinesterases in the cerebral cortex of Alzheimer’s disease. Brain Res. 1989, 498, 185–189. [Google Scholar] [CrossRef]
- Geula, C.; Mesulam, M.M. Cholinesterases and the pathology of Alzheimer disease. Alzheimer Dis. Assoc. Disord. 1995, 9 (Suppl. 2), 23–28. [Google Scholar] [CrossRef]
- Arendt, T.; Brückner, M.K.; Lange, M.; Bigl, V. Changes in acetylcholinesterase and butyrylcholinesterase in Alzheimer’s disease resemble embryonic development—A study of molecular forms. Neurochem. Int. 1992, 21, 381–396. [Google Scholar] [CrossRef]
- Wright, C.I.; Geula, C.; Mesulam, M.M. Neurological cholinesterases in the normal brain and in Alzheimer’s disease: Relationship to plaques, tangles, and patterns of selective vulnerability. Ann. Neurol. 1993, 34, 373–384. [Google Scholar] [CrossRef]
- Lockridge, O.; Bartels, C.F.; Vaughan, T.A.; Wong, C.K.; Norton, S.E.; Johnson, L.L. Complete amino acid sequence of human serum cholinesterase. J. Biol. Chem. 1987, 262, 549–557. [Google Scholar] [CrossRef]
- Vellom, D.C.; Radić, Z.; Li, Y.; Pickering, N.A.; Camp, S.; Taylor, P. Amino acid residues controlling acetylcholinesterase and butyrylcholinesterase specificity. Biochemistry 1993, 32, 12–17. [Google Scholar] [CrossRef]
- Masson, P.; Xie, W.; Froment, M.T.; Levitsky, V.; Fortier, P.L.; Albaret, C.; Lockridge, O. Interaction between the peripheral site residues of human butyrylcholinesterase, D70 and Y332, in binding and hydrolysis of substrates. Biochim. Biophys. Acta 1999, 1433, 281–293. [Google Scholar] [CrossRef]
- Edmondson, D.E.; Bhattacharyya, A.K.; Walker, M.C. Spectral and kinetic studies of imine product formation in the oxidation of p-(N,N-dimethylamino)benzylamine analogues by monoamine oxidase B. Biochemistry 1993, 32, 5196–5202. [Google Scholar] [CrossRef] [PubMed]
- Fowler, C.J.; Benedetti, M.S. The metabolism of dopamine by both forms of monoamine oxidase in the rat brain and its inhibition by cimoxatone. J. Neurochem. 1983, 40, 1534–1541. [Google Scholar] [CrossRef] [PubMed]
- Hall, D.W.; Logan, B.W.; Parsons, G.H. Further studies on the inhibition of monoamine oxidase by M and B 9302 (clorgyline). I. Substrate specificity in various mammalian species. Biochem. Pharmacol. 1969, 18, 1447–1454. [Google Scholar] [CrossRef]
- Chiba, K.; Trevor, A.; Castagnoli, N. Metabolism of the neurotoxic tertiary amine, MPTP, by brain monoamine oxidase. Biochem. Biophys. Res. Commun. 1984, 120, 574–578. [Google Scholar] [CrossRef]
- Ma, J.; Yoshimura, M.; Yamashita, E.; Nakagawa, A.; Ito, A.; Tsukihara, T. Structure of rat monoamine oxidase A and its specific recognitions for substrates and inhibitors. J. Mol. Biol. 2004, 338, 103–114. [Google Scholar] [CrossRef]
- Binda, C.; Newton-Vinson, P.; Hubálek, F.; Edmondson, D.E.; Mattevi, A. Structure of human monoamine oxidase B, a drug target for the treatment of neurological disorders. Nat. Struct. Biol. 2002, 9, 22–26. [Google Scholar] [CrossRef]
- Binda, C.; Li, M.; Hubalek, F.; Restelli, N.; Edmondson, D.E.; Mattevi, A. Insights into the mode of inhibition of human mitochondrial monoamine oxidase B from high-resolution crystal structures. Proc. Natl. Acad. Sci. USA 2003, 100, 9750–9755. [Google Scholar] [CrossRef] [Green Version]
- Nandigama, R.K.; Miller, J.R.; Edmondson, D.E. Loss of serotonin oxidation as a component of the altered substrate specificity in the Y444F mutant of recombinant human liver MAO A. Biochemistry 2001, 40, 14839–14846. [Google Scholar] [CrossRef]
- Geha, R.M.; Chen, K.; Wouters, J.; Ooms, F.; Shih, J.C. Analysis of Conserved Active Site Residues in Monoamine Oxidase A and B and Their Three-dimensional Molecular Modeling. J. Biol. Chem. 2002, 277, 17209–17216. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Binda, C.; Mattevi, A.; Edmondson, D.E. Functional role of the “aromatic cage” in human monoamine oxidase B: Structures and catalytic properties of Tyr435 mutant proteins. Biochemistry 2006, 45, 4775–4784. [Google Scholar] [CrossRef]
- Son, S.-Y.; Ma, J.; Kondou, Y.; Yoshimura, M.; Yamashita, E.; Tsukihara, T. Structure of human monoamine oxidase A at 2.2-A resolution: The control of opening the entry for substrates/inhibitors. Proc. Natl. Acad. Sci. USA 2008, 105, 5739–5744. [Google Scholar] [CrossRef] [Green Version]
- Nastasă, C.; Tamaian, R.; Oniga, O.; Tiperciuc, B. 5-Arylidene(chromenyl-methylene)-thiazolidinediones: Potential New Agents against Mutant Oncoproteins K-Ras, N-Ras and B-Raf in Colorectal Cancer and Melanoma. Med. Kaunas Lith. 2019, 55, 85. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Rizvi, S.M.D.; Shaikh, S.; Naaz, D.; Shakil, S.; Ahmad, A.; Haneef, M.; Abuzenadah, A.M. Kinetics and Molecular Docking Study of an Anti-diabetic Drug Glimepiride as Acetylcholinesterase Inhibitor: Implication for Alzheimer’s Disease-Diabetes Dual Therapy. Neurochem. Res. 2016, 41, 1475–1482. [Google Scholar] [CrossRef]
- Shaker, B.; Yu, M.-S.; Lee, J.; Lee, Y.; Jung, C.; Na, D. User guide for the discovery of potential drugs via protein structure prediction and ligand docking simulation. J. Microbiol. Seoul Korea 2020, 58, 235–244. [Google Scholar] [CrossRef]
- Svobodova, B.; Mezeiova, E.; Hepnarova, V.; Hrabinova, M.; Muckova, L.; Kobrlova, T.; Jun, D.; Soukup, O.; Jimeno, M.L.; Marco-Contelles, J.; et al. Exploring Structure-Activity Relationship in Tacrine-Squaramide Derivatives as Potent Cholinesterase Inhibitors. Biomolecules 2019, 9, 379. [Google Scholar] [CrossRef] [Green Version]
- Jung, H.A.; Roy, A.; Choi, J.S. In vitro monoamine oxidase A and B inhibitory activity and molecular docking simulations of fucoxanthin. Fish. Sci. 2016, 1, 123–132. [Google Scholar] [CrossRef]
- Rabbani, N.; Tabrez, S.; Islam, B.U.; Rehman, M.T.; Alsenaidy, A.M.; AlAjmi, M.F.; Khan, R.A.; Alsenaidy, M.A.; Khan, M.S. Characterization of colchicine binding with normal and glycated albumin: In vitro and molecular docking analysis. J. Biomol. Struct. Dyn. 2018, 36, 3453–3462. [Google Scholar] [CrossRef]
- Rehman, M.T.; Shamsi, H.; Khan, A.U. Insight into the binding mechanism of imipenem to human serum albumin by spectroscopic and computational approaches. Mol. Pharm. 2014, 11, 1785–1797. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Cherukupalli, S.; Jing, L.; Liu, X.; Zhan, P. Fsp3: A new parameter for drug-likeness. Drug Discov. Today 2020, 25, 1839–1845. [Google Scholar] [CrossRef] [PubMed]
- Openmolecules.org. Available online: http://www.openmolecules.org/datawarrior/download.html (accessed on 7 December 2020).
- AlAjmi, M.F.; Rehman, M.T.; Hussain, A.; Rather, G.M. Pharmacoinformatics approach for the identification of Polo-like kinase-1 inhibitors from natural sources as anti-cancer agents. Int. J. Biol. Macromol. 2018, 116, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Brańka, A.C. Nosé-Hoover chain method for nonequilibrium molecular dynamics simulation. Phys. Rev. E 2000, 61, 4769–4773. [Google Scholar] [CrossRef] [PubMed]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Tripathi, S.K.; Muttineni, R.; Singh, S.K. Extra precision docking, free energy calculation and molecular dynamics simulation studies of CDK2 inhibitors. J. Theor. Biol. 2013, 334, 87–100. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No. | ID Number | Targets/ Formula | Docking Energy (kcal/mol) | |||
|---|---|---|---|---|---|---|
| AChE (1acj) | BChE (4bds) | MAO-A (2z5x) | MAO-B (2v5z) | |||
| 1 | F0870-0001 | C24H15NO6 | −12.9 | −12.6 | −11.5 | −13.6 |
| 2 | F1094-0205 | C26H23NO4 | −12.9 | −11 | −10.8 | −12.6 |
| 3 | F3293-0320 | C22H13NO7 | −12.4 | −11.1 | −12.3 | −13.4 |
| 4 | F1094-0201 | C26H19NO4 | −12.3 | −11.2 | −12.3 | −11.5 |
| 5 | F0850-4777 | C24H18O5 | −12.2 | −10.7 | −13.6 | −12.5 |
| 6 | F3385-6048 | C27H21NO8 | −12.2 | −11.1 | −13.2 | −12.6 |
| 7 | F1094-0200 | C25H17NO4 | −12.1 | −11.2 | −11 | −13.2 |
| 8 | F1865-0198 | C23H15NO6 | −12 | −10.9 | −12.6 | −13.3 |
| 9 | F3139-1101 | C24H16O4 | −12 | −10.3 | −12.4 | −13.6 |
| 10 | F3139-1218 | C26H18O6 | −11.8 | −10.4 | −12.9 | −13.3 |
| 11 | Tacrine | C13H14N2 | −8.5 | −8.4 | ND | ND |
| 12 | Harmine | C13H12N2O | ND | ND | −8.7 | ND |
| 13 | Safinamide | C17H19FN2O2 | ND | ND | ND | −9.5 |
| Pharmacokinetics | Physicochemical Properties | Drug-Likeness | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| S. No. | ID Number | BBB-P | GI-A | MW | Clog-P | HBA | HBD | RB | TPSA | L-V | FSP3 |
| 1 | F0870-0001 | NO | High | 413.37 | 3.29 | 7 | 2 | 3 | 113.77 | 0 | 0.04 |
| 2 | F1094-0205 | YES | High | 413.47 | 6.32 | 5 | 0 | 3 | 59.75 | 0 | 0.3 |
| 3 | F3293-0320 | NO | High | 403.34 | 3.25 | 7 | 0 | 6 | 119.4 | 0 | 0.04 |
| 4 | F1094-0201 | YES | High | 409.43 | 5.79 | 5 | 0 | 2 | 59.75 | 0 | 0.15 |
| 5 | F0850-4777 | YES | High | 386.39 | 4.51 | 5 | 0 | 5 | 65.74 | 0 | 0.08 |
| 6 | F3385-6048 | NO | High | 487.46 | 4.99 | 9 | 0 | 6 | 96.67 | 0 | 0.18 |
| 7 | F1094-0200 | YES | High | 395.4 | 5.48 | 5 | 0 | 2 | 59.75 | 0 | 0.12 |
| 8 | F1865-0198 | NO | High | 401.37 | 5.34 | 6 | 0 | 5 | 102.37 | 0 | 0.04 |
| 9 | F3139-1101 | YES | High | 368.38 | 5.12 | 4 | 0 | 5 | 56.51 | 0 | 0 |
| 10 | F3139-1218 | NO | High | 426.42 | 4.45 | 6 | 0 | 5 | 78.88 | 0 | 0.07 |
| S. No. | Compound | Mutagenic | Tumorigenic | Reproductive Effect | Irritant |
|---|---|---|---|---|---|
| 1 | F0850-4777 | None | None | None | None |
| 2 | F0870-0001 | None | None | Low | None |
| 3 | F1094-0200 | None | None | High | None |
| 4 | F1094-0201 | None | None | High | None |
| 5 | F1094-0205 | None | None | High | None |
| 6 | F1865-0198 | None | None | High | None |
| 7 | F3139-1101 | None | None | None | High |
| 8 | F3139-1218 | None | None | High | None |
| 9 | F3293-0320 | None | None | None | None |
| 10 | F3385-6048 | None | None | None | None |
| Proteins | ΔEMM | ΔGSolv or ΔGSolGB | ΔGSelf-contact | ΔGH-bond | ΔGSA or ΔGSol_Lipo | ΔGPacking | ΔG or ΔGBind | ||
|---|---|---|---|---|---|---|---|---|---|
| ΔGCoulomb | ΔGvdW | ΔGCovalent | |||||||
| AChE | 1.25 ± 0.87 | −19.24 ± 1.52 | 0.65 ± 0.05 | 6.18 ± 0.54 | 0 | −0.16 ± 0.04 | −15.81 ± 1.22 | −3.22 ± 0.28 | −30.35 ± 3.28 |
| BChE | −0.54 ± 0.04 | −20.17 ± 1.41 | 1.16 ± 0.06 | 9.65 ± 0.69 | 0 | 0 | −13.49 ± 1.07 | 0 | −23.39 ± 3.07 |
| MAO-A | −6.14 ± 0.39 | −17.21 ± 1.19 | 3.79 ± 0.06 | 12.71 ± 1.06 | 0 | −1.20 ± 0.03 | −11.65 ± 0.08 | −0.94 ± 0.03 | −20.64 ± 2.93 |
| MAO-B | −3.97 ± 0.23 | −17.80 ± 1.14 | −0.05 ± 0.01 | 9.39 ± 0.57 | 0 | −0.18 v | −16.28 ± 1.09 | −0.49 ± 0.02 | −29.38 ± 2.99 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iqbal, D.; Rehman, M.T.; Bin Dukhyil, A.; Rizvi, S.M.D.; Al Ajmi, M.F.; Alshehri, B.M.; Banawas, S.; Khan, M.S.; Alturaiki, W.; Alsaweed, M. High-Throughput Screening and Molecular Dynamics Simulation of Natural Product-like Compounds against Alzheimer’s Disease through Multitarget Approach. Pharmaceuticals 2021, 14, 937. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14090937
Iqbal D, Rehman MT, Bin Dukhyil A, Rizvi SMD, Al Ajmi MF, Alshehri BM, Banawas S, Khan MS, Alturaiki W, Alsaweed M. High-Throughput Screening and Molecular Dynamics Simulation of Natural Product-like Compounds against Alzheimer’s Disease through Multitarget Approach. Pharmaceuticals. 2021; 14(9):937. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14090937
Chicago/Turabian StyleIqbal, Danish, Md Tabish Rehman, Abdulaziz Bin Dukhyil, Syed Mohd Danish Rizvi, Mohamed F. Al Ajmi, Bader Mohammed Alshehri, Saeed Banawas, M. Salman Khan, Wael Alturaiki, and Mohammed Alsaweed. 2021. "High-Throughput Screening and Molecular Dynamics Simulation of Natural Product-like Compounds against Alzheimer’s Disease through Multitarget Approach" Pharmaceuticals 14, no. 9: 937. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14090937