
Asymmetric Total Syntheses of Both Enantiomers of Plymuthipyranone B and Its Unnatural Analogues: Evaluation of anti-MRSA Activity and Its Chiral Discrimination


Abstract
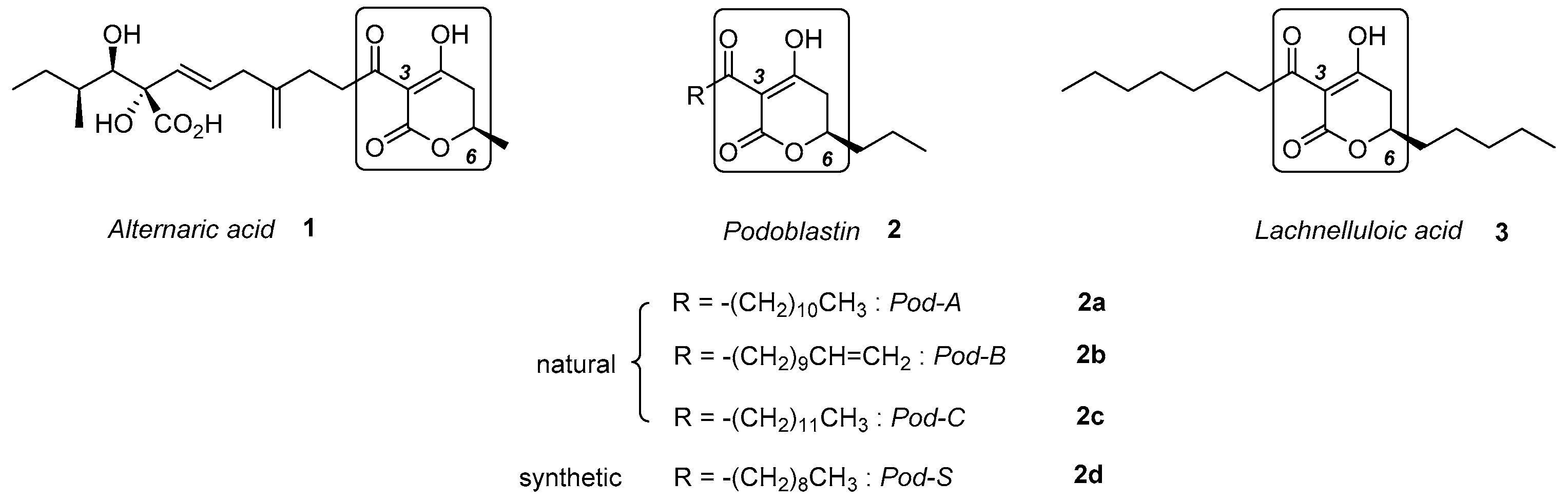
:1. Introduction
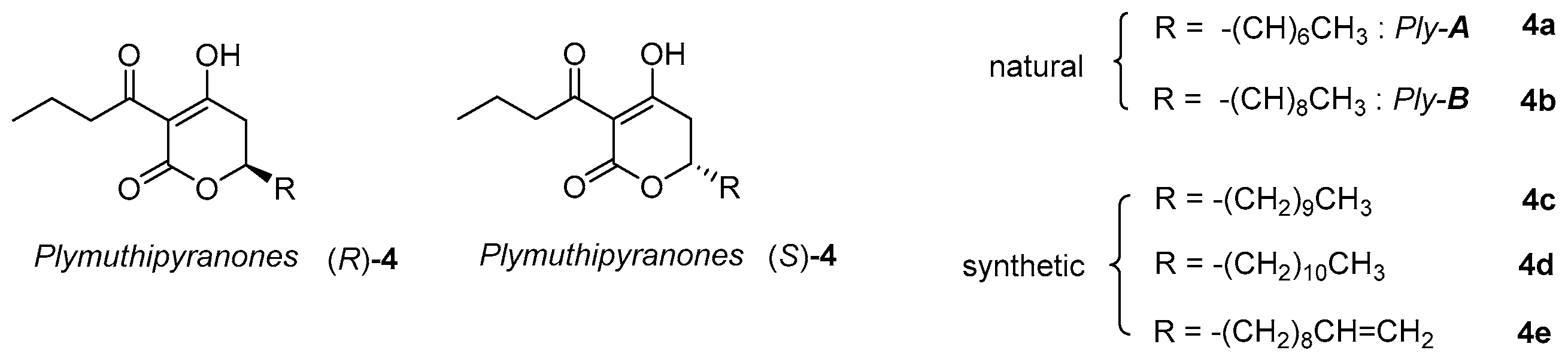
2. Results and Discussion
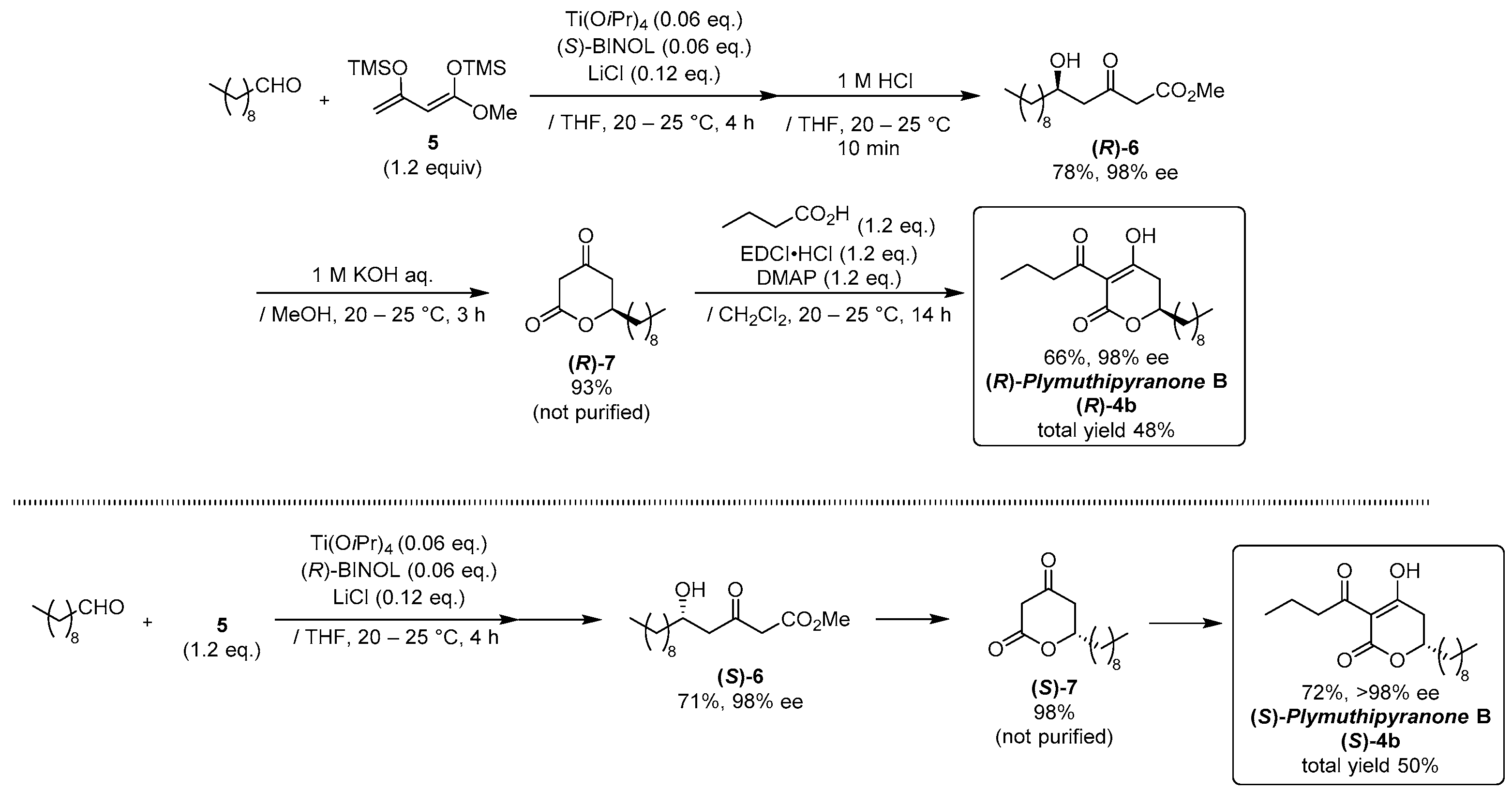
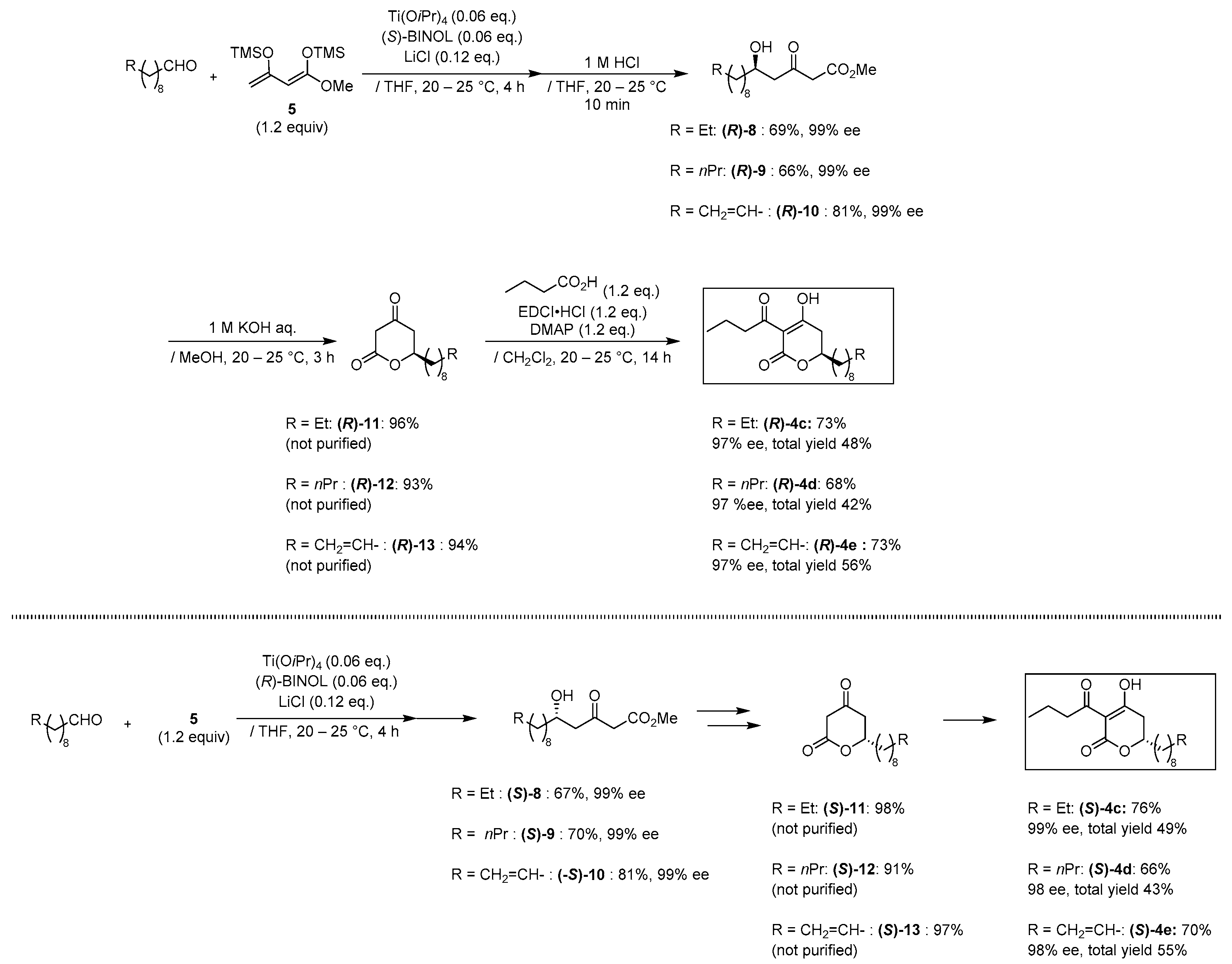
2.1. Synthesis of Three Sets of Natural Plymuthipyranone B and Two Sets of Synthetic Analogues
2.2. Antibacterial Evaluation against MRSA
3. Materials and Methods
3.1. Synthesis
























3.2. Bioassay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Davies-Coleman, M.T.; Rivett, D.E.A. Naturally Occurring 6-Substituted 5,6-Dihydro-α-pyrones. In Progress in the Chemistry of Organic Natural Products; Herz, W., Grisebach, H., Kirby, G.W., Tamm, C., Eds.; Springer: New York, NY, USA, 1989; Volume 55, pp. 1–35. ISBN 978-3-7091-9004-3. [Google Scholar]
- Brian, P.W.; Curtis, P.J.; Hemming, H.G.; Unwin, C.H.; Wright, J.M. Alternaric Acid, a Biologically Active Metabolic Product of the Fungus Alternaria solani. Nature 1949, 164, 534. [Google Scholar] [CrossRef]
- Tabuchi, H.; Ichihara, A. Structures and stereochemistries of new compounds related to alternaric acid. J. Chem. Soc. Perkin Trans. 1994, 1, 125. [Google Scholar] [CrossRef]
- Tabuchi, H.; Hamamoto, T.; Miki, S.; Tejima, T.; Ichihara, A. Total Synthesis and Stereochemistry of Alternaric Acid. J. Org. Chem. 1994, 59, 4749–4759. [Google Scholar] [CrossRef]
- Trost, B.M.; Probst, G.D.; Schoop, A. Ruthenium-Catalyzed Alder Ene Type Reactions. A Formal Synthesis of Alternaric Acid. J. Am. Chem. Soc. 1998, 120, 9228–9236. [Google Scholar] [CrossRef]
- Slade, M.C.; Johnson, J.S. Alternaric acid: Formal synthesis and related studies. Beilstein J. Org. Chem. 2013, 9, 166–172. [Google Scholar] [CrossRef] [Green Version]
- Nagase, R.; Oguni, Y.; Ureshino, S.; Mura, H.; Misaki, T.; Tanabe, Y. Asymmetric Ti-crossed Claisen condensation: Application to concise asymmetric total synthesis of alternaric acid. Chem. Commun. 2013, 49, 7001. [Google Scholar] [CrossRef]
- Sugi, M.; Nagase, R.; Misaki, T.; Nakatsuji, H.; Tanabe, Y. Asymmetric Total Synthesis of (-)-Azaspirene by Utilizing Ti-Claisen Condensation and Ti-Direct Aldol Reaction. Eur. J. Org. Chem. 2016, 2016, 4834–4841. [Google Scholar] [CrossRef]
- Miyakado, M.; Inoue, S.; Tanabe, Y.; Watanabe, K.; Ohno, N.; Yoshioka, H.; Mabry, T.J. Podoblastin A, B and C. New antifungal 3-acyl-4-hydroxy-5,6-dihydro-2-pyrones obtained from Podophyllum Peltatum L. Chem. Lett. 1982, 11, 1539. [Google Scholar] [CrossRef] [Green Version]
- Tanabe, Y.; Miyakado, M.; Ohno, N.; Yoshioka, H. A new 3-acyl-4-hydroxy-2-pyrone synthesis and its application to total synthesis of (±) podoblastin A, B and C. Chem. Lett. 1982, 11, 1543–1546. [Google Scholar] [CrossRef] [Green Version]
- Kawakami, H.; Hirokawa, S.; Asaoka, M.; Takei, H. New Method for the Synthesis of Podoblastin Derivatives and 3-Acyltetronic Acids. Chem. Lett. 1987, 16, 85. [Google Scholar] [CrossRef]
- Ichimoto, I.; Machiya, K.; Kirihata, M.; Ueda, H. Stereoselective Synthesis of Podoblastins and Their Antiblast Activity. J. Pestic. Sci. 1988, 13, 605–613. [Google Scholar] [CrossRef] [Green Version]
- Ayer, W.A.; Villar, J.D.F. Metabolites of Lachnellulafuscosanguinea (Rehm). Part 1. The isolation, structure determination, and synthesis of lachnelluloic acid. Can. J. Chem. 1985, 63, 1161–1165. [Google Scholar] [CrossRef] [Green Version]
- Mineeva, I.V. Asymmetric synthesis of (+)-(S)-Massoia lactone, pheromone of Idea leuconoe. Formal total synthesis of valilactone and lachnelluloic acid. Russ. J. Org. Chem. 2013, 49, 1647–1654. [Google Scholar] [CrossRef]
- Fujiwara, T.; Tsutsumi, T.; Nakata, K.; Nakatsuji, H.; Tanabe, Y. Asymmetric Total Syntheses of Two 3-Acyl-5,6- dihydro-2H-pyrones: (R)-Podoblastin-S and (R)- Lachnelluloic Acid with Verification of the Absolute Configuration of (−)-Lachnelluloic Acid. Molecules 2017, 22, 69. [Google Scholar] [CrossRef]
- Bjerketorp, J.; Levenfors, J.J.; Sahlberg, C.; Nord, C.L.; Andersson, P.F.; Guss, B.; Öberg, B.; Broberg, A. Antibacterial 3,6-Disubstituted 4-Hydroxy-5,6-dihydro-2H-pyran-2-ones from Serratia plymuthica MF371-2. J. Nat. Prod. 2017, 80, 2997–3002. [Google Scholar] [CrossRef]
- Broberg, A.; Andersson, P.; Levenfors, J.; Bjerketorp, J.; Sahlberg, C. Preparation of Substituted Dihydropyranones and Methods of Treating Bacterial Infections. WO Patent 2017095319 A1, 8 June 2017. [Google Scholar]
- Tanabe, Y.; Suzukamo, G.; Komuro, Y.; Imanishi, N.; Morooka, S.; Enomoto, M.; Kojima, A.; Sanemitsu, Y.; Mizutani, M. Structure-activity relationship of optically active 2-(3-pyridyl)thiazolidin-4-ones as a PAF antagonists. Tetrahedron Lett. 1991, 32, 379–382. [Google Scholar] [CrossRef]
- Tanabe, Y.; Yamamoto, H.; Murakami, M.; Yanagi, K.; Kubota, Y.; Okumura, H.; Sanemitsu, Y.; Suzukamo, G. Synthetic study of the highly potent and selective anti-platelet activating factor thiazolidin-4-one agents and related compounds. J. Chem. Soc. Perkin Trans. 1995, 1, 935–947. [Google Scholar] [CrossRef]
- Nishii, Y.; Wakimura, K.-I.; Tsuchiya, T.; Nakamura, S.; Tanabe, Y. Synthesis and Stereostructure-activity Relationship of a Synthetic Pyrethroid, 2-chloro-1-methyl-3-phenylcyclopropylmethyl-3-phenoxybenzyl ether. J. Chem. Soc. Perkin Trans. 1996, 1, 1243–1249. [Google Scholar] [CrossRef]
- Nishii, Y.; Maruyama, N.; Wakasugi, K.; Tanabe, Y. Synthesis and stereostructure–activity relationship of three asymmetric center pyrethroids: 2-methyl-3-phenylcyclopropyl-methyl 3-phenoxybenzyl ether and cyanohydrin ester. Bioorg. Med. Chem. 2001, 9, 33–39. [Google Scholar] [CrossRef]
- Taniguchi, T.; Taketomo, Y.; Moriyama, M.; Matsuo, N.; Tanabe, Y. Synthesis and Stereostructure-Activity Relationship of Novel Pyrethroids Possessing Two Asymmetric Centers on a Cyclopropane Ring. Molecules 2019, 24, 1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawamoto, M.; Moriyama, M.; Ashida, Y.; Matsuo, N.; Tanabe, Y. Total Syntheses of All Six Chiral Natural Pyrethrins: Accurate Determination of the Physical Properties, Their Insecticidal Activities, and Evaluation of Synthetic Methods. J. Org. Chem. 2020, 85, 2984–2999. [Google Scholar] [CrossRef]
- Moriyama, M.; Nakata, K.; Fujiwara, T.; Tanabe, Y. Divergent Asymmetric Total Synthesis of All Four Pestalotin Diastereomers from (R)-Glycidol. Molecules 2020, 25, 394. [Google Scholar] [CrossRef] [Green Version]
- Soriente, A.; De Rosa, M.; Villano, R.; Scettri, A. Enantioselective aldol condensation of 1,3-bis-(trimethylsilyloxy)-1-methoxy-buta-1,3-diene promoted by chiral Ti(IV)/BINOL complex. Tetrahedron Asymmetry 2000, 11, 2255–2258. [Google Scholar] [CrossRef]
- Soriente, A.; De Rosa, M.; Stanzione, M.; Villano, R.; Scettri, A. An efficient asymmetric aldol reaction of Chan’s diene promoted by chiral Ti(IV)–BINOL complex. Tetrahedron Asymmetry 2001, 12, 959–963. [Google Scholar] [CrossRef]
- Villano, R.; Rosaria, M.; De Rosa, M.; Soriente, A.; Stanzione, M.; Scettri, A. Pronounced asymmetric amplification in the aldol condensation of Chan’s diene promoted by a Ti(IV)/BINOL complex. Tetrahedron Asymmetry 2004, 15, 2421–2424. [Google Scholar] [CrossRef]
- Xu, Q.; Yu, J.; Han, F.; Hu, J.; Chen, W.; Yang, L. Achiral additives dramatically enhance enantioselectivities in the BINOL–Ti(IV) complex catalyzed aldol condensations of aldehydes with Chan’s diene. Tetrahedron Asymmetry 2010, 21, 156–158. [Google Scholar] [CrossRef]
- Chan, T.-H.; Brownbridge, P. Reaction of electrophiles with 1,3-bis(trimethylsiloxy)-1-methoxybuta-1,3-diene, a dianion equivalent of methyl acetoacetate. J. Chem. Soc. Chem. Commun. 1979, 578–579. [Google Scholar] [CrossRef]
- Chan, T.-H.; Brownbridge, P. Chemistry of enol silyl ethers. 5. A novel cycloaromatization reaction. Regiocontrolled synthesis of substituted methyl salicylates. J. Am. Chem. Soc. 1980, 102, 3534–3538. [Google Scholar] [CrossRef]
- Langer, P. Cyclization Reactions of 1,3-Bis-Silyl Enol Ethers and Related Masked Dianions: A Review. Synthesis 2002, 441–459. [Google Scholar] [CrossRef]
- Garnero, J.; Caperos, J.; Anwander, A.; Jacot-Guillarmod, A. Flavoring substances: Design of 6-alkyl- (and 6,6-dialkyl-) 5,6-dihydro-2-pyrone. Dev. Food Sci. 1988, 18, 903–913. [Google Scholar]
- Clinical and Laboratory Standards Institute. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standard, 10th ed.; CLSI Document M07-A10; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2015. [Google Scholar]
- Clinical and Laboratory Standards Institute. Performance Standards for Antimicrobial Susceptibility Testing, 27th ed.; CLSI Supplement M100; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2017. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MIC (µg/mL) | MIC (µg/mL) | ||||
|---|---|---|---|---|---|
| ATCC 43300 | ATCC 33591 | ATCC 43300 | ATCC 33591 | ||
 plymuthipyranone B (R)-4b | 4 | 8 |  plymuthipyranone B (S)-4b | 16 | 16 |
 (R)-4c | 4 | 8 |  (S)-4c | 2 | 4 |
 (R)-4d | 2 | 4 |  (S)-4d | 4 | 4 |
 (R)-4e | 16 | 16 |  (S)-4e | 16 | 16 |
| vancomycin | 1 | 1 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moriyama, M.; Liu, X.; Enoki, Y.; Matsumoto, K.; Tanabe, Y. Asymmetric Total Syntheses of Both Enantiomers of Plymuthipyranone B and Its Unnatural Analogues: Evaluation of anti-MRSA Activity and Its Chiral Discrimination. Pharmaceuticals 2021, 14, 938. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14090938
Moriyama M, Liu X, Enoki Y, Matsumoto K, Tanabe Y. Asymmetric Total Syntheses of Both Enantiomers of Plymuthipyranone B and Its Unnatural Analogues: Evaluation of anti-MRSA Activity and Its Chiral Discrimination. Pharmaceuticals. 2021; 14(9):938. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14090938
Chicago/Turabian StyleMoriyama, Mizuki, Xiaoxi Liu, Yuki Enoki, Kazuaki Matsumoto, and Yoo Tanabe. 2021. "Asymmetric Total Syntheses of Both Enantiomers of Plymuthipyranone B and Its Unnatural Analogues: Evaluation of anti-MRSA Activity and Its Chiral Discrimination" Pharmaceuticals 14, no. 9: 938. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14090938