
Treatment of Cardiovascular Disease in Rheumatoid Arthritis: A Complex Challenge with Increased Atherosclerotic Risk



Abstract
:
1. Introduction
2. Methods
3. Background
3.1. Causes of Increased ASCVD Risk in RA
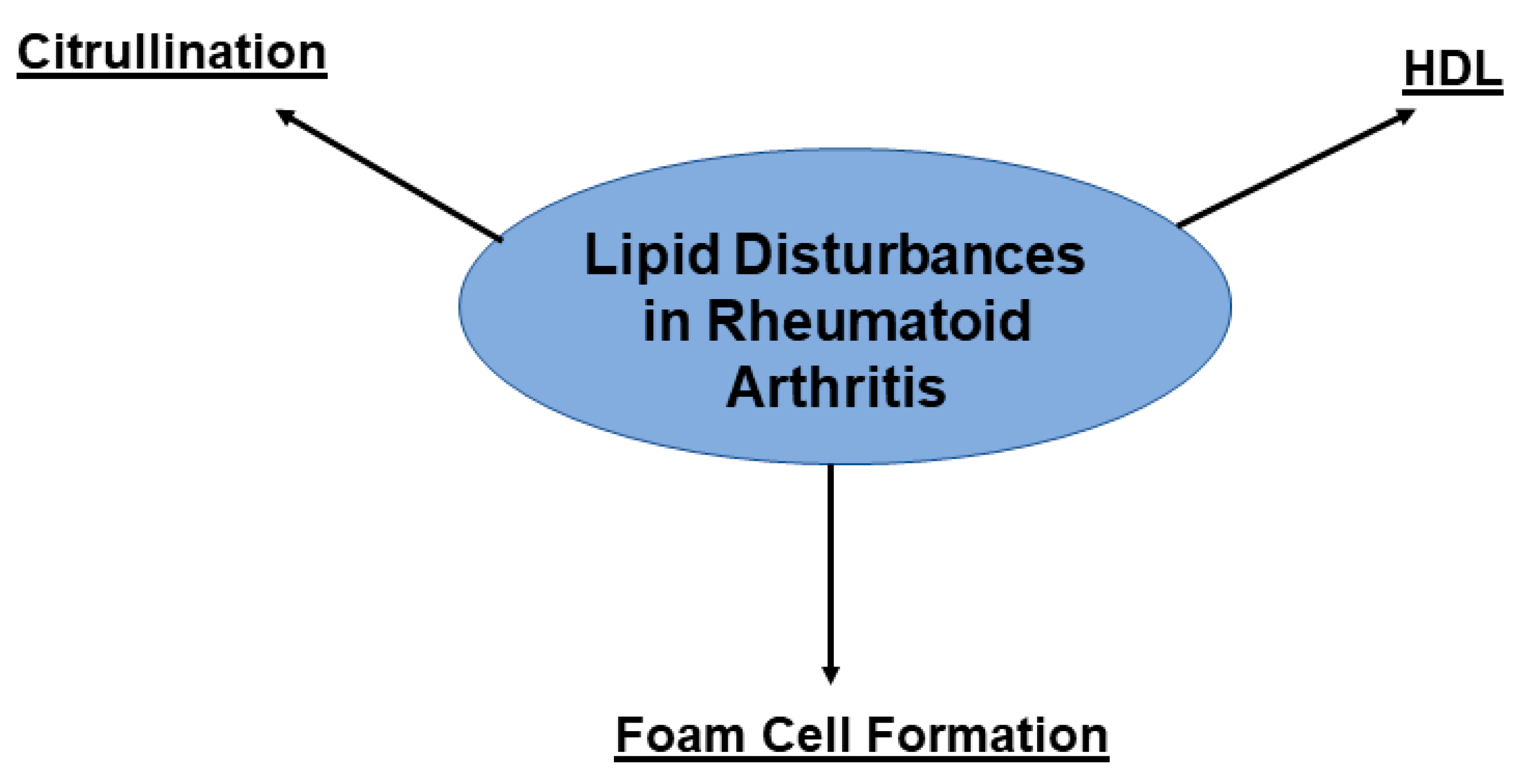
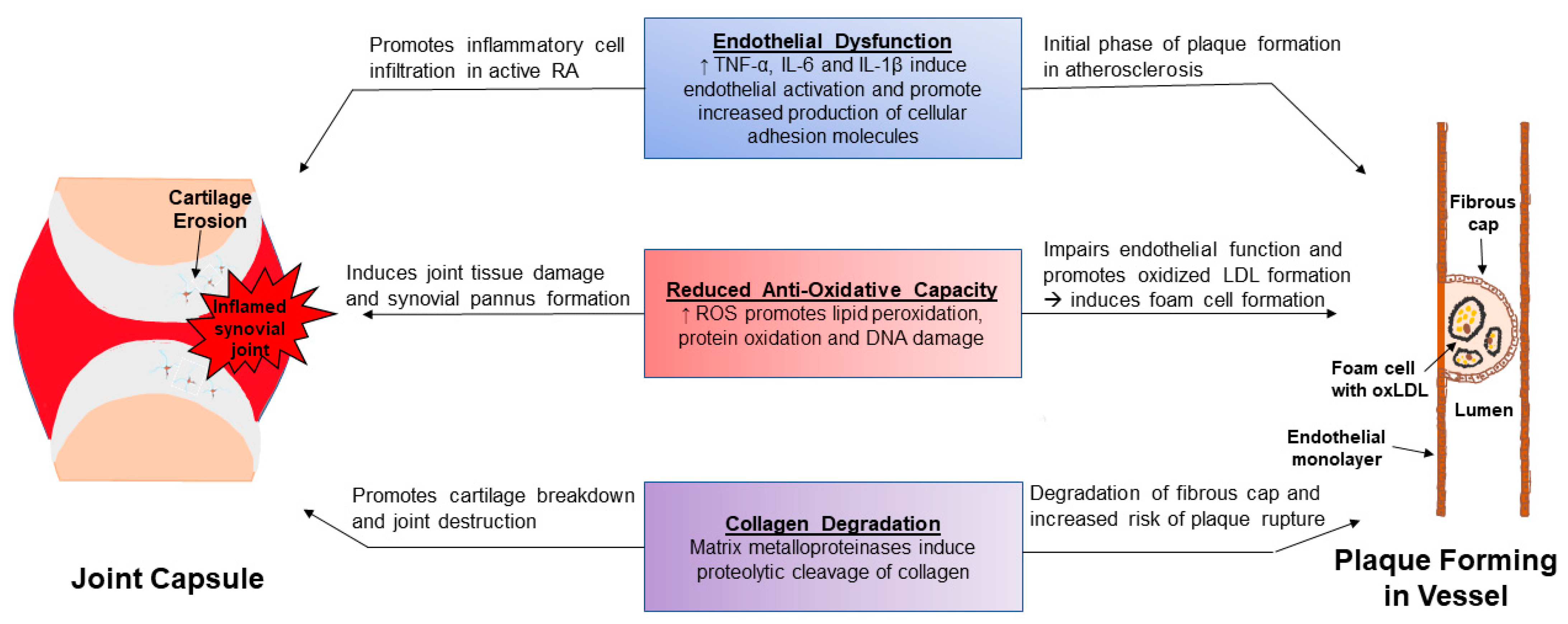
3.2. RA and CVD: Shared Pathways
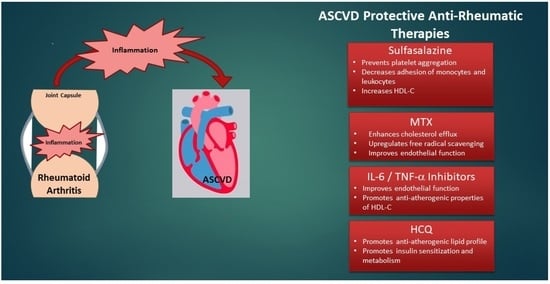
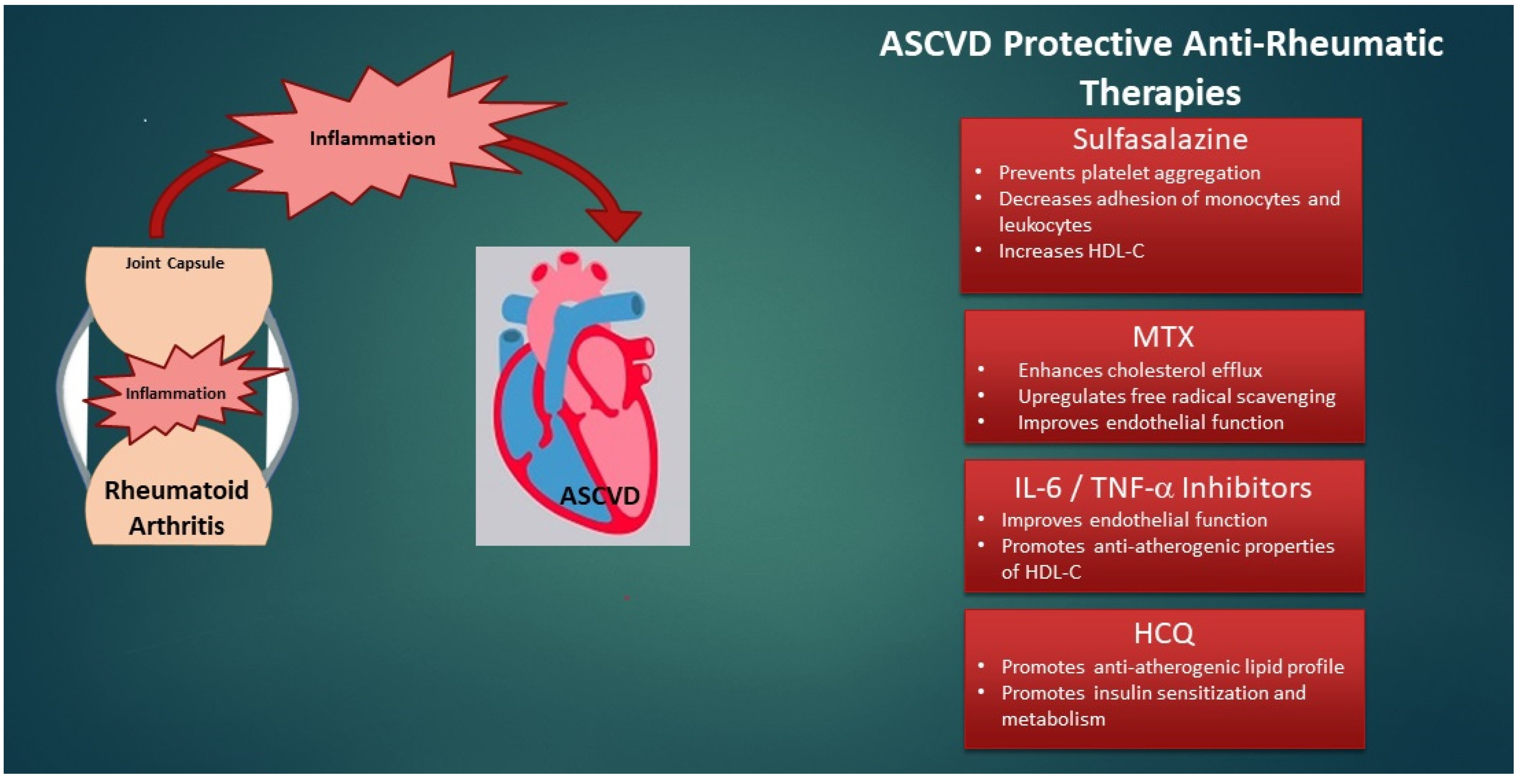
4. Treatment of RA—Does It Address CVD Risk?
5. ASCVD Management in RA
5.1. Cardiovascular-Specific Use of Statins
5.2. Drugs That Treat RA and Their Impact on ASCVD
5.2.1. Methotrexate
5.2.2. Sulfasalazine
5.2.3. TNF-α Inhibitors
5.2.4. IL-6 Inhibitors
5.2.5. JAK Kinase Inhibitors
5.2.6. HCQ: A Potential Double Agent
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gibofsky, A. Epidemiology, pathophysiology, and diagnosis of rheumatoid arthritis: A synopsis. Am. J. Manag. Care 2014, 20 (Suppl. 7), S128–S135. [Google Scholar]
- Safiri, S.; Kolahi, A.A.; Hoy, D.; Smith, E.; Bettampadi, D.; Mansournia, M.A.; Almasi-Hashiani, A.; Ashrafi-Asgarabad, A.; Moradi-Lakeh, M.; Qorbani, M.; et al. Global, regional and national burden of rheumatoid arthritis 1990–2017: A systematic analysis of the global burden of disease study 2017. Ann. Rheum. Dis. 2019, 78, 1463–1471. [Google Scholar] [CrossRef]
- Tedeschi, S.K.; Bermas, B.; Costenbader, K.H. Sexual disparities in the incidence and course of SLE and RA. Clin. Immunol. 2013, 149, 211–218. [Google Scholar] [CrossRef]
- Hunter, T.M.; Boytsov, N.N.; Zhang, X.; Schroeder, K.; Michaud, K.; Araujo, A.B. Prevalence of rheumatoid arthritis in the United States adult population in healthcare claims databases, 2004–2014. Rheumatol. Int. 2017, 37, 1551–1557. [Google Scholar] [CrossRef]
- McInnes, I.B.; Schett, G. Pathogenetic insights from the treatment of rheumatoid arthritis. Lancet 2017, 389, 2328–2337. [Google Scholar] [CrossRef] [Green Version]
- García-Patos, V. Rheumatoid nodule. Semin. Cutan. Med. Surg. 2007, 26, 100–107. [Google Scholar] [CrossRef]
- Yunt, Z.X.; Solomon, J.J. Lung Disease in Rheumatoid Arthritis. Rheum. Dis. Clin. N. Am. 2015, 41, 225–236. [Google Scholar] [CrossRef] [Green Version]
- Prete, M.; Racanelli, V.; Digiglio, L.; Vacca, A.; Dammacco, F.; Perosa, F. Extra-articular manifestations of rheumatoid arthritis: An update. Autoimmun. Rev. 2011, 11, 123–131. [Google Scholar] [CrossRef]
- Toms, T.E.; Symmons, D.P.; Kitas, G.D. Dyslipidaemia in rheumatoid arthritis: The role of inflammation, drugs, lifestyle and genetic factors. Curr. Vasc. Pharmacol. 2010, 8, 301–326. [Google Scholar] [CrossRef]
- Holmqvist, M.E.; Wedrén, S.; Jacobsson, L.T.; Klareskog, L.; Nyberg, F.; Rantapää-Dahlqvist, S.; Alfredsson, L.; Askling, J. Rapid increase in myocardial infarction risk following diagnosis of rheumatoid arthritis amongst patients diagnosed between 1995 and 2006. J. Intern. Med. 2010, 268, 578–585. [Google Scholar] [CrossRef] [Green Version]
- Koren Krajnc, M.; Hojs, R.; Holc, I.; Knez, Ž.; Pahor, A. Accelerated atherosclerosis in premenopausal women with rheumatoid arthritis—15-year follow-up. Bosn. J. Basic Med. Sci. 2021, 21, 477–483. [Google Scholar] [CrossRef]
- Skeoch, S.; Bruce, I.N. Atherosclerosis in rheumatoid arthritis: Is it all about inflammation? Nat. Rev. Rheumatol. 2015, 11, 390–400. [Google Scholar] [CrossRef]
- del Rincón, I.D.; Williams, K.; Stern, M.P.; Freeman, G.L.; Escalante, A. High incidence of cardiovascular events in a rheumatoid arthritis cohort not explained by traditional cardiac risk factors. Arthritis Rheum. 2001, 44, 2737–2745. [Google Scholar] [CrossRef]
- Björsenius, I.; Rantapää-Dahlqvist, S.; Berglin, E.; Södergren, A. Extent of atherosclerosis after 11-year prospective follow-up in patients with early rheumatoid arthritis was affected by disease severity at diagnosis. Scand. J. Rheumatol. 2020, 49, 443–451. [Google Scholar] [CrossRef]
- Aviña-Zubieta, J.A.; Choi, H.K.; Sadatsafavi, M.; Etminan, M.; Esdaile, J.M.; Lacaille, D. Risk of cardiovascular mortality in patients with rheumatoid arthritis: A meta-analysis of observational studies. Arthritis Rheum. 2008, 59, 1690–1697. [Google Scholar] [CrossRef]
- Semb, A.G.; Kvien, T.K.; Aastveit, A.H.; Jungner, I.; Pedersen, T.R.; Walldius, G.; Holme, I. Lipids, myocardial infarction and ischaemic stroke in patients with rheumatoid arthritis in the Apolipoprotein-related Mortality RISk (AMORIS) Study. Ann. Rheum. Dis. 2010, 69, 1996–2001. [Google Scholar] [CrossRef]
- Lindhardsen, J.; Ahlehoff, O.; Gislason, G.H.; Madsen, O.R.; Olesen, J.B.; Torp-Pedersen, C.; Hansen, P.R. The risk of myocardial infarction in rheumatoid arthritis and diabetes mellitus: A Danish nationwide cohort study. Ann. Rheum. Dis. 2011, 70, 929–934. [Google Scholar] [CrossRef] [Green Version]
- Cross, M.; Smith, E.; Hoy, D.; Carmona, L.; Wolfe, F.; Vos, T.; Williams, B.; Gabriel, S.; Lassere, M.; Johns, N.; et al. The global burden of rheumatoid arthritis: Estimates from the global burden of disease 2010 study. Ann. Rheum. Dis. 2014, 73, 1316–1322. [Google Scholar] [CrossRef]
- Ghosh-Swaby, O.R.; Kuriya, B. Awareness and perceived risk of cardiovascular disease among individuals living with rheumatoid arthritis is low: Results of a systematic literature review. Arthritis Res. Ther. 2019, 21, 33. [Google Scholar] [CrossRef] [Green Version]
- England, B.R.; Thiele, G.M.; Anderson, D.R.; Mikuls, T.R. Increased cardiovascular risk in rheumatoid arthritis: Mechanisms and implications. BMJ 2018, 361, k1036. [Google Scholar] [CrossRef]
- Navarro-Millán, I.; Yang, S.; DuVall, S.L.; Chen, L.; Baddley, J.; Cannon, G.W.; Delzell, E.S.; Zhang, J.; Safford, M.M.; Patkar, N.M.; et al. Association of hyperlipidaemia, inflammation and serological status and coronary heart disease among patients with rheumatoid arthritis: Data from the National Veterans Health Administration. Ann. Rheum. Dis. 2016, 75, 341–347. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, L.; Delzell, E.; Muntner, P.; Hillegass, W.B.; Safford, M.M.; Millan, I.Y.; Crowson, C.S.; Curtis, J.R. The association between inflammatory markers, serum lipids and the risk of cardiovascular events in patients with rheumatoid arthritis. Ann. Rheum. Dis. 2014, 73, 1301–1308. [Google Scholar] [CrossRef]
- Liao, K.P.; Liu, J.; Lu, B.; Solomon, D.H.; Kim, S.C. Association between lipid levels and major adverse cardiovascular events in rheumatoid arthritis compared to non-rheumatoid arthritis patients. Arthritis Rheumatol. 2015, 67, 2004–2010. [Google Scholar] [CrossRef] [Green Version]
- Brites, F.; Martin, M.; Guillas, I.; Kontush, A. Antioxidative activity of high-density lipoprotein (HDL): Mechanistic insights into potential clinical benefit. BBA Clin. 2017, 8, 66–77. [Google Scholar] [CrossRef]
- Alisik, T.; Alisik, M.; Nacir, B.; Ayhan, F.F.; Genc, H.; Erel, O. Evaluation of dysfunctional high-density lipoprotein levels with myeloperoxidase/paraoxonase-1 ratio in rheumatoid arthritis. Int. J. Clin. Pract. 2021, 75, e14172. [Google Scholar] [CrossRef]
- Parada-Turska, J.; Wójcicka, G.; Beltowski, J. Paraoxonase 1 Phenotype and Protein N-Homocysteinylation in Patients with Rheumatoid Arthritis: Implications for Cardiovascular Disease. Antioxidants 2020, 9, 899. [Google Scholar] [CrossRef]
- Charles-Schoeman, C.; Watanabe, J.; Lee, Y.Y.; Furst, D.E.; Amjadi, S.; Elashoff, D.; Park, G.; McMahon, M.; Paulus, H.E.; Fogelman, A.M.; et al. Abnormal function of high-density lipoprotein is associated with poor disease control and an altered protein cargo in rheumatoid arthritis. Arthritis Rheum. 2009, 60, 2870–2879. [Google Scholar] [CrossRef] [Green Version]
- Charles-Schoeman, C.; Lee, Y.Y.; Shahbazian, A.; Gorn, A.H.; Fitzgerald, J.; Ranganath, V.K.; Taylor, M.; Ragavendra, N.; McMahon, M.; Elashoff, D.; et al. Association of paraoxonase 1 gene polymorphism and enzyme activity with carotid plaque in rheumatoid arthritis. Arthritis Rheum. 2013, 65, 2765–2772. [Google Scholar] [CrossRef] [Green Version]
- Xie, B.; He, J.; Liu, Y.; Liu, T.; Liu, C. A meta-analysis of HDL cholesterol efflux capacity and concentration in patients with rheumatoid arthritis. Lipids Health Dis. 2021, 20, 18. [Google Scholar] [CrossRef]
- Charles-Schoeman, C.; Lee, Y.Y.; Grijalva, V.; Amjadi, S.; FitzGerald, J.; Ranganath, V.K.; Taylor, M.; McMahon, M.; Paulus, H.E.; Reddy, S.T. Cholesterol efflux by high density lipoproteins is impaired in patients with active rheumatoid arthritis. Ann. Rheum. Dis. 2012, 71, 1157–1162. [Google Scholar] [CrossRef]
- Liao, K.P.; Playford, M.P.; Frits, M.; Coblyn, J.S.; Iannaccone, C.; Weinblatt, M.E.; Shadick, N.S.; Mehta, N.N. The association between reduction in inflammation and changes in lipoprotein levels and HDL cholesterol efflux capacity in rheumatoid arthritis. J. Am. Heart. Assoc. 2015, 4, e001588. [Google Scholar] [CrossRef] [Green Version]
- Ronda, N.; Favari, E.; Borghi, M.O.; Ingegnoli, F.; Gerosa, M.; Chighizola, C.; Zimetti, F.; Adorni, M.P.; Bernini, F.; Meroni, P.L. Impaired serum cholesterol efflux capacity in rheumatoid arthritis and systemic lupus erythematosus. Ann. Rheum. Dis. 2014, 73, 609–615. [Google Scholar] [CrossRef]
- Voloshyna, I.; Modayil, S.; Littlefield, M.J.; Belilos, E.; Belostocki, K.; Bonetti, L.; Rosenblum, G.; Carsons, S.E.; Reiss, A.B. Plasma from rheumatoid arthritis patients promotes pro-atherogenic cholesterol transport gene expression in THP-1 human macrophages. Exp. Biol. Med. 2013, 238, 1192–1197. [Google Scholar] [CrossRef] [Green Version]
- Turgunova, L.G.; Shalygina, A.A.; Zalkalns, J.P.; Klyuyev, D.A.; Akhmaltdinova, L.L.; Dosmagambetova, R.S. Assessment of Adipokines, CXCL16 Chemokine Levels in Patients with Rheumatoid Arthritis Combined with Metabolic Syndrome. Clin. Med. Insights Arthritis Musculoskelet. Disord. 2021, 14, 1179544120985860. [Google Scholar] [CrossRef]
- Dragoljevic, D.; Kraakman, M.J.; Nagareddy, P.R.; Ngo, D.; Shihata, W.; Kammoun, H.L.; Whillas, A.; Lee, M.K.S.; Al-Sharea, A.; Pernes, G.; et al. Defective cholesterol metabolism in haematopoietic stem cells promotes monocyte-driven atherosclerosis in rheumatoid arthritis. Eur. Heart J. 2018, 39, 2158–2167. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Khan, M.S.; Akhter, F.; Husain, F.M.; Ahmad, S.; Chen, L. The non-enzymatic glycation of LDL proteins results in biochemical alterations—A correlation study of Apo B100-AGE with obesity and rheumatoid arthritis. Int. J. Biol. Macromol. 2019, 122, 195–200. [Google Scholar] [CrossRef]
- Kim, J.Y.; Lee, E.Y.; Park, J.K.; Song, Y.W.; Kim, J.R.; Cho, K.H. Patients with Rheumatoid Arthritis Show Altered Lipoprotein Profiles with Dysfunctional High-Density Lipoproteins that Can Exacerbate Inflammatory and Atherogenic Process. PLoS ONE 2016, 11, e0164564. [Google Scholar] [CrossRef]
- de Groot, L.; Hinkema, H.; Westra, J.; Smit, A.J.; Kallenberg, C.G.; Bijl, M.; Posthumus, M.D. Advanced glycation endproducts are increased in rheumatoid arthritis patients with controlled disease. Arthritis Res. Ther. 2011, 13, R205. [Google Scholar] [CrossRef] [Green Version]
- Rajamohan, A.; Heit, B.; Cairns, E.; Barra, L. Citrullinated and homocitrullinated low-density lipoprotein in rheumatoid arthritis. Scand. J. Rheumatol. 2021, 50, 343–350. [Google Scholar] [CrossRef]
- Govindan, K.P.; Basha, S.; Ramesh, V.; Kumar, C.N.; Swathi, S. A comparative study on serum lipoprotein (a) and lipid profile between rheumatoid arthritis patients and normal subjects. J. Pharm. Bioallied Sci. 2015, 7, S22–S25. [Google Scholar] [CrossRef]
- Yang, X.; Chang, Y.; Wei, W. Endothelial dysfunction and inflammation: Immunity in rheumatoid arthritis. Mediators Inflamm. 2016, 2016, 6813016. [Google Scholar] [CrossRef] [Green Version]
- Salem, H.R.; Zahran, E.S. Vascular cell adhesion molecule-1 in rheumatoid arthritis patients: Relation to disease activity, oxidative stress, and systemic inflammation. Saudi Med. J. 2021, 42, 620–628. [Google Scholar] [CrossRef]
- Gimbrone, M.A.; Garcia-Cardena, G., Jr. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, R.; Williams, J.; Sime, K.; Jin, H.S.; Thompson, C.; Jordan, L.; Lang, D.; Halcox, J.P.; Ellins, E.; Jones, G.W.; et al. The role of interleukin-6 trans-signalling on cardiovascular dysfunction in inflammatory arthritis. Rheumatology 2021, 60, 2852–2861. [Google Scholar] [CrossRef]
- Totoson, P.; Maguin-Gaté, K.; Prati, C.; Wendling, D.; Demougeot, C. Mechanisms of endothelial dysfunction in rheumatoid arthritis: Lessons from animal studies. Arthritis Res. Ther. 2014, 16, 202. [Google Scholar] [CrossRef] [Green Version]
- Su, J.B. Vascular endothelial dysfunction and pharmacological treatment. World J. Cardiol. 2015, 7, 719–741. [Google Scholar] [CrossRef]
- Akhmedov, A.; Crucet, M.; Simic, B.; Kraler, S.; Bonetti, N.R.; Ospelt, C.; Distler, O.; Ciurea, A.; Liberale, L.; Jauhiainen, M.; et al. TNFα induces endothelial dysfunction in rheumatoid arthritis via LOX-1 and arginase 2: Reversal by monoclonal TNFα antibodies. Cardiovasc. Res. 2021, cvab005. [Google Scholar] [CrossRef]
- Lassere, M.N.; Rappo, J.; Portek, I.J.; Sturgess, A.; Edmonds, J.P. How many life years are lost in patients with rheumatoid arthritis? Secular cause-specific and all-cause mortality in rheumatoid arthritis, and their predictors in a long-term Australian cohort study. Intern. Med. J. 2013, 43, 66–72. [Google Scholar] [CrossRef]
- van den Hoek, J.; Boshuizen, H.C.; Roorda, L.D.; Tijhuis, G.J.; Nurmohamed, M.T.; van den Bos, G.A.M.; Dekker, J. Mortality in patients with rheumatoid arthritis: A 15-year prospective cohort study. Rheumatol. Int. 2017, 37, 487–493. [Google Scholar] [CrossRef] [Green Version]
- Sokka, T.; Abelson, B.; Pincus, T. Mortality in rheumatoid arthritis: 2008 update. Clin. Exp. Rheumatol. 2008, 26 (Suppl. 51), S35–S61. [Google Scholar] [PubMed]
- Solomon, D.H.; Reed, G.W.; Kremer, J.M.; Curtis, J.R.; Farkouh, M.E.; Harrold, L.R.; Hochberg, M.C.; Tsao, P.; Greenberg, J.D. Disease activity in rheumatoid arthritis and the risk of cardiovascular events. Arthritis Rheumatol. 2015, 67, 1449–1455. [Google Scholar] [CrossRef]
- Chang, K.; Yang, S.M.; Kim, S.H.; Han, K.H.; Park, S.J.; Shin, J.I. Smoking and rheumatoid arthritis. Int. J. Mol. Sci. 2014, 15, 22279–22295. [Google Scholar] [CrossRef] [Green Version]
- Sandoo, A.; Chanchlani, N.; Hodson, J.; Smith, J.P.; Douglas, K.M.; Kitas, G.D. Classical cardiovascular disease risk factors associate with vascular function and morphology in rheumatoid arthritis: A six-year prospective study. Arthritis Res. Ther. 2013, 15, R203. [Google Scholar] [CrossRef] [Green Version]
- Chung, C.P.; Giles, J.T.; Kronmal, R.A.; Post, W.S.; Gelber, A.C.; Petri, M.; Szklo, M.; Detrano, R.; Budoff, M.J.; Blumenthal, R.S.; et al. Progression of coronary artery atherosclerosis in rheumatoid arthritis: Comparison with participants from the Multi-Ethnic Study of Atherosclerosis. Arthritis Res. Ther. 2013, 15, R134. [Google Scholar] [CrossRef] [Green Version]
- Jafri, K.; Bartels, C.M.; Shin, D.; Gelfand, J.M.; Ogdie, A. Incidence and Management of Cardiovascular Risk Factors in Psoriatic Arthritis and Rheumatoid Arthritis: A Population-Based Study. Arthritis Care Res. 2017, 69, 51–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, J.; Alemao, E.; Reynolds, K.; Kawabata, H.; Solomon, D.H.; Liao, K.P.; Niu, F.; Cheetham, T.C. Cardiovascular Outcomes Associated with Lowering Low-density Lipoprotein Cholesterol in Rheumatoid Arthritis and Matched Nonrheumatoid Arthritis. J. Rheumatol. 2016, 43, 1989–1996. [Google Scholar] [CrossRef]
- Willerson, J.T.; Ridker, P.M. Inflammation as a cardiovascular risk factor. Circulation 2004, 109 (Suppl. 1), II2–II10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansson, G.K. Inflammation, atherosclerosis, and coronary artery disease. N. Engl. J. Med. 2005, 352, 1685–1695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geovanini, G.R.; Libby, P. Atherosclerosis and inflammation: Overview and updates. Clin. Sci. 2018, 132, 1243–1252. [Google Scholar] [CrossRef]
- Interleukin-6 Receptor Mendelian Randomisation Analysis (IL6R MR) Consortium; Swerdlow, D.I.; Holmes, M.V.; Kuchenbaecker, K.B.; Engmann, J.E.; Shah, T.; Sofat, R.; Guo, Y.; Chung, C.; Peasey, A.; et al. The interleukin-6 receptor as a target for prevention of coronary heart disease: A mendelian randomisation analysis. Lancet 2012, 379, 1214–1224. [Google Scholar] [CrossRef] [Green Version]
- Ku, I.A.; Imboden, J.B.; Hsue, P.Y.; Ganz, P. Rheumatoid arthritis: Model of systemic inflammation driving atherosclerosis. Circ. J. 2009, 73, 977–985. [Google Scholar] [CrossRef] [Green Version]
- Larsen, B.A.; Laughlin, G.A.; Cummins, K.; Barrett-Connor, E.; Wassel, C.L. Adipokines and severity and progression of coronary artery calcium: Findings from the Rancho Bernardo Study. Atherosclerosis 2017, 265, 1–6. [Google Scholar] [CrossRef]
- López-Mejías, R.; González-Gay, M.A. IL-6: Linking chronic inflammation and vascular calcification. Nat. Rev. Rheumatol. 2019, 15, 457–459. [Google Scholar] [CrossRef]
- Rho, Y.H.; Chung, C.P.; Oeser, A.; Solus, J.; Asanuma, Y.; Sokka, T.; Pincus, T.; Raggi, P.; Gebretsadik, T.; Shintani, A.; et al. Inflammatory mediators and premature coronary atherosclerosis in rheumatoid arthritis. Arthritis Rheum. 2009, 61, 1580–1585. [Google Scholar] [CrossRef] [Green Version]
- Shobeiri, N.; Bendeck, M.P. Interleukin-1β Is a Key Biomarker and Mediator of Inflammatory Vascular Calcification. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 179–180. [Google Scholar] [CrossRef] [Green Version]
- Libby, P.; Ridker, P.M.; Hansson, G.K. Progress and challenges in translating the biology of atherosclerosis. Nature 2011, 473, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.T.; Sun, Y.H.; Zong, S.H.; Xiang, Y.B. Serum levels of IL-6 and TNF-α may correlate with activity and severity of rheumatoid arthritis. Med. Sci. Monit. 2015, 21, 4030–4038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popa, C.; van Tits, L.J.; Barrera, P.; Lemmers, H.L.; van den Hoogen, F.H.; van Riel, P.L.; Radstake, T.R.; Netea, M.G.; Roest, M.; Stalenhoef, A.F. Anti-inflammatory therapy with tumour necrosis factor alpha inhibitors improves high-density lipoprotein cholesterol antioxidative capacity in rheumatoid arthritis patients. Ann. Rheum. Dis. 2009, 68, 868–872. [Google Scholar] [CrossRef]
- Venetsanopoulou, A.I.; Pelechas, E.; Voulgari, P.V.; Drosos, A.A. The lipid paradox in rheumatoid arthritis: The dark horse of the augmented cardiovascular risk. Rheumatol. Int. 2020, 40, 1181–1191. [Google Scholar] [CrossRef] [PubMed]
- Bartoloni, E.; Alunno, A.; Bistoni, O.; Gerli, R. How early is the atherosclerotic risk in rheumatoid arthritis? Autoimmun. Rev. 2010, 9, 701–707. [Google Scholar] [CrossRef]
- Page, M.J.; Bester, J.; Pretorius, E. The inflammatory effects of TNF-α and complement component 3 on coagulation. Sci. Rep. 2018, 8, 1812. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Tang, M.; Huang, D.; Jiang, W.; Li, M.; Ji, H.; Park, J.; Xu, B.; Atchison, L.J.; Truskey, G.A.; et al. Real-time observation of leukocyte-endothelium interactions in tissue-engineered blood vessel. Lab Chip 2018, 18, 2047–2054. [Google Scholar] [CrossRef]
- Barbati, C.; Colasanti, T.; Vomero, M.; Ceccarelli, F.; Celia, A.I.; Perricone, C.; Spinelli, F.R.; Conti, F.; Valesini, G.; Alessandri, C. Up-regulation of autophagy by etanercept treatment results in TNF-induced apoptosis reduction in EA.hy926 endothelial cell line. Clin. Exp. Rheumatol. 2021, 39, 606–611. [Google Scholar] [PubMed]
- Mussbacher, M.; Salzmann, M.; Brostjan, C.; Hoesel, B.; Schoergenhofer, C.; Datler, H.; Hohensinner, P.; Basílio, J.; Petzelbauer, P.; Assinger, A.; et al. Cell Type-Specific Roles of NF-κB Linking Inflammation and Thrombosis. Front. Immunol. 2019, 10, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, Z.; Nallasamy, P.; Liu, D.; Shah, H.; Li, J.Z.; Chitrakar, R.; Si, H.; McCormick, J.; Zhu, H.; Zhen, W.; et al. Luteolin protects against vascular inflammation in mice and TNF-alpha-induced monocyte adhesion to endothelial cells via suppressing IKBα/NF-κB signaling pathway. J. Nutr. Biochem. 2015, 26, 293–302. [Google Scholar] [CrossRef] [Green Version]
- Gunter, S.; Solomon, A.; Tsang, L.; Woodiwiss, A.J.; Robinson, C.; Millen, A.M.; Norton, G.R.; Dessein, P.H. Apelin concentrations are associated with altered atherosclerotic plaque stability mediator levels and atherosclerosis in rheumatoid arthritis. Atherosclerosis 2017, 256, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Karpouzas, G.A.; Malpeso, J.; Choi, T.Y.; Li, D.; Munoz, S.; Budoff, M.J. Prevalence, extent and composition of coronary plaque in patients with rheumatoid arthritis without symptoms or prior diagnosis of coronary artery disease. Ann. Rheum. Dis. 2014, 73, 1797–1804. [Google Scholar] [CrossRef] [PubMed]
- Mai, W.; Liao, Y. Targeting IL-1β in the Treatment of Atherosclerosis. Front. Immunol. 2020, 11, 589654. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.; Hollan, I.; Curran, S.A.; Riggio, M.P.; Mikkelsen, K.; Almdahl, S.M.; Aukrust, P.; McInnes, I.B.; Goodyear, C.S. Brief Report: Proatherogenic Cytokine Microenvironment in the Aortic Adventitia of Patients with Rheumatoid Arthritis. Arthritis Rheumatol. 2016, 68, 1361–1366. [Google Scholar] [CrossRef]
- Winchester, R.; Giles, J.T.; Nativ, S.; Downer, K.; Zhang, H.Z.; Bag-Ozbek, A.; Zartoshti, A.; Bokhari, S.; Bathon, J.M. Association of Elevations of Specific T Cell and Monocyte Subpopulations in Rheumatoid Arthritis with Subclinical Coronary Artery Atherosclerosis. Arthritis Rheumatol. 2016, 68, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Hot, A.; Lenief, V.; Miossec, P. Combination of IL-17 and TNFα induces a pro-inflammatory, pro-coagulant and pro-thrombotic phenotype in human endothelial cells. Ann. Rheum. Dis. 2012, 71, 768–776. [Google Scholar] [CrossRef]
- Ramji, D.P.; Davies, T.S. Cytokines in atherosclerosis: Key players in all stages of disease and promising therapeutic targets. Cytokine Growth Factor Rev. 2015, 26, 673–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, I.; Moon, S.O.; Kim, S.H.; Kim, H.J.; Koh, Y.S.; Koh, G.Y. Vascular endothelial growth factor expression of intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1), and E-selectin through nuclear factor-kappa B activation in endothelial cells. J. Biol. Chem. 2001, 276, 7614–7620. [Google Scholar] [CrossRef] [Green Version]
- Pamukcu, B.; Lip, G.Y.; Shantsila, E. The nuclear factor—kappa B pathway in atherosclerosis: A potential therapeutic target for atherothrombotic vascular disease. Thromb. Res. 2011, 128, 117–123. [Google Scholar] [CrossRef]
- Li, Q.; Verma, I.M. NF-kappaB regulation in the immune system. Nat. Rev. Immunol. 2002, 2, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.Y.; Kim, D.H.; Lee, E.K.; Chung, K.W.; Chung, S.; Lee, B.; Seo, A.Y.; Chung, J.H.; Jung, Y.S.; Im, E.; et al. Redefining Chronic Inflammation in Aging and Age-Related Diseases: Proposal of the Senoinflammation Concept. Aging Dis. 2019, 10, 367–382. [Google Scholar] [CrossRef] [Green Version]
- Shen, C.; Sun, X.G.; Liu, N.; Mu, Y.; Hong, C.C.; Wei, W.; Zheng, F. Increased serum amyloid A and its association with autoantibodies, acute phase reactants and disease activity in patients with rheumatoid arthritis. Mol. Med. Rep. 2015, 11, 1528–1534. [Google Scholar] [CrossRef] [Green Version]
- Sack, G.H., Jr. Serum amyloid A (SAA) proteins. Subcell. Biochem. 2020, 94, 421–436. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Ohkawa, R.; Yoshimoto, A.; Yano, K.; Ichimura, N.; Nishimori, M.; Okubo, S.; Yatomi, Y.; Tozuka, M. Effects of serum amyloid A on the structure and antioxidant ability of high-density lipoprotein. Biosci. Rep. 2016, 36, e00369. [Google Scholar] [CrossRef] [Green Version]
- Green, M.M.; Gough, A.A.; Devlin, J.; Smith, J.; Astin, P.; Taylor, D.; Emery, P. Serum MMP-3 and MMP-1 and progression of joint damage in early rheumatoid arthritis. Rheumatology 2003, 42, 83–88. [Google Scholar] [CrossRef] [Green Version]
- Fiedorczyk, M.; Klimiuk, P.A.; Sierakowski, S.; Gindzienska-Sieskiewicz, E.; Chwiecko, J. Serum matrix metalloproteinases and tissue inhibitors of metalloproteinases in patients with early rheumatoid arthritis. J. Rheumatol. 2006, 33, 1523–1529. [Google Scholar]
- Tchetverikov, I.; Lard, L.R.; DeGroot, J.; Verzijl, N.; TeKoppele, J.M.; Breedveld, F.C.; Huizinga, T.W.; Hanemaaijer, R. Matrix metalloproteinases-3, -8, -9 as markers of disease activity and joint damage progression in early rheumatoid arthritis. Ann. Rheum. Dis. 2003, 62, 1094–1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newby, A.C. Metalloproteinases and vulnerable atherosclerotic plaques. Trends Cardiovasc. Med. 2007, 17, 253–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olejarz, W.; Łacheta, D.; Kubiak-Tomaszewska, G. Matrix Metalloproteinases as Biomarkers of Atherosclerotic Plaque Instability. Int. J. Mol. Sci. 2020, 21, 3946. [Google Scholar] [CrossRef]
- Tousoulis, D.; Oikonomou, E.; Economou, E.K.; Crea, F.; Kaski, J.C. Inflammatory cytokines in atherosclerosis: Current therapeutic approaches. Eur. Heart J. 2016, 37, 1723–1732. [Google Scholar] [CrossRef] [Green Version]
- Bennett, M.R.; Sinha, S.; Owens, G.K. Vascular smooth muscle cells in atherosclerosis. Circ. Res. 2016, 118, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Espié, P.; He, Y.; Koo, P.; Sickert, D.; Dupuy, C.; Chokoté, E.; Schuler, R.; Mergentaler, H.; Ristov, J.; Milojevic, J.; et al. First-in-human clinical trial to assess pharmacokinetics, pharmacodynamics, safety, and tolerability of iscalimab, an anti-CD40 monoclonal antibody. Am. J. Transplant. 2020, 20, 463–473. [Google Scholar] [CrossRef]
- Peters, A.L.; Stunz, L.L.; Bishop, G.A. CD40 and autoimmunity: The dark side of a great activator. Semin. Immunol. 2009, 21, 293–300. [Google Scholar] [CrossRef] [Green Version]
- Antoniades, C.; Bakogiannis, C.; Tousoulis, D.; Antonopoulos, A.S.; Stefanadis, C. The CD40/CD40 ligand system: Linking inflammation with atherothrombosis. J. Am. Coll. Cardiol. 2009, 54, 669–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mälarstig, A.; Lindahl, B.; Wallentin, L.; Siegbahn, A. Soluble CD40L levels are regulated by the -3459 A>G polymorphism and predict myocardial infarction and the efficacy of antithrombotic treatment in non-ST elevation acute coronary syndrome. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1667–1673. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Yan, J.; Yang, P.; Du, R.; Chen, G. The relationship between CD40 gene polymorphism and unstable coronary atherosclerotic plaques. Clin. Cardiol. 2010, 33, E55–E60. [Google Scholar] [CrossRef]
- Michel, N.A.; Zirlik, A.; Wolf, D. CD40L and Its Receptors in Atherothrombosis-An Update. Front. Cardiovasc. Med. 2017, 4, 40. [Google Scholar] [CrossRef] [Green Version]
- Mateen, S.; Moin, S.; Khan, A.Q.; Zafar, A.; Fatima, N. Increased reactive oxygen species formation and oxidative stress in rheumatoid arthritis. PLoS ONE 2016, 11, e0152925. [Google Scholar] [CrossRef] [Green Version]
- Quiñonez-Flores, C.M.; González-Chávez, S.A.; Del Río Nájera, D.; Pacheco-Tena, C. Oxidative Stress Relevance in the Pathogenesis of the Rheumatoid Arthritis: A Systematic Review. Biomed. Res. Int. 2016, 2016, 6097417. [Google Scholar] [CrossRef] [Green Version]
- Harrison, D.; Griendling, K.K.; Landmesser, U.; Hornig, B.; Drexler, H. Role of oxidative stress in atherosclerosis. Am. J. Cardiol. 2003, 91, 7A–11A. [Google Scholar] [CrossRef]
- Sarban, S.; Kocyigit, A.; Yazar, M.; Isikan, U.E. Plasma total antioxidant capacity, lipid peroxidation, and erythrocyte antioxidant enzyme activities in patients with rheumatoid arthritis and osteoarthritis. Clin. Biochem. 2005, 38, 981–986. [Google Scholar] [CrossRef]
- Oğul, Y.; Gür, F.; Cengiz, M.; Gür, B.; Sarı, R.A.; Kızıltunç, A. Evaluation of oxidant and intracellular anti-oxidant activity in rheumatoid arthritis patients: In vivo and in silico studies. Int. Immunopharmacol. 2021, 97, 107654. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tabas, I. Emerging roles of mitochondria ROS in atherosclerotic lesions: Causation or association? J. Atheroscler. Thromb. 2014, 21, 381–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kattoor, A.J.; Pothineni, N.V.K.; Palagiri, D.; Mehta, J.L. Oxidative stress in atherosclerosis. Curr. Atheroscler. Rep. 2017, 19, 42. [Google Scholar] [CrossRef]
- Datta, S.; Kundu, S.; Ghosh, P.; De, S.; Ghosh, A.; Chatterjee, M. Correlation of oxidant status with oxidative tissue damage in patients with rheumatoid arthritis. Clin. Rheumatol. 2014, 33, 1557–1564. [Google Scholar] [CrossRef]
- Marchio, P.; Guerra-Ojeda, S.; Vila, J.M.; Aldasoro, M.; Victor, V.M.; Mauricio, M.D. Targeting early atherosclerosis: A focus on oxidative stress and inflammation. Oxid. Med. Cell Longev. 2019, 2019, 8563845. [Google Scholar] [CrossRef] [PubMed]
- Herlitz-Cifuentes, H.; Vejar, C.; Flores, A.; Jara, P.; Bustos, P.; Castro, I.; Poblete, E.; Saez, K.; Opazo, M.; Gajardo, J.; et al. Plasma from Patients with Rheumatoid Arthritis Reduces Nitric Oxide Synthesis and Induces Reactive Oxygen Species in A Cell-Based Biosensor. Biosensors 2019, 9, 32. [Google Scholar] [CrossRef] [Green Version]
- Solomon, D.H.; Bitton, A.; Katz, J.N.; Radner, H.; Brown, E.M.; Fraenkel, L. Review: Treat to target in rheumatoid arthritis: Fact, fiction, or hypothesis? Arthritis Rheumatol. 2014, 66, 775–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bugatti, S.; De Stefano, L.; Manzo, A.; Sakellariou, G.; Xoxi, B.; Montecucco, C. Limiting factors to Boolean remission differ between autoantibody-positive and -negative patients in early rheumatoid arthritis. Ther. Adv. Musculoskelet. Dis. 2021, 13, 1759720X211011826. [Google Scholar] [CrossRef]
- Smolen, J.S.; Landewé, R.; Bijlsma, J.; Burmester, G.; Chatzidionysiou, K.; Dougados, M.; Nam, J.; Ramiro, S.; Voshaar, M.; van Vollenhoven, R.; et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2016 update. Ann. Rheum. Dis. 2017, 76, 960–977. [Google Scholar] [CrossRef]
- Wasserman, A.M. Diagnosis and management of rheumatoid arthritis. Am. Fam. Physician 2011, 84, 1245–1252. [Google Scholar] [PubMed]
- Burmester, R.; Pope, J.E. Novel treatment strategies in rheumatoid arthritis. Lancet 2017, 389, 2338–2348. [Google Scholar] [CrossRef]
- Abbasi, M.; Mousavi, M.J.; Jamalzehi, S.; Alimohammadi, R.; Bezvan, M.H.; Mohammadi, H.; Aslani, S. Strategies toward rheumatoid arthritis therapy; the old and the new. J. Cell Physiol. 2019, 234, 10018–10031. [Google Scholar] [CrossRef]
- Kavanaugh, A.; Wells, A.F. Benefits and risks of low-dose glucocorticoid treatment in the patient with rheumatoid arthritis. Rheumatology 2014, 53, 1742–1751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giles, J.T.; Rist, P.M.; Liao, K.P.; Tawakol, A.; Fayad, Z.A.; Mani, V.; Paynter, N.P.; Ridker, P.M.; Glynn, R.J.; Lu, F.; et al. Testing the Effects of Disease-Modifying Antirheumatic Drugs on Vascular Inflammation in Rheumatoid Arthritis: Rationale and Design of the TARGET Trial. ACR Open Rheumatol. 2021, 3, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Liao, K.P. Cardiovascular disease in patients with rheumatoid arthritis. Trends Cardiovasc. Med. 2017, 27, 136–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masson, W.; Rossi, E.; Alvarado, R.N.; Cornejo-Peña, G.; Damonte, J.I.; Fiorini, N.; Mora-Crespo, L.M.; Tobar-Jaramillo, M.A.; Scolnik, M. Rheumatoid Arthritis, Statin Indication and Lipid Goals: Analysis According to Different Recommendations. Rheumatol. Clin. 2021. [Google Scholar] [CrossRef]
- Chhibber, A.; Hansen, S.; Biskupiak, J. Statin use and mortality in rheumatoid arthritis: An incident user cohort study. J. Manag. Care Spec. Pharm. 2021, 27, 296–305. [Google Scholar] [CrossRef]
- Abeles, A.M.; Pillinger, M.H. Statins as anti-inflammatory and immunomodulatory agents: A future in rheumatologic therapy? Arthritis Rheum. 2006, 54, 393–407. [Google Scholar] [CrossRef]
- Asher, J.; Houston, M. Statins and C-reactive protein levels. J. Clin. Hypertens. 2007, 9, 622–628. [Google Scholar] [CrossRef] [PubMed]
- Koushki, K.; Shahbaz, S.K.; Mashayekhi, K.; Sadeghi, M.; Zayeri, Z.D.; Taba, M.Y.; Banach, M.; Al-Rasadi, K.; Johnston, T.P.; Sahebkar, A. Anti-inflammatory Action of Statins in Cardiovascular Disease: The Role of Inflammasome and Toll-Like Receptor Pathways. Clinic. Rev. Allerg. Immunol. 2021, 60, 175–199. [Google Scholar] [CrossRef] [PubMed]
- Oesterle, A.; Laufs, U.; Liao, J.K. Pleiotropic effects of statins on the cardiovascular system. Circ. Res. 2017, 120, 229–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oesterle, A.; Liao, J.K. The Pleiotropic effects of statins—From coronary artery disease and stroke to atrial fibrillation and ventricular tachyarrhythmia. Curr. Vasc. Pharmacol. 2019, 17, 222–232. [Google Scholar] [CrossRef] [PubMed]
- van Boheemen, L.; Turk, S.; Beers-Tas, M.V.; Bos, W.; Marsman, D.; Griep, E.N.; Starmans-Kool, M.; Popa, C.D.; van Sijl, A.; Boers, M.; et al. Atorvastatin is unlikely to prevent rheumatoid arthritis in high risk individuals: Results from the prematurely stopped STAtins to Prevent Rheumatoid Arthritis (STAPRA) trial. RMD Open 2021, 7, e001591. [Google Scholar] [CrossRef] [PubMed]
- Sramek, M.; Neradil, J.; Veselska, R. Much more than you expected: The non-DHFR-mediated effects of methotrexate. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 499–503. [Google Scholar] [CrossRef] [PubMed]
- Pang, Z.; Wang, G.; Ran, N.; Lin, H.; Wang, Z.; Guan, X.; Yuan, Y.; Fang, K.; Liu, J.; Wang, F. Inhibitory Effect of Methotrexate on Rheumatoid Arthritis Inflammation and Comprehensive Metabolomics Analysis Using Ultra-Performance Liquid Chromatography-Quadrupole Time of Flight-Mass Spectrometry (UPLC-Q/TOF-MS). Int. J. Mol. Sci. 2018, 19, 2894. [Google Scholar] [CrossRef] [Green Version]
- Singh, J.A.; Saag, K.G.; Bridges, S.L., Jr.; Akl, E.A.; Bannuru, R.R.; Sullivan, M.C.; Vaysbrot, E.; McNaughton, C.; Osani, M.; Shmerling, R.H.; et al. 2015 American College of Rheumatology guideline for the treatment of rheumatoid arthritis. Arthritis Care Res. 2016, 68, 1–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smolen, J.S.; Landewé, R.B.M.; Bijlsma, J.W.J.; Burmester, G.R.; Dougados, M.; Kerschbaumer, A.; McInnes, I.B.; Sepriano, A.; van Vollenhoven, R.F.; de Wit, M.; et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2019 update. Ann. Rheum. Dis. 2020, 79, 685–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, H.K.; Hernán, M.A.; Seeger, J.D.; Robins, J.M.; Wolfe, F. Methotrexate and mortality in patients with rheumatoid arthritis: A prospective study. Lancet 2002, 359, 1173–1177. [Google Scholar] [CrossRef]
- Verhoeven, F.; Prati, C.; Chouk, M.; Demougeot, C.; Wendling, D. Methotrexate and cardiovascular risk in rheumatic diseases: A comprehensive review. Expert Rev. Clin. Pharmacol. 2021, 1–8. [Google Scholar] [CrossRef]
- Sun, K.J.; Liu, L.L.; Hu, J.H.; Chen, Y.Y.; Xu, D.Y. Methotrexate can prevent cardiovascular events in patients with rheumatoid arthritis: An updated meta-analysis. Medicine 2021, 100, e24579. [Google Scholar] [CrossRef]
- Coomes, E.; Chan, E.S.; Reiss, A.B. Methotrexate in atherogenesis and cholesterol metabolism. Cholesterol 2011, 2011, 503028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cronstein, B.N.; Aune, T.M. Methotrexate and its mechanisms of action in inflammatory arthritis. Nat. Rev. Rheumatol. 2020, 16, 145–154. [Google Scholar] [CrossRef]
- Reiss, A.B.; Cronstein, B.N. Regulation of foam cells by adenosine. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 879–886. [Google Scholar] [CrossRef] [Green Version]
- Reiss, A.B.; Carsons, S.E.; Anwar, K.; Rao, S.; Edelman, S.D.; Zhang, H.; Fernandez, P.; Cronstein, B.N.; Chan, E.S. Atheroprotective effects of methotrexate on reverse cholesterol transport proteins and foam cell transformation in human THP-1 monocyte/macrophages. Arthritis Rheum. 2008, 58, 3675–3683. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.Y.; Chih, H.M.; Lan, J.L.; Chang, H.Y.; Chen, W.W.; Chiang, E.P. Blood lipid profiles and peripheral blood mononuclear cell cholesterol metabolism gene expression in patients with and without methotrexate treatment. BMC Med. 2011, 9, 4. [Google Scholar] [CrossRef] [Green Version]
- Reiss, A.B.; Grossfeld, D.; Kasselman, L.J.; Renna, H.A.; Vernice, N.A.; Drewes, W.; Konig, J.; Carsons, S.E.; DeLeon, J. Adenosine and the Cardiovascular System. Am. J. Cardiovasc. Drugs 2019, 19, 449–464. [Google Scholar] [CrossRef] [PubMed]
- Atzeni, F.; Rodríguez-Carrio, J.; Popa, C.D.; Nurmohamed, M.T.; Szűcs, G.; Szekanecz, Z. Cardiovascular effects of approved drugs for rheumatoid arthritis. Nat. Rev. Rheumat. 2021, 17, 270–290. [Google Scholar] [CrossRef] [PubMed]
- Chaabane, S.; Messedi, M.; Akrout, R.; Ben Hamad, M.; Turki, M.; Marzouk, S.; Keskes, L.; Bahloul, Z.; Rebai, A.; Ayedi, F.; et al. Association of hyperhomocysteinemia with genetic variants in key enzymes of homocysteine metabolism and methotrexate toxicity in rheumatoid arthritis patients. Inflamm. Res. 2018, 67, 703–710. [Google Scholar] [CrossRef]
- Johnson, T.M.; Sayles, H.R.; Baker, J.F.; George, M.D.; Roul, P.; Zheng, C.; Sauer, B.; Liao, K.P.; Anderson, D.R.; Mikuls, T.R.; et al. Investigating changes in disease activity as a mediator of cardiovascular risk reduction with methotrexate use in rheumatoid arthritis. Ann. Rheum. Dis. 2021, annrheumdis-2021-220125. [Google Scholar] [CrossRef] [PubMed]
- Stenson, W.F.; Lobos, E.A. Inhibition of platelet thromboxane synthetase by sulphasalazine. Biochem. Pharmacol. 1983, 32, 2205–2209. [Google Scholar] [CrossRef]
- MacMullan, P.A.; Madigan, A.M.; Paul, N.; Peace, A.J.; Alagha, A.; Nolan, K.B.; McCarthy, G.M.; Kenny, D. Sulfasalazine and its metabolites inhibit platelet function in patients with inflammatory arthritis. Clin. Rheumatol. 2016, 35, 447–455. [Google Scholar] [CrossRef]
- Day, A.L.; Singh, J.A. Cardiovascular Disease Risk in Older Adults and Elderly Patients with Rheumatoid Arthritis: What Role Can Disease-Modifying Antirheumatic Drugs Play in Cardiovascular Risk Reduction? Drugs Aging 2019, 36, 493–510. [Google Scholar] [CrossRef]
- Tabit, C.E.; Holbrook, M.; Shenouda, S.M.; Dohadwala, M.M.; Widlansky, M.E.; Frame, A.A.; Kim, B.H.; Duess, M.A.; Kluge, M.A.; Levit, A.; et al. Effect of sulfasalazine on inflammation and endothelial function in patients with established coronary artery disease. Vasc. Med. 2012, 17, 101–107. [Google Scholar] [CrossRef] [Green Version]
- Wahl, C.; Liptay, S.; Adler, G.; Schmid, R.M. Sulfasalazine: A potent and specific inhibitor of nuclear factor kappa B. J. Clin. Investig. 1998, 101, 1163–1174. [Google Scholar] [CrossRef] [Green Version]
- Pateras, I.; Giaginis, C.; Tsigris, C.; Patsouris, E.; Theocharis, S. NF-κB signaling at the crossroads of inflammation and atherogenesis: Searching for new therapeutic links. Expert Opin. Ther. Targets 2014, 18, 1089–1101. [Google Scholar] [CrossRef]
- Park, Y.B.; Choi, H.K.; Kim, M.Y.; Lee, W.K.; Song, J.; Kim, D.K.; Lee, S.K. Effects of antirheumatic therapy on serum lipid levels in patients with rheumatoid arthritis: A prospective study. Am. J. Med. 2002, 113, 188–193. [Google Scholar] [CrossRef]
- Atzeni, F.; Turiel, M.; Caporali, R.; Cavagna, L.; Tomasoni, L.; Sitia, S.; Sarzi-Puttini, P. The effect of pharmacological therapy on the cardiovascular system of patients with systemic rheumatic diseases. Autoimmun. Rev. 2010, 9, 835–839. [Google Scholar] [CrossRef] [PubMed]
- Karimifar, M.; Sepehrifar, M.S.; Moussavi, H.; Sepehrifar, M.B.; Mottaghi, P.; Siavash, M.; Karimifar, M. The effects of conventional drugs in the treatment of rheumatoid arthritis on the serum lipids. J. Res. Med. Sci. 2018, 23, 105. [Google Scholar] [CrossRef] [PubMed]
- Naranjo, A.; Sokka, T.; Descalzo, M.A.; Calvo-Alén, J.; Hørslev-Petersen, K.; Luukkainen, R.K.; Combe, B.; Burmester, G.R.; Devlin, J.; Ferraccioli, G.; et al. Cardiovascular disease in patients with rheumatoid arthritis: Results from the QUEST-RA study. Arthritis Res. Ther. 2008, 10, R30. [Google Scholar] [CrossRef] [Green Version]
- Zanten, J.; Sandoo, A.; Metsios, G.S.; Kalinoglou, A.; Ntoumanis, N.; Kitas, G.D. Comparison of the effects of exercise and anti-TNF treatment on cardiovascular health in rheumatoid arthritis: Results from two controlled trials. Rheumatol. Int. 2019, 39, 219–225. [Google Scholar] [CrossRef] [Green Version]
- Barnabe, C.; Martin, B.J.; Ghali, W.A. Systematic review and meta-analysis: Anti-tumor necrosis factor α therapy and cardiovascular events in rheumatoid arthritis. Arthritis Care Res. 2011, 63, 522–529. [Google Scholar] [CrossRef]
- Ntusi, N.A.B.; Francis, J.M.; Sever, E.; Liu, A.; Piechnik, S.K.; Ferreira, V.M.; Matthews, P.M.; Robson, M.D.; Wordsworth, P.B.; Neubauer, S.; et al. Anti-TNF modulation reduces myocardial inflammation and improves cardiovascular function in systemic rheumatic diseases. Int. J. Cardiol. 2018, 270, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Ait-Oufella, H.; Libby, P.; Tedgui, A. Anticytokine Immune Therapy and Atherothrombotic Cardiovascular Risk. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1510–1519. [Google Scholar] [CrossRef]
- Thalayasingam, N.; Isaacs, J.D. Anti-TNF therapy. Best Pract. Res. Clin. Rheumatol. 2011, 25, 549–567. [Google Scholar] [CrossRef]
- Low, A.S.; Symmons, D.P.; Lunt, M.; Mercer, L.K.; Gale, C.P.; Watson, K.D.; Dixon, W.G.; Hyrich, K.L.; British Society for Rheumatology Biologics Register for Rheumatoid Arthritis, BSRBR-RA; BSRBR Control Centre Consortium. Relationship between exposure to tumour necrosis factor inhibitor therapy and incidence and severity of myocardial infarction in patients with rheumatoid arthritis. Ann. Rheum. Dis. 2017, 76, 654–660. [Google Scholar] [CrossRef] [Green Version]
- Végh, E.; Kerekes, G.; Pusztai, A.; Hamar, A.; Szamosi, S.; Váncsa, A.; Bodoki, L.; Pogácsás, L.; Balázs, F.; Hodosi, K.; et al. Effects of 1-year anti-TNF-α therapy on vascular function in rheumatoid arthritis and ankylosing spondylitis. Rheumatol. Int. 2020, 40, 427–436. [Google Scholar] [CrossRef] [Green Version]
- Pusztai, A.; Hamar, A.; Horváth, Á.; Gulyás, K.; Végh, E.; Bodnár, N.; Kerekes, G.; Czókolyová, M.; Szamosi, S.; Bodoki, L.; et al. Soluble Vascular Biomarkers in Rheumatoid Arthritis and Ankylosing Spondylitis: Effects of 1-year Antitumor Necrosis Factor-α Therapy. J. Rheumatol. 2021, 48, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Greco, T.P.; Conti-Kelly, A.M.; Anthony, J.R.; Greco, T., Jr.; Doyle, R.; Boisen, M.; Kojima, K.; Matsuura, E.; Lopez, L.R. Oxidized-LDL/beta2-glycoprotein I complexes are associated with disease severity and increased risk for adverse outcomes in patients with acute coronary syndromes. Am. J. Clin. Pathol. 2010, 133, 737–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karpouzas, G.A.; Bui, V.L.; Ronda, N.; Hollan, I.; Ormseth, S.R. Biologics and atherosclerotic cardiovascular risk in rheumatoid arthritis: A review of evidence and mechanistic insights. Expert Rev. Clin. Immunol. 2021, 17, 355–374. [Google Scholar] [CrossRef] [PubMed]
- Grimble, R.F. Inflammatory status and insulin resistance. Curr. Opin. Clin. Nutr. Metab. Care 2002, 5, 551–559. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.P.; Oeser, A.; Solus, J.F.; Gebretsadik, T.; Shintani, A.; Avalos, I.; Sokka, T.; Raggi, P.; Pincus, T.; Stein, C.M. Inflammation-associated insulin resistance: Differential effects in rheumatoid arthritis and systemic lupus erythematosus define potential mechanisms. Arthritis Rheum. 2008, 58, 2105–2112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avouac, J.; Elhai, M.; Forien, M.; Sellam, J.; Eymard, F.; Molto, A.; Banal, F.; Damiano, J.; Dieudé, P.; Larger, E.; et al. Influence of inflammatory and non-inflammatory rheumatic disorders on the clinical and biological profile of type-2 diabetes. Rheumatology 2021, 60, 3598–3606. [Google Scholar] [CrossRef] [PubMed]
- Wijbrandts, C.A.; Leuven, S.I.; Boom, H.D.; Gerlag, D.M.; Stroes, E.G.; Kastelein, J.J.; Tak, P.P. Sustained changes in lipid profile and macrophage migration inhibitory factor levels after anti-tumor necrosis factor therapy in rheumatoid arthritis. Ann. Rheum. Dis. 2009, 68, 1316–1321. [Google Scholar] [CrossRef] [PubMed]
- Min, H.K.; Kim, H.R.; Lee, S.H.; Shin, K.; Kim, H.A.; Park, S.H.; Kwok, S.K. Four-year follow-up of atherogenicity in rheumatoid arthritis patients: From the nationwide Korean College of Rheumatology Biologics Registry. Clin. Rheumatol. 2021, 40, 3105–3113. [Google Scholar] [CrossRef] [PubMed]
- Cacciapaglia, F.; Anelli, M.G.; Rinaldi, A.; Serafino, L.; Covelli, M.; Scioscia, C.; Iannone, F.; Lapadula, G. Lipid profile of rheumatoid arthritis patients treated with anti-tumor necrosis factor-alpha drugs changes according to disease activity and predicts clinical response. Drug Dev. Res. 2014, 75 (Suppl. 1), S77–S80. [Google Scholar] [CrossRef]
- Daïen, C.I.; Duny, Y.; Barnetche, T.; Daurès, J.P.; Combe, B.; Morel, J. Effect of TNF inhibitors on lipid profile in rheumatoid arthritis: A systematic review with meta-analysis. Ann. Rheum. Dis. 2012, 71, 862–868. [Google Scholar] [CrossRef] [PubMed]
- Cacciapaglia, F.; Anelli, M.G.; Rinaldi, A.; Fornaro, M.; Lopalco, G.; Scioscia, C.; Lapadula, G.; Iannone, F. Lipids and Atherogenic Indices Fluctuation in Rheumatoid Arthritis Patients on Long-Term Tocilizumab Treatment. Mediat. Inflamm. 2018, 2018, 2453265. [Google Scholar] [CrossRef]
- Jones, G.; Sebba, A.; Gu, J.; Lowenstein, M.B.; Calvo, A.; Gomez-Reino, J.J.; Siri, D.A.; Tomsic, M.; Alecock, E.; Woodworth, T.; et al. Comparison of tocilizumab monotherapy versus methotrexate monotherapy in patients with moderate to severe rheumatoid arthritis: The AMBITION study. Ann. Rheum. Dis. 2010, 69, 88–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawashiri, S.Y.; Kawakami, A.; Yamasaki, S.; Imazato, T.; Iwamoto, N.; Fujikawa, K.; Aramaki, T.; Tamai, M.; Nakamura, H.; Ida, H.; et al. Effects of the anti-interleukin-6 receptor antibody, tocilizumab, on serum lipid levels in patients with rheumatoid arthritis. Rheumatol. Int. 2011, 31, 451–456. [Google Scholar] [CrossRef]
- Smolen, J.S.; Beaulieu, A.; Rubbert-Roth, A.; Ramos-Remus, C.; Rovensky, J.; Alecock, E.; Woodworth, T.; Alten, R.; OPTION Investigators. Effect of interleukin-6 receptor inhibition with tocilizumab in patients with rheumatoid arthritis (OPTION study): A double-blind, placebo-controlled, randomised trial. Lancet 2008, 371, 987–997. [Google Scholar] [CrossRef]
- Zhang, J.; Xie, F.; Yun, H.; Chen, L.; Muntner, P.; Levitan, E.B.; Safford, M.M.; Kent, S.T.; Osterman, M.T.; Lewis, J.D.; et al. Comparative effects of biologics on cardiovascular risk among older patients with rheumatoid arthritis. Ann. Rheum. Dis. 2016, 75, 1813–1818. [Google Scholar] [CrossRef]
- Kim, S.C.; Solomon, D.H.; Rogers, J.R.; Gale, S.; Klearman, M.; Sarsour, K.; Schneeweiss, S. Cardiovascular Safety of Tocilizumab Versus Tumor Necrosis Factor Inhibitors in Patients With Rheumatoid Arthritis: A Multi-Database Cohort Study. Arthritis Rheumatol. 2017, 69, 1154–1164. [Google Scholar] [CrossRef] [Green Version]
- Castagné, B.; Viprey, M.; Martin, J.; Schott, A.M.; Cucherat, M.; Soubrier, M. Cardiovascular safety of tocilizumab: A systematic review and network meta-analysis. PLoS ONE 2019, 14, e0220178. [Google Scholar] [CrossRef]
- Cacciapaglia, F.; Perniola, S.; Venerito, V.; Anelli, M.G.; Härdfeldt, J.; Fornaro, M.; Moschetta, A.; Iannone, F. The Impact of Biologic Drugs on High-Density Lipoprotein Cholesterol Efflux Capacity in Rheumatoid Arthritis Patients. J. Clin. Rheumatol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Ormseth, M.J.; Yancey, P.G.; Solus, J.F.; Bridges, S.L., Jr.; Curtis, J.R.; Linton, M.F.; Fazio, S.; Davies, S.S.; Roberts, L.J., 2nd; Vickers, K.C.; et al. Effect of Drug Therapy on Net Cholesterol Efflux Capacity of High-Density Lipoprotein-Enriched Serum in Rheumatoid Arthritis. Arthritis Rheumatol. 2016, 68, 2099–2105. [Google Scholar] [CrossRef] [PubMed]
- Anastasius, M.; Luquain-Costaz, C.; Kockx, M.; Jessup, W.; Kritharides, L. A critical appraisal of the measurement of serum “cholesterol efflux capacity” and its use as surrogate marker of risk of cardiovascular disease. Biochim. Biophys. Acta. Mol. Cell. Biol Lipids 2018, 1863, 1257–1273. [Google Scholar] [CrossRef]
- Pierini, F.S.; Botta, E.; Soriano, E.R.; Martin, M.; Boero, L.; Meroño, T.; Saez, M.S.; Lozano Chiappe, E.; Cerda, O.; Brites, F. Effect of Tocilizumab on LDL and HDL Characteristics in Patients with Rheumatoid Arthritis. An Observational Study. Rheumatol. Ther. 2021, 8, 803–815. [Google Scholar] [CrossRef]
- Ferraz-Amaro, I.; Hernández-Hernández, M.V.; Tejera-Segura, B.; Delgado-Frías, E.; Macía-Díaz, M.; Machado, J.D.; Diaz-González, F. Effect of IL-6 Receptor Blockade on Proprotein Convertase Subtilisin/Kexin Type-9 and Cholesterol Efflux Capacity in Rheumatoid Arthritis Patients. Horm. Metab. Res. 2019, 51, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Bechman, K.; Yates, M.; Galloway, J.B. The new entries in the therapeutic armamentarium: The small molecule JAK inhibitors. Pharmacol. Res. 2019, 147, 104392. [Google Scholar] [CrossRef]
- Moura, R.A.; Fonseca, J.E. JAK inhibitors and modulation of B cell immune responses in rheumatoid arthritis. Front. Med. 2020, 7, 607725. [Google Scholar] [CrossRef] [PubMed]
- Strand, V.; Goncalves, J.; Isaacs, J.D. Immunogenicity of biologic agents in rheumatology. Nat. Rev. Rheumatol. 2021, 17, 81–97. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Ogata, F.; Tsunoda, R. Risk of venous thromboembolism associated with Janus kinase inhibitors for rheumatoid arthritis: Case presentation and literature review. Clin. Rheumatol. 2021, 40, 4457–4471. [Google Scholar] [CrossRef] [PubMed]
- Charles-Schoeman, C.; Wicker, P.; Gonzalez-Gay, M.A.; Boy, M.; Zuckerman, A.; Soma, K.; Geier, J.; Kwok, K.; Riese, R. Cardiovascular safety findings in patients with rheumatoid arthritis treated with tofacitinib, an oral Janus kinase inhibitor. Semin. Arthritis Rheum. 2016, 46, 261–271. [Google Scholar] [CrossRef]
- Charles-Schoeman, C.; DeMasi, R.; Valdez, H.; Soma, K.; Hwang, L.J.; Boy, M.G.; Biswas, P.; McInnes, I.B. Risk Factors for Major Adverse Cardiovascular Events in Phase III and Long-Term Extension Studies of Tofacitinib in Patients with Rheumatoid Arthritis. Arthritis Rheumatol. 2019, 71, 1450–1459. [Google Scholar] [CrossRef] [Green Version]
- Wollenhaupt, J.; Lee, E.B.; Curtis, J.R.; Silverfield, J.; Terry, K.; Soma, K.; Mojcik, C.; DeMasi, R.; Strengholt, S.; Kwok, K.; et al. Safety and efficacy of tofacitinib for up to 9.5 years in the treatment of rheumatoid arthritis: Final results of a global, open-label, long-term extension study. Arthritis Res. Ther. 2019, 21, 89. [Google Scholar] [CrossRef] [Green Version]
- Conaghan, P.G.; Mysler, E.; Tanaka, Y.; Da Silva-Tillmann, B.; Shaw, T.; Liu, J.; Ferguson, R.; Enejosa, J.V.; Cohen, S.; Nash, P.; et al. Upadacitinib in Rheumatoid Arthritis: A Benefit-Risk Assessment Across a Phase III Program. Drug Saf. 2021, 44, 515–530. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Huang, Y.; Xiao, S.; Sun, X.; Fan, Y.; Zhang, Z. Impact of janus kinase inhibitors on risk of cardiovascular events in patients with rheumatoid arthritis: Systematic review and meta-analysis of randomised controlled trials. Ann. Rheum. Dis. 2019, 78, 1048–1054. [Google Scholar] [CrossRef]
- Xie, W.; Zhang, Z. Tofacitinib in cardiovascular outcomes: Friend or foe? Rheumatology 2020, 59, 1797–1798. [Google Scholar] [CrossRef] [Green Version]
- Rainsford, K.D.; Parke, A.L.; Clifford-Rashotte, M.; Kean, W.F. Therapy and pharmacological properties of hydroxychloroquine and chloroquine in treatment of systemic lupus erythematosus, rheumatoid arthritis and related diseases. Inflammopharmacology 2015, 23, 231–269. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.L.; Chang, C.C.; Kor, C.T.; Yang, T.H.; Chiu, P.F.; Tarng, D.C.; Hsu, C.C. Hydroxychloroquine Use and Risk of CKD in Patients with Rheumatoid Arthritis. Clin. J. Am. Soc. Nephrol. 2018, 13, 702–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Zvi, I.; Kivity, S.; Langevitz, P.; Shoenfeld, Y. Hydroxychloroquine: From malaria to autoimmunity. Clin. Rev. Allergy Immunol. 2012, 42, 145–153. [Google Scholar] [CrossRef]
- Schrezenmeier, E.; Dörner, T. Mechanisms of action of hydroxychloroquine and chloroquine: Implications for rheumatology. Nat. Rev. Rheumatol. 2020, 16, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Shi, N.; Zhang, S.; Silverman, G.; Li, M.; Cai, J.; Niu, H. Protective effect of hydroxychloroquine on rheumatoid arthritis-associated atherosclerosis. Anim. Model Exp. Med. 2019, 2, 98–106. [Google Scholar] [CrossRef]
- Hu, C.; Lu, L.; Wan, J.P.; Wen, C. The Pharmacological Mechanisms and Therapeutic Activities of Hydroxychloroquine in Rheumatic and Related Diseases. Curr. Med. Chem. 2017, 24, 2241–2249. [Google Scholar] [CrossRef]
- Fan, H.W.; Ma, Z.X.; Chen, J.; Yang, X.Y.; Cheng, J.L.; Li, Y.B. Pharmacokinetics and Bioequivalence Study of Hydroxychloroquine Sulfate Tablets in Chinese Healthy Volunteers by LC-MS/MS. Rheumatol. Ther. 2015, 2, 183–195. [Google Scholar] [CrossRef] [Green Version]
- Wallace, D.J.; Gudsoorkar, V.S.; Weisman, M.H.; Venuturupalli, S.R. New insights into mechanisms of therapeutic effects of antimalarial agents in SLE. Nat. Rev. Rheumatol. 2012, 8, 522–533. [Google Scholar] [CrossRef]
- Eugenia Schroeder, M.; Russo, S.; Costa, C.; Hori, J.; Tiscornia, I.; Bollati-Fogolín, M.; Zamboni, D.S.; Ferreira, G.; Cairoli, E.; Hill, M. Pro-inflammatory Ca++-activated K+ channels are inhibited by hydroxychloroquine. Sci. Rep. 2017, 15, 1892. [Google Scholar] [CrossRef]
- Silva, J.C.; Mariz, H.A.; Rocha, L.F., Jr.; Oliveira, P.S.; Dantas, A.T.; Duarte, A.L.; Pitta, I.; Galdino, S.L.; Pitta, M.G. Hydroxychloroquine decreases Th17-related cytokines in systemic lupus erythematosus and rheumatoid arthritis patients. Clinics 2013, 68, 766–771. [Google Scholar] [CrossRef]
- Mercer, E.; Rekedal, L.; Garg, R.; Lu, B.; Massarotti, E.M.; Solomon, D.H. Hydroxychloroquine improves insulin sensitivity in obese non-diabetic individuals. Arthritis Res. Ther. 2012, 14, R135. [Google Scholar] [CrossRef] [Green Version]
- Sharma, T.S.; Wasko, M.C.; Tang, X.; Vedamurthy, D.; Yan, X.; Cote, J.; Bili, A. Hydroxychloroquine Use Is Associated with Decreased Incident Cardiovascular Events in Rheumatoid Arthritis Patients. J. Am. Heart Assoc. 2016, 5, e002867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wasko, M.C.; Hubert, H.B.; Lingala, V.B.; Elliott, J.R.; Luggen, M.E.; Fries, J.F.; Ward, M.M. Hydroxychloroquine and risk of diabetes in patients with rheumatoid arthritis. JAMA 2007, 298, 187–193. [Google Scholar] [CrossRef] [Green Version]
- Guevara, M.; Ng, B. Positive effect of hydroxychloroquine on lipid profiles of patients with rheumatoid arthritis: A Veterans Affair cohort. Eur. J. Rheumatol. 2020, 8, 62–66. [Google Scholar] [CrossRef] [PubMed]
- Mangoni, A.A.; Woodman, R.J.; Piga, M.; Cauli, A.; Fedele, A.L.; Gremese, E.; Erre, G.L.; EDRA Study Group. Patterns of Anti-Inflammatory and Immunomodulating Drug Usage and Microvascular Endothelial Function in Rheumatoid Arthritis. Front. Cardiovasc. Med. 2021, 8, 681327. [Google Scholar] [CrossRef]
- Kedia, A.K.; Mohansundaram, K.; Goyal, M.; Ravindran, V. Safety of long-term use of four common conventional disease modifying anti-rheumatic drugs in rheumatoid arthritis. J. R. Coll. Physicians Edinb. 2021, 51, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Doyno, C.; Sobieraj, D.M.; Baker, W.L. Toxicity of chloroquine and hydroxychloroquine following therapeutic use or overdose. Clin. Toxicol. 2021, 59, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Rempenault, C.; Combe, B.; Barnetche, T.; Gaujoux-Viala, C.; Lukas, C.; Morel, J.; Hua, C. Metabolic and cardiovascular benefits of hydroxychloroquine in patients with rheumatoid arthritis: A systematic review and meta-analysis. Ann. Rheum. Dis. 2018, 77, 98–103. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| DMARDs | ||
|---|---|---|
| Name of Drug | Mechanism of Action | |
| Anti-rheumatic Properties | Atheroprotective Properties | |
| Methotrexate | Inhibits dihydrofolate reductase and several immune pathways involved in purine and pyrimidine synthesis. | Enhances macrophage cholesterol efflux and prevents foams cell differentiation and activation. Upregulates free radical scavenging; improves endothelial function. |
| Sulfasalazine | Reduces production of inflammatory cytokines, likely through inhibition of NF-κB activation. | Prevents arachidonic acid-mediated platelet aggregation, decreases adhesion of monocytes and leukocytes, and increases HDL-C. |
| Hydroxychloroquine | Interferes with toll-like receptor signaling, reduces calcium signaling in B and T cells and matrix metalloprotease activity | Positively impacts insulin sensitization, promotes anti-atherogenic lipid profile. Anti-thrombotic and anticoagulant properties. |
| Tumor Necrosis Factor (TNF)-α Inhibitors | ||
| Etanercept Infliximab Adalimumab | Biologics that inactivate TNF-α. Etanercept is a fusion protein of human immunoglobulin 1 Fc domain and TNF-α receptor. Infliximab is a mouse-human chimeric anti-human TNF-α antibody Adalimumab is a human anti-human TNF-α antibody | TNF-α promotes numerous inflammatory responses associated with atherosclerosis, including induction of vascular adhesion and monocyte/macrophage proliferation. TNF-α impacts lipid metabolism by stimulating liver triglyceride production. |
| IL-6 Inhibitors | ||
| Tocilizumab | Inhibits IL-6 which contributes to inflammation and antibody production through its action on T cells, B cells, monocytes and neutrophils | Decreases inflammatory proteins such as serum amyloid A, and restores the anti-atherogenic function of HDL by increasing HDL cholesterol efflux capacity. |
| JAK Kinase Inhibitors | ||
| Tofacitinib | Small molecules that target the JAK-STAT signaling pathway. Reduce expression of cytokine related genes. | Risk of adverse cardiovascular events still being evaluated. Many studies show no difference compared to placebo or biologic |
| Upadacitinib | More JAK1 selective | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmed, S.; Jacob, B.; Carsons, S.E.; De Leon, J.; Reiss, A.B. Treatment of Cardiovascular Disease in Rheumatoid Arthritis: A Complex Challenge with Increased Atherosclerotic Risk. Pharmaceuticals 2022, 15, 11. https://0-doi-org.brum.beds.ac.uk/10.3390/ph15010011
Ahmed S, Jacob B, Carsons SE, De Leon J, Reiss AB. Treatment of Cardiovascular Disease in Rheumatoid Arthritis: A Complex Challenge with Increased Atherosclerotic Risk. Pharmaceuticals. 2022; 15(1):11. https://0-doi-org.brum.beds.ac.uk/10.3390/ph15010011
Chicago/Turabian StyleAhmed, Saba, Benna Jacob, Steven E. Carsons, Joshua De Leon, and Allison B. Reiss. 2022. "Treatment of Cardiovascular Disease in Rheumatoid Arthritis: A Complex Challenge with Increased Atherosclerotic Risk" Pharmaceuticals 15, no. 1: 11. https://0-doi-org.brum.beds.ac.uk/10.3390/ph15010011