
Epigenetic Modulation of Gremlin-1/NOTCH Pathway in Experimental Crescentic Immune-Mediated Glomerulonephritis

 , , , , , , and
, , , , , , and 
Abstract
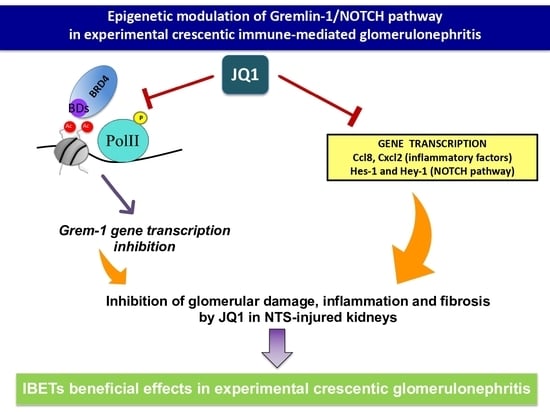
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
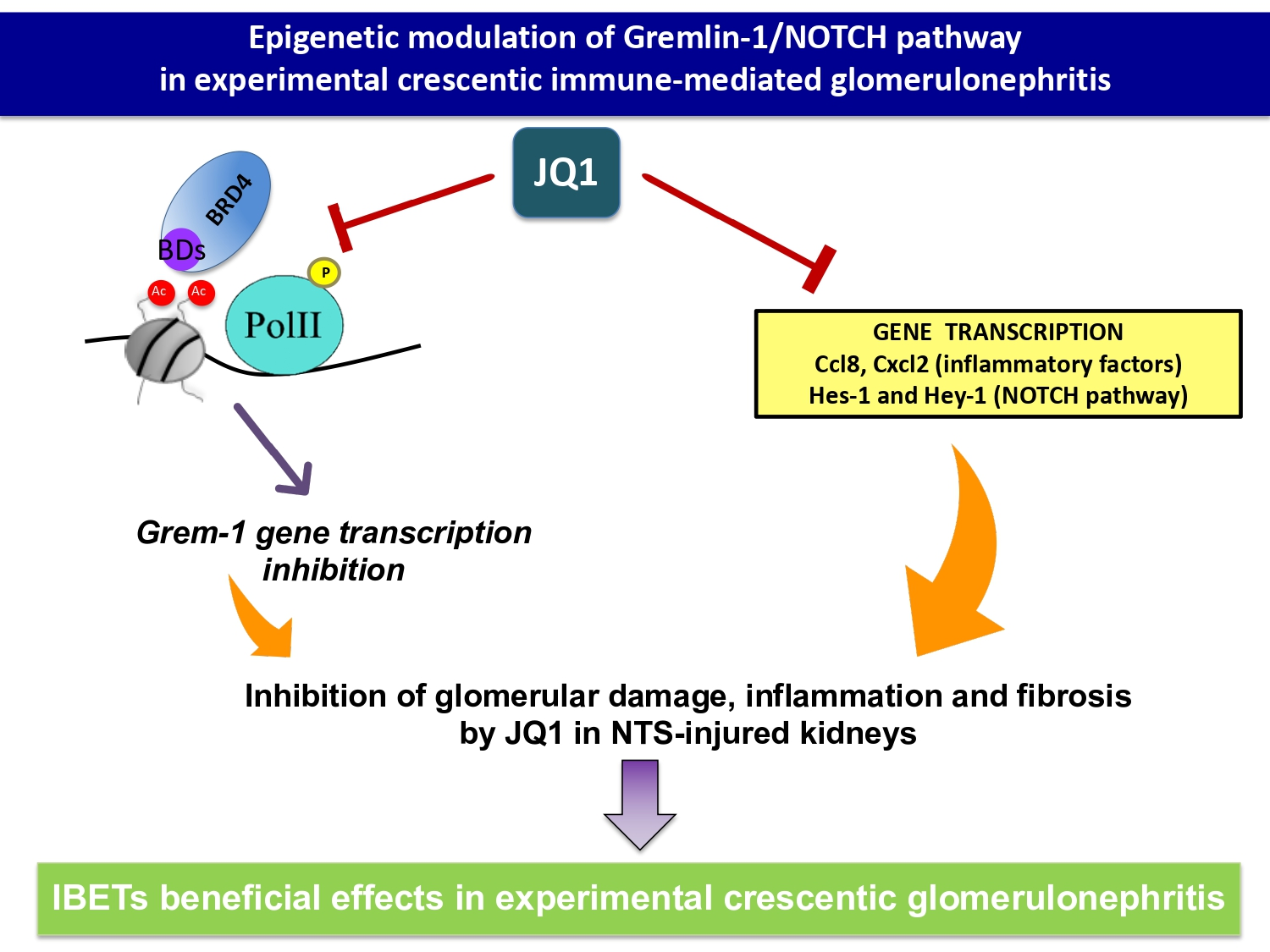
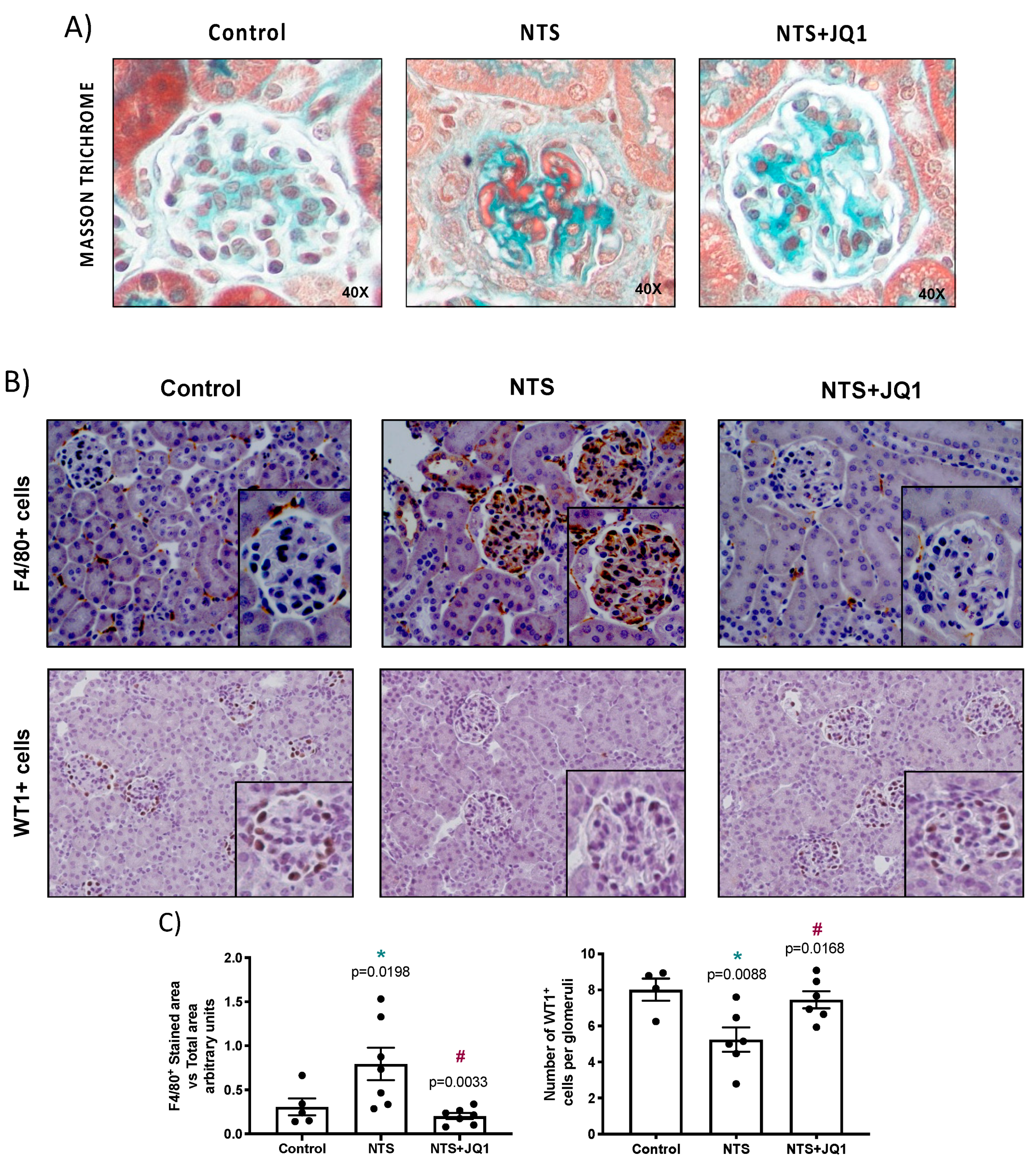
2.1. BET Inhibition Ameliorates Renal Damage in Experimental Nephrotoxic Nephritis
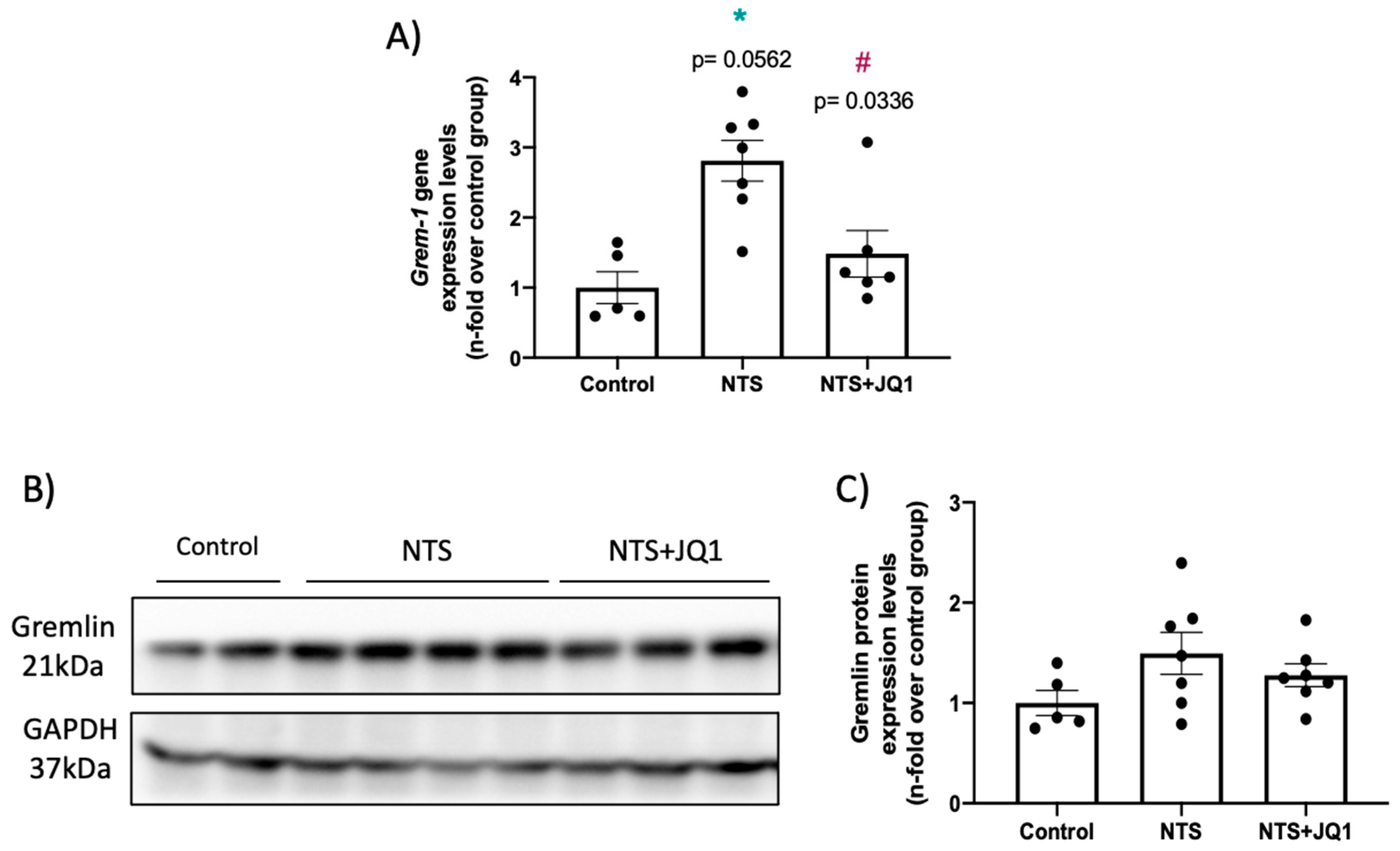
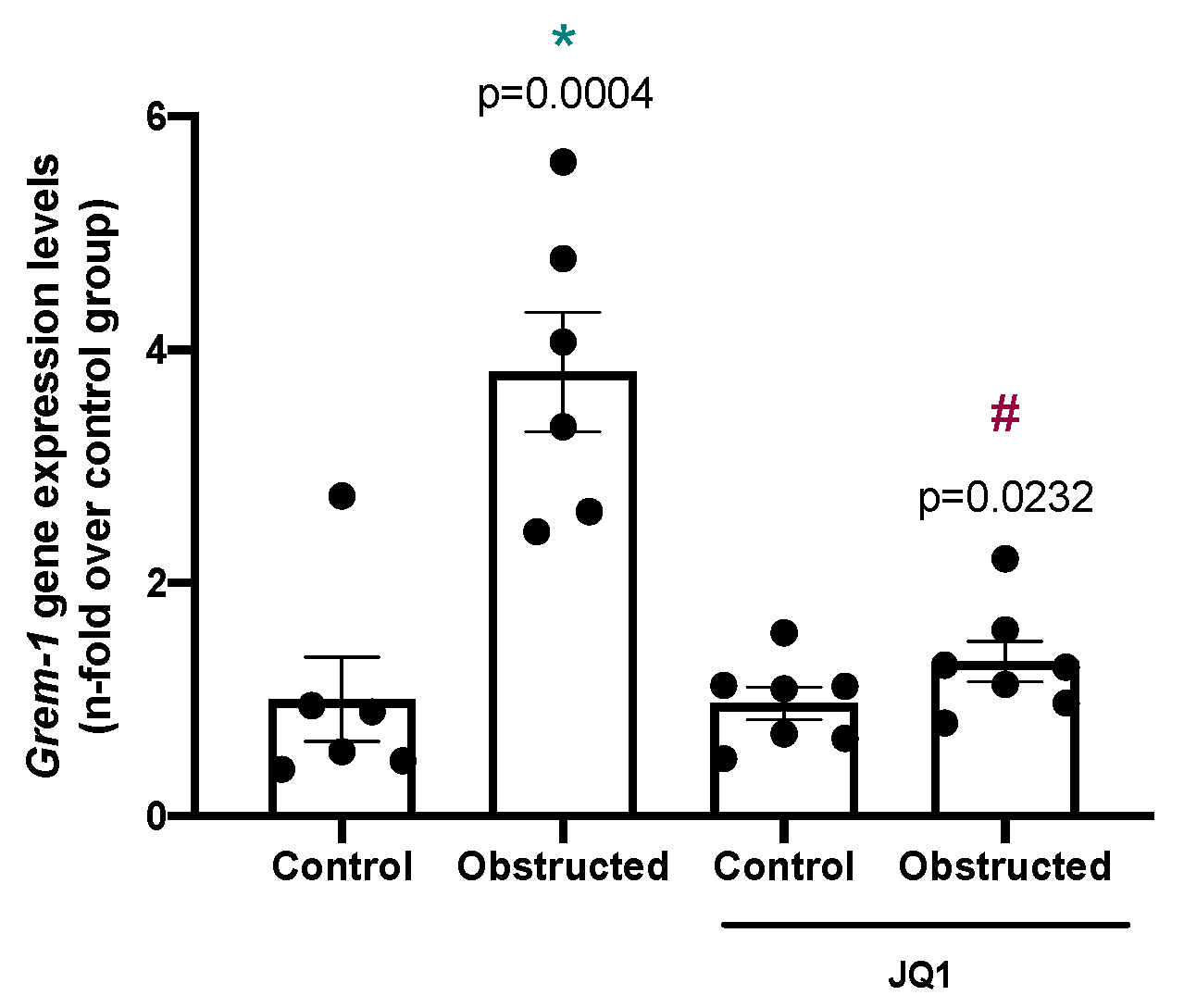
2.2. Gremlin-1 Is Overexpressed in Experimental Nephrotoxic Nephritis in Mice: Effect of BET Inhibition
2.3. Gremlin-1 Is One of the Specific Targets of BET Inhibition
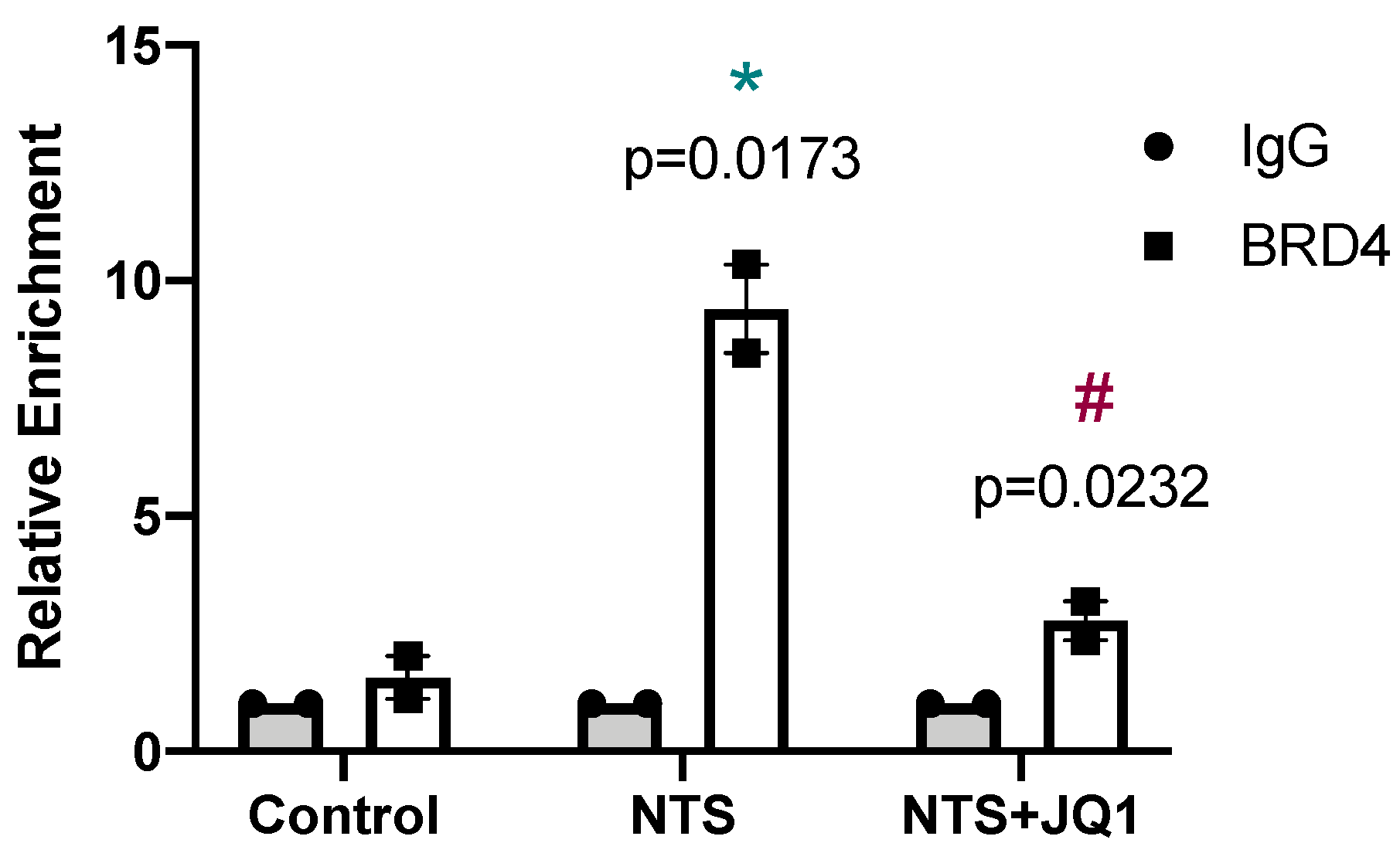
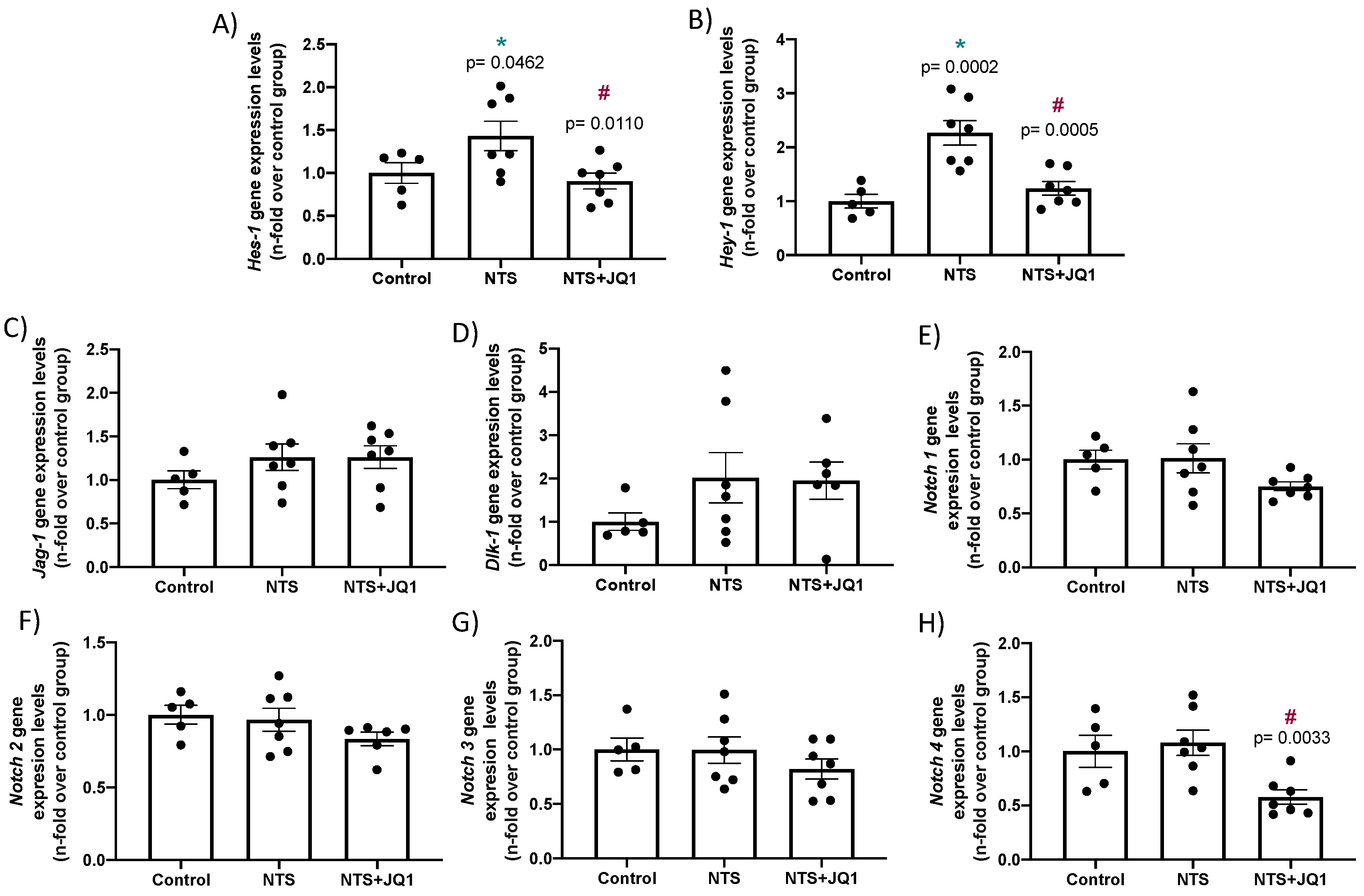
2.4. JQ1 Inhibits the NOTCH Pathway in Experimental Nephrotoxic Nephritis
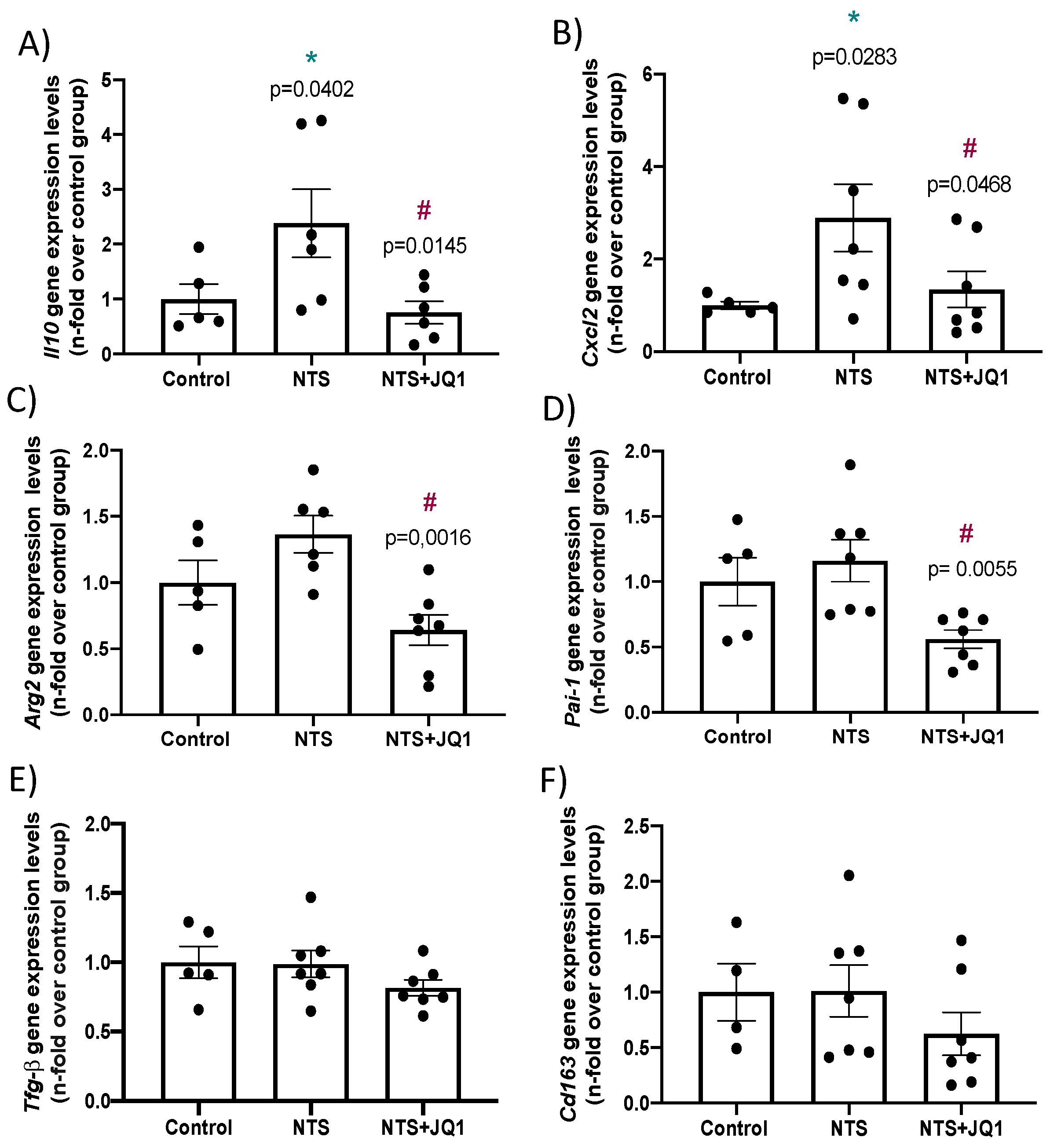
2.5. Differential Gene Expression in Experimental Nephrotoxic Nephritis: Impact of BET Inhibition
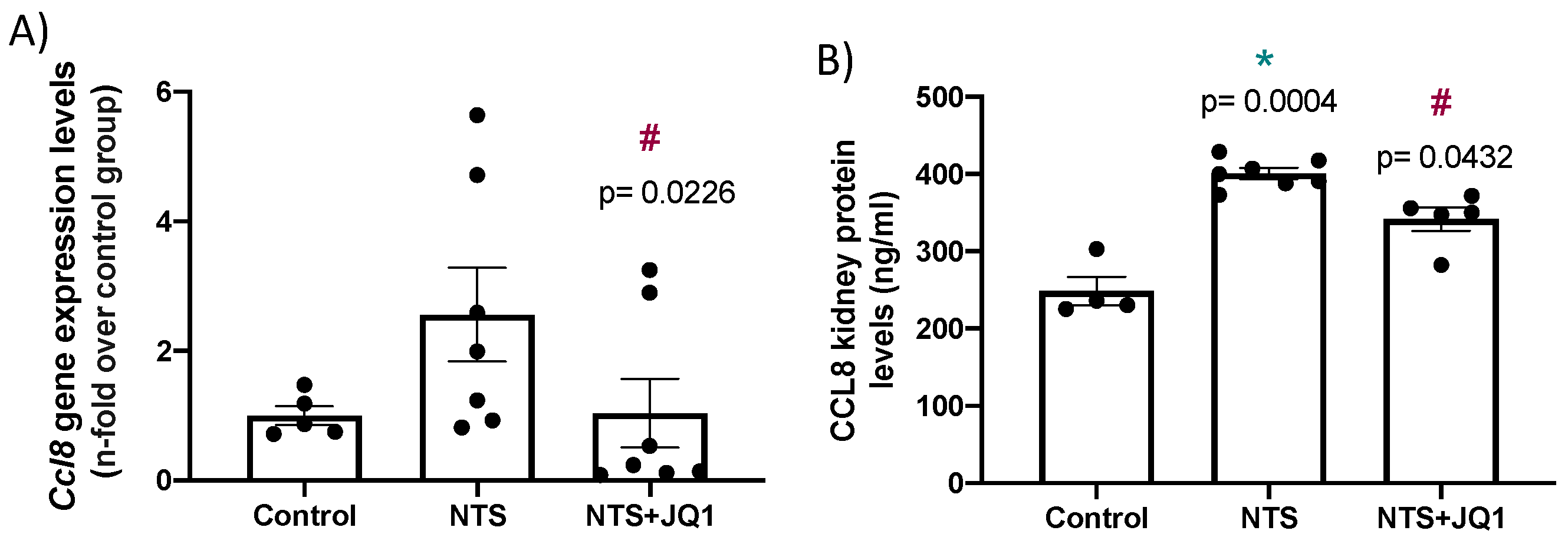
2.6. BET Inhibition Diminished the Renal Expression of the Chemokine Ccl-8 in Experimental Anti-glomerular Basement Membrane Nephritis
2.7. BET Inhibition Downregulates Gremlin-1 Expression in Other Models of Renal Fibrosis
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Experimental Models
4.3. Histology and Immunohistochemistry
4.4. Gene Expression Studies
4.5. Protein Studies
4.6. Chromatin Immunoprecipitation
- Forward 5′-GACCAATGGAGAGACGCAGT-3′
- Reverse 5′-GTTCTTCGCTGTGGACGAGT-3′
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stevens, P.E.; Levin, A. Kidney Disease: Improving Global Outcomes Chronic Kidney Disease Guideline Development Work Group Members. Evaluation and Management of Chronic Kidney Disease: Synopsis of the Kidney Disease: Improving Global Outcomes 2012 Clinical Practice Guideline. Ann. Intern. Med. 2013, 158, 825–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foreman, K.J.; Marquez, N.; Dolgert, A.; Fukutaki, K.; Fullman, N.; McGaughey, M.; Pletcher, M.A.; Smith, A.E.; Tang, K.; Yuan, C.-W.; et al. Forecasting life expectancy, years of life lost, and all-Cause and cause-Specific mortality for 250 causes of death: Reference and alternative scenarios for 2016-40 for 195 countries and territories. Lancet 2018, 392, 2052–2090. [Google Scholar] [CrossRef] [Green Version]
- Ortiz, A.; Asociación Información Enfermedades Renales Genéticas (AIRG-E); European Kidney Patients’ Federation (EKPF); Federación Nacional de Asociaciones para la Lucha Contra las Enfermedades del Riñón (ALCER); Fundación Renal Íñigo Álvarez de Toledo (FRIAT); Red de Investigación Renal (REDINREN); Resultados en Salud 2040 (RICORS2040); Sociedad Española de Nefrología (SENEFRO) Council; Sociedad Española de Trasplante (SET) Council; Organización Nacional de Trasplantes (ONT). RICORS2040: The need for collaborative research in chronic kidney disease. RICORS2040: The need for collaborative research in chronic kidney disease. Clin. Kidney J. 2021, 1–16. [Google Scholar] [CrossRef]
- Parmar, M.S.; Bashir, K. Crescentric glomerulonephritis. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/books/NBK430727/ (accessed on 18 January 2022).
- Jennette, J.C.; Nachman, P.H. ANCA Glomerulonephritis and Vasculitis. Clin. J. Am. Soc. Nephrol. 2017, 12, 1680–1691. [Google Scholar] [CrossRef] [PubMed]
- Pusey, C.D.; McAdoo, S.P. Antiglomerular Basement Membrane Disease. Semin. Respir. Crit. Care Med. 2018, 39, 494–503. [Google Scholar] [CrossRef]
- Smyth, L.; McKay, G.; Maxwell, A.P.; McKnight, A.J. DNA hypermethylation and DNA hypomethylation is present at different loci in chronic kidney disease. Epigenetics 2013, 9, 366–376. [Google Scholar] [CrossRef] [Green Version]
- Biswas, S.; Rao, C.M. Epigenetic tools (The Writers, The Readers and The Erasers) and their implications in cancer therapy. Eur. J. Pharmacol. 2018, 837, 8–24. [Google Scholar] [CrossRef]
- Sanchez, R.; Zhou, M.-M. The role of human bromodomains in chromatin biology and gene transcription. Curr. Opin. Drug Discov. Dev. 2009, 12, 659–665. [Google Scholar]
- Zou, Z.; Huang, B.; Wu, X.; Zhang, H.; Qi, J.; Bradner, J.E.; Nair, S.S.; Chen, L.-F. Brd4 maintains constitutively active NF-κB in cancer cells by binding to acetylated RelA. Oncogene 2014, 33, 2395–2404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicodeme, E.; Jeffrey, K.; Schaefer, U.; Beinke, S.; Dewell, S.; Chung, C.-W.; Chandwani, R.; Marazzi, I.; Wilson, P.; Coste, H.; et al. Suppression of inflammation by a synthetic histone mimic. Nature 2010, 468, 1119–1123. [Google Scholar] [CrossRef]
- Morgado-Pascual, J.L.; Rayego-Mateos, S.; Tejedor, L.; Suarez-Alvarez, B.; Ruiz-Ortega, M. Bromodomain and Extraterminal Proteins as Novel Epigenetic Targets for Renal Diseases. Front. Pharmacol. 2019, 10, 1315. [Google Scholar] [CrossRef]
- Hénique, C.; Papista, C.; Guyonnet, L.; Lenoir, O.; Tharaux, P.-L. Update on crescentic glomerulonephritis. Semin. Immunopathol. 2014, 36, 479–490. [Google Scholar] [CrossRef]
- Jennette, J.C.; Falk, R.J.; Hu, P.; Xiao, H. Pathogenesis of Antineutrophil Cytoplasmic Autoantibody–Associated Small-Vessel Vasculitis. Annu. Rev. Pathol. Mech. Dis. 2013, 8, 139–160. [Google Scholar] [CrossRef] [Green Version]
- Falk, R.J.; Jennette, J.C. ANCA Disease: Where Is This Field Heading? J. Am. Soc. Nephrol. 2010, 21, 745–752. [Google Scholar] [CrossRef] [Green Version]
- Droguett, A.; Valderrama, G.; Burgos, M.E.; Carpio, D.; Saka, C.; Egido, J.; Ruiz-Ortega, M.; Mezzano, S. Gremlin, A Potential Urinary Biomarker of Anca-Associated Crescentic Glomerulonephritis. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Mezzano, S.; Droguett, A.; Lavoz, C.; Krall, P.; Egido, J.; Ruiz-Ortega, M. Gremlin and renal diseases: Ready to jump the fence to clinical utility? Nephrol. Dial. Transplant. 2017, 33, 735–741. [Google Scholar] [CrossRef]
- Brazil, D.P.; Church, R.H.; Surae, S.; Godson, C.; Martin, F. BMP signalling: Agony and antagony in the family. Trends Cell Biol. 2015, 25, 249–264. [Google Scholar] [CrossRef] [Green Version]
- Lappin, D.W.; Hensey, C.; McMahon, R.; Godson, C.; Brady, H.R. Gremlins, glomeruli and diabetic nephropathy. Curr. Opin. Nephrol. Hypertens. 2000, 9, 469–472. [Google Scholar] [CrossRef]
- Lavoz, C.; Poveda, J.; Marquez-Exposito, L.; Rayego-Mateos, S.; Rodrigues-Diez, R.R.; Ortiz, A.; Egido, J.; Mezzano, S.; Ruiz-Ortega, M. Gremlin activates the Notch pathway linked to renal inflammation. Clin. Sci. 2018, 132, 1097–1115. [Google Scholar] [CrossRef] [PubMed]
- Lavoz, C.; Alique, M.; Díez, R.R.; Pato, J.; Keri, G.; Mezzano, S.; Egido, J.; Ruiz-Ortega, M. Gremlin regulates renal inflammation via the vascular endothelial growth factor receptor 2 pathway. J. Pathol. 2015, 236, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Marquez-Exposito, L.; Lavoz, C.; Díez, R.R.; Rayego-Mateos, S.; Orejudo, M.; Cantero-Navarro, E.; Ortiz, A.; Egido, J.; Selgas, R.; Mezzano, S.; et al. Gremlin Regulates Tubular Epithelial to Mesenchymal Transition via VEGFR2: Potential Role in Renal Fibrosis. Front. Pharmacol. 2018, 9, 1195. [Google Scholar] [CrossRef] [PubMed]
- Marquez-Exposito, L.; Cantero-Navarro, E.; Díez, R.R.; Orejudo, M.; Tejera-Muñoz, A.; Tejedor, L.; Rayego-Mateos, S.; Rández-Garbayo, J.; Santos-Sanchez, L.; Mezzano, S.; et al. Molecular Regulation of Notch Signaling by Gremlin. Adv. Exp. Med. Biol. 2020, 1227, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.W.; Roxburgh, S.A.; McGettigan, P.; Berthier, C.C.; Higgins, D.G.; Kretzler, M.; Cohen, C.D.; Mezzano, S.; Brazil, D.P.; Martin, F. Co-regulation of Gremlin and Notch signalling in diabetic nephropathy. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2008, 1782, 10–21. [Google Scholar] [CrossRef] [Green Version]
- Murea, M.; Park, J.-K.; Sharma, S.; Kato, H.; Gruenwald, A.; Niranjan, T.; Si, H.; Thomas, D.B.; Pullman, J.M.; Melamed, M.L.; et al. Expression of Notch pathway proteins correlates with albuminuria, glomerulosclerosis, and renal function. Kidney Int. 2010, 78, 514–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirin, Y.; Susztak, K. Notch in the kidney: Development and disease. J. Pathol. 2011, 226, 394–403. [Google Scholar] [CrossRef] [Green Version]
- Lavoz, C.; Droguett, A.; Burgos, M.E.; Carpio, D.; Ortiz, A.; Egido, J.; Mezzano, S.; Ruizortega, M. Estudio traslacional de la vía Notch en nefropatía hipertensiva. Nefrología 2014, 34, 369–376. [Google Scholar] [CrossRef]
- Marquez-Exposito, L.; Cantero-Navarro, E.; Lavoz, C.; Fierro-Fernández, M.; Poveda, J.; Rayego-Mateos, S.; Rodrigues-Diez, R.R.; Morgado-Pascual, J.L.; Orejudo, M.; Mezzano, S.; et al. Análisis de la vía Notch como una posible diana terapéutica en la patología renal. Nefrología 2018, 38, 466–475. [Google Scholar] [CrossRef]
- Fortini, M.E. Notch Signaling: The Core Pathway and Its Posttranslational Regulation. Dev. Cell 2009, 16, 633–647. [Google Scholar] [CrossRef] [Green Version]
- Bollée, G.; Flamant, M.; Schordan, S.; Fligny, C.; Rumpel, E.; Milon, M.; Schordan, E.; Sabaa, N.; Vandermeersch, S.; Galaup, A.; et al. Epidermal growth factor receptor promotes glomerular injury and renal failure in rapidly progressive crescentic glomerulonephritis. Nat. Med. 2011, 17, 1242–1250. [Google Scholar] [CrossRef] [Green Version]
- Lazareth, H.; Henique, C.; Lenoir, O.; Puelles, V.G.; Flamant, M.; Bollée, G.; Fligny, C.; Camus, M.; Guyonnet, L.; Millien, C.; et al. The tetraspanin CD9 controls migration and proliferation of parietal epithelial cells and glomerular disease progression. Nat. Commun. 2019, 10, 1–17. [Google Scholar] [CrossRef]
- Suarez-Alvarez, B.; Morgado-Pascual, J.L.; Rayego-Mateos, S.; Rodriguez, R.M.; Rodrigues-Diez, R.; Cannata-Ortiz, P.; Sanz, A.B.; Egido, J.; Tharaux, P.-L.; Ortiz, A.; et al. Inhibition of Bromodomain and Extraterminal Domain Family Proteins Ameliorates Experimental Renal Damage. J. Am. Soc. Nephrol. 2016, 28, 504–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.; Park, J.; Qiu, C.; Chung, K.W.; Li, S.-Y.; Sirin, Y.; Han, S.H.; Taylor, V.; Zimber-Strobl, U.; Susztak, K. Jagged1/Notch2 controls kidney fibrosis via Tfam-mediated metabolic reprogramming. PLoS Biol. 2018, 16, e2005233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bielesz, B.; Sirin, Y.; Si, H.; Niranjan, T.; Gruenwald, A.; Ahn, S.; Kato, H.; Pullman, J.; Gessler, M.; Haase, V.H.; et al. Epithelial Notch signaling regulates interstitial fibrosis development in the kidneys of mice and humans. J. Clin. Investig. 2010, 120, 4040–4054. [Google Scholar] [CrossRef] [Green Version]
- Marquez-Exposito, L.; Rodrigues-Diez, R.R.; Rayego-Mateos, S.; Fierro-Fernandez, M.; Rodrigues-Diez, R.; Orejudo, M.; Santos-Sanchez, L.; Blanco, E.M.; Laborda, J.; Mezzano, S.; et al. Deletion of delta-like 1 homologue accelerates renal inflammation by modulating the Th17 immune response. FASEB J. 2021, 35, e21213. [Google Scholar] [CrossRef] [PubMed]
- Brix, S.R.; Stege, G.; Disteldorf, E.; Hoxha, E.; Krebs, C.; Krohn, S.; Otto, B.; Klätschke, K.; Herden, E.; Heymann, F.; et al. CC Chemokine Ligand 18 in ANCA-Associated Crescentic GN. J. Am. Soc. Nephrol. 2015, 26, 2105–2117. [Google Scholar] [CrossRef] [Green Version]
- Cantero-Navarro, E.; Rayego-Mateos, S.; Orejudo, M.; Tejedor-Santamaria, L.; Tejera-Muñoz, A.; Sanz, A.B.; Marquez-Exposito, L.; Marchant, V.; Santos-Sanchez, L.; Egido, J.; et al. Role of Macrophages and Related Cytokines in Kidney Disease. Front. Med. 2021, 8, 688060. [Google Scholar] [CrossRef] [PubMed]
- Islam, S.A.; Ling, M.; Leung, J.; Shreffler, W.G.; Luster, A.D. Identification of human CCR8 as a CCL18 receptor. J. Exp. Med. 2013, 210, 1889–1898. [Google Scholar] [CrossRef]
- Mezzano, S.; Droguett, A.; Burgos, M.E.; Aros, C.; Ardiles, L.; Flores, C.; Carpio, D.; Carvajal, G.; Ruiz-Ortega, M.; Egido, J. Expression of gremlin, a bone morphogenetic protein antagonist, in glomerular crescents of pauci-immune glomerulonephritis. Nephrol. Dial. Transplant. 2007, 22, 1882–1890. [Google Scholar] [CrossRef] [Green Version]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET Bromodomain Inhibition as a Therapeutic Strategy to Target c-Myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef] [Green Version]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [Green Version]
- Bandukwala, H.S.; Gagnon, J.; Togher, S.; Greenbaum, J.A.; Lamperti, E.D.; Parr, N.J.; Molesworth, A.M.H.; Smithers, N.; Lee, K.; Witherington, J.; et al. Selective inhibition of CD4+ T-cell cytokine production and autoimmunity by BET protein and c-Myc inhibitors. Proc. Natl. Acad. Sci. USA 2012, 109, 14532–14537. [Google Scholar] [CrossRef] [Green Version]
- Mele, D.A.; Salmeron, A.; Ghosh, S.; Huang, H.-R.; Bryant, B.M.; Lora, J.M. BET bromodomain inhibition suppresses TH17-mediated pathology. J. Exp. Med. 2013, 210, 2181–2190. [Google Scholar] [CrossRef] [Green Version]
- Dolan, V.; Murphy, M.; Sadlier, D.; Lappin, D.; Doran, P.; Godson, C.; Martin, F.; O’Meara, Y.; Schmid, H.; Henger, A.; et al. Expression of Gremlin, a Bone Morphogenetic Protein Antagonist, in Human Diabetic Nephropathy. Am. J. Kidney Dis. 2005, 45, 1034–1039. [Google Scholar] [CrossRef] [PubMed]
- Carvajal, G.; Droguett, A.; Burgos, M.; Aros, C.; Ardiles, L.; Flores, C.; Carpio, D.; Ruiz-Ortega, M.; Egido, J.; Mezzano, S. Gremlin: A Novel Mediator of Epithelial Mesenchymal Transition and Fibrosis in Chronic Allograft Nephropathy. Transplant. Proc. 2008, 40, 734–739. [Google Scholar] [CrossRef]
- Roxburgh, S.A.; Kattla, J.J.; Curran, S.P.; O’Meara, Y.M.; Pollock, C.A.; Goldschmeding, R.; Godson, C.; Martin, F.; Brazil, D.P. Allelic Depletion of grem1 Attenuates Diabetic Kidney Disease. Diabetes 2009, 58, 1641–1650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues-Díez, R.; Lavoz, C.; Carvajal, G.; Rayego-Mateos, S.; Rodrigues-Diez, R.R.; Arduan, A.O.; Egido, J.; Mezzano, S.; Ruiz-Ortega, M. Gremlin Is a Downstream Profibrotic Mediator of Transforming Growth Factor-Beta in Cultured Renal Cells. Nephron Exp. Nephrol. 2012, 122, 62–74. [Google Scholar] [CrossRef]
- Droguett, A.; Krall, P.; Burgos, M.E.; Valderrama, G.; Carpio, D.; Ardiles, L.; Díez, R.R.; Kerr, B.; Walz, K.; Ruiz-Ortega, M.; et al. Tubular Overexpression of Gremlin Induces Renal Damage Susceptibility in Mice. PLoS ONE 2014, 9, e101879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchant, V.; Droguett, A.; Valderrama, G.; Burgos, M.E.; Carpio, D.; Kerr, B.; Ruiz-Ortega, M.; Egido, J.; Mezzano, S. Tubular overexpression of Gremlin in transgenic mice aggravates renal damage in diabetic nephropathy. Am. J. Physiol. Physiol. 2015, 309, F559–F568. [Google Scholar] [CrossRef] [Green Version]
- Church, R.H.; Ali, I.; Tate, M.; Lavin, D.; Krishnakumar, A.; Kok, H.M.; Hombrebueno, J.R.; Dunne, P.D.; Bingham, V.; Goldschmeding, R.; et al. Gremlin1 plays a key role in kidney development and renal fibrosis. Am. J. Physiol. Physiol. 2017, 312, F1141–F1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, M.; Crean, J.; Brazil, D.P.; Sadlier, D.; Martin, F.; Godson, C. Regulation and consequences of differential gene expression in diabetic kidney disease. Biochem. Soc. Trans. 2008, 36, 941–945. [Google Scholar] [CrossRef] [Green Version]
- Kane, R.; Stevenson, L.; Godson, C.; Stitt, A.; O’Brien, C. Gremlin gene expression in bovine retinal pericytes exposed to elevated glucose. Br. J. Ophthalmol. 2005, 89, 1638–1642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMahon, R.; Murphy, M.; Clarkson, M.; Taal, M.; Mackenzie, H.S.; Godson, C.; Martin, F.; Brady, H.R. IHG-2, a Mesangial Cell Gene Induced by High Glucose, Is Human gremlin Gremlin. Regulation by Extracellular Glucose Concentration, Cyclic Mechanical Strain, and Transforming Growth Factor-Beta1. J. Biol. Chem. 2000, 275, 9901–9904. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Li, Y.; Liu, S.; Shi, Y.; Chi, Y.; Liu, G.; Shan, T. Gremlin aggravates hyperglycemia-induced podocyte injury by a TGFβ/smad dependent signaling pathway. J. Cell. Biochem. 2013, 114, 2101–2113. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Huang, H.; Li, Y.; Liu, M.; Shi, Y.; Chi, Y.; Zhang, T. Gremlin induces cell proliferation and extra cellular matrix accumulation in mouse mesangial cells exposed to high glucose via the ERK1/2 pathway. BMC Nephrol. 2013, 14, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Shi, Y.; Wada, J.; Malakauskas, S.M.; Liu, M.; Ren, Y.; Du, C.; Duan, H.; Li, Y.; Li, Y.; et al. In Vivo Delivery of Gremlin siRNA Plasmid Reveals Therapeutic Potential against Diabetic Nephropathy by Recovering Bone Morphogenetic Protein-7. PLoS ONE 2010, 5, e11709. [Google Scholar] [CrossRef] [Green Version]
- Lavoz, C.; Rodrigues-Diez, R.R.; Plaza, A.; Carpio, D.; Egido, J.; Ruiz-Ortega, M.; Mezzano, S. VEGFR2 Blockade Improves Renal Damage in an Experimental Model of Type 2 Diabetic Nephropathy. J. Clin. Med. 2020, 9, 302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chenivesse, C.; Tsicopoulos, A. CCL18–Beyond chemotaxis. Cytokine 2018, 109, 52–56. [Google Scholar] [CrossRef]
- Little, A.C.; Pathanjeli, P.; Wu, Z.; Bao, L.; Goo, L.E.; Yates, J.; Oliver, C.R.; Soellner, M.B.; Merajver, S.D. IL-4/IL-13 Stimulated Macrophages Enhance Breast Cancer Invasion Via Rho-GTPase Regulation of Synergistic VEGF/CCL-18 Signaling. Front. Oncol. 2019, 9, 456. [Google Scholar] [CrossRef] [Green Version]
- Møller, H.J. Soluble CD163. Scand. J. Clin. Lab. Investig. 2011, 72, 1–13. [Google Scholar] [CrossRef]
- Yokoe, Y.; Tsuboi, N.; Imaizumi, T.; Kitagawa, A.; Karasawa, M.; Ozeki, T.; Endo, N.; Sawa, Y.; Kato, S.; Katsuno, T.; et al. Clinical impact of urinary CD11b and CD163 on the renal outcomes of anti-neutrophil cytoplasmic antibody-associated glomerulonephritis. Nephrol. Dial. Transplant. 2021, 36, 1452–1463. [Google Scholar] [CrossRef]
- Villacorta, J.; Lucientes, L.; Goicoechea, E.; Acevedo, M.; Cavero, T.; Sanchez-Camara, L.; Díaz-Crespo, F.; Gimenez-Moyano, S.; García-Bermejo, L.; Fernandez-Juarez, G. Urinary soluble CD163 as a biomarker of disease activity and relapse in antineutrophil cytoplasm antibody-associated glomerulonephritis. Clin. Kidney J. 2021, 14, 212–219. [Google Scholar] [CrossRef]
- El Machhour, F.; Keuylian, Z.; Kavvadas, P.; Dussaule, J.-C.; Chatziantoniou, C. Activation of Notch3 in Glomeruli Promotes the Development of Rapidly Progressive Renal Disease. J. Am. Soc. Nephrol. 2014, 26, 1561–1575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, Z.; Li, X.; Wang, H.; Chen, G.; Feng, Z.; Wu, Y.; Yin, H.; Zhao, G.; Deng, Z.; Zhao, C.; et al. BRD4 regulates self-renewal ability and tumorigenicity of glioma-initiating cells by enrichment in the Notch1 promoter region. Clin. Transl. Med. 2020, 10, e181. [Google Scholar] [CrossRef]
- Lai, J.; Liu, Z.; Zhao, Y.; Ma, C.; Huang, H. Anticancer Effects of I-BET151, an Inhibitor of Bromodomain and Extra-Terminal Domain Proteins. Front. Oncol. 2021, 11, 3492. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.; Wu, Q.; Kim, L.; E Miller, T.; Liau, B.B.; Mack, S.C.; Yang, K.; Factor, D.C.; Fang, X.; Huang, Z.; et al. RBPJ maintains brain tumor–initiating cells through CDK9-mediated transcriptional elongation. J. Clin. Investig. 2016, 126, 2757–2772. [Google Scholar] [CrossRef] [Green Version]
- Ferrandino, F.; Grazioli, P.; Bellavia, D.; Campese, A.F.; Screpanti, I.; Felli, M.P. Notch and NF-κB: Coach and Players of Regulatory T-Cell Response in Cancer. Front. Immunol. 2018, 9, 2165. [Google Scholar] [CrossRef] [Green Version]
- Saito, T.; Tanaka, S. Molecular mechanisms underlying osteoarthritis development: Notch and NF-κB. Arthritis Res. Ther. 2017, 19, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Hajmirza, A.; Emadali, A.; Gauthier, A.; Casasnovas, O.; Gressin, R.; Callanan, M.B. BET Family Protein BRD4: An Emerging Actor in NFκB Signaling in Inflammation and Cancer. Biomedicines 2018, 6, 16. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Qi, J.; Bradner, J.E.; Xiao, G.; Chen, L.-F. Bromodomain and Extraterminal (BET) Protein Inhibition Suppresses Human T Cell Leukemia Virus 1 (HTLV-1) Tax Protein-mediated Tumorigenesis by Inhibiting Nuclear Factor κB (NF-κB) Signaling. J. Biol. Chem. 2013, 288, 36094–36105. [Google Scholar] [CrossRef] [Green Version]
- Huang, B.; Yang, X.-D.; Zhou, M.-M.; Ozato, K.; Chen, L.-F. Brd4 Coactivates Transcriptional Activation of NF-κB via Specific Binding to Acetylated RelA. Mol. Cell. Biol. 2009, 29, 1375–1387. [Google Scholar] [CrossRef] [Green Version]
- Garg, P. A Review of Podocyte Biology. Am. J. Nephrol. 2018, 47 (Suppl. 1), 3–13. [Google Scholar] [CrossRef]
- Cellesi, F.; Li, M.; Rastaldi, M.P. Podocyte injury and repair mechanisms. Curr. Opin. Nephrol. Hypertens. 2015, 24, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Niranjan, T.; Bielesz, B.; Gruenwald, A.; Ponda, M.P.; Kopp, J.B.; Thomas, D.B.; Susztak, K. The Notch pathway in podocytes plays a role in the development of glomerular disease. Nat. Med. 2008, 14, 290–298. [Google Scholar] [CrossRef]
- Waters, A.M.; Wu, M.Y.; Onay, T.; Scutaru, J.; Liu, J.; Lobe, C.G.; Quaggin, S.E.; Piscione, T.D. Ectopic Notch Activation in Developing Podocytes Causes Glomerulosclerosis. J. Am. Soc. Nephrol. 2008, 19, 1139–1157. [Google Scholar] [CrossRef] [Green Version]
- Schiffer, M.; Bitzer, M.; Roberts, I.S.; Kopp, J.B.; Dijke, P.T.; Mundel, P.; Bottinger, E.P. Apoptosis in podocytes induced by TGF-β and Smad7. J. Clin. Investig. 2001, 108, 807–816. [Google Scholar] [CrossRef] [PubMed]
- Asfahani, R.I.; Tahoun, M.M.; Miller-Hodges, E.V.; Bellerby, J.; Virasami, A.K.; Sampson, R.D.; Moulding, D.; Sebire, N.J.; Hohenstein, P.; Scambler, P.J.; et al. Activation of podocyte Notch mediates early Wt1 glomerulopathy. Kidney Int. 2018, 93, 903–920. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Peng, R.; Ren, Y.; Apparsundaram, S.; Deguzman, J.; Bauer, C.M.; Hoffman, A.F.; Hamilton, S.; Liang, Z.; Zeng, H.; et al. BET Bromodomain Proteins Mediate Downstream Signaling Events following Growth Factor Stimulation in Human Lung Fibroblasts and Are Involved in Bleomycin-Induced Pulmonary Fibrosis. Mol. Pharmacol. 2013, 83, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Peng, R.; Phillips, J.E.; Deguzman, J.; Ren, Y.; Apparsundaram, S.; Luo, Q.; Bauer, C.M.; Fuentes, M.E.; DeMartino, J.A.; et al. Assessment of Brd4 Inhibition in Idiopathic Pulmonary Fibrosis Lung Fibroblasts and in Vivo Models of Lung Fibrosis. Am. J. Pathol. 2013, 183, 470–479. [Google Scholar] [CrossRef]
- Wang, J.; Zhou, F.; Lin, Z.; Mei, H.; Wang, Y.; Ma, H.; Shi, L.; Huang, A.; Zhang, T.; Wu, G. Pharmacological targeting of BET proteins attenuates radiation-induced lung fibrosis. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, M.; Wang, H.-X.; Chen, W.-J. BET-inhibition by JQ1 alleviates streptozotocin-induced diabetic cardiomyopathy. Toxicol. Appl. Pharmacol. 2018, 352, 9–18. [Google Scholar] [CrossRef]
- Duan, Q.; McMahon, S.; Anand, P.; Shah, H.; Thomas, S.; Salunga, H.T.; Huang, Y.; Zhang, R.; Sahadevan, A.; Lemieux, M.E.; et al. BET bromodomain inhibition suppresses innate inflammatory and profibrotic transcriptional networks in heart failure. Sci. Transl. Med. 2017, 9, eaah5084. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.; Mu, J.; Gong, Y.; Lu, C.; Zhao, Y.; He, T.; Qin, Z. Brd4 inhibition attenuates unilateral ureteral obstruction-induced fibrosis by blocking TGF-β-mediated Nox4 expression. Redox Biol. 2017, 11, 390–402. [Google Scholar] [CrossRef] [PubMed]
- Xiong, C.; Masucci, M.V.; Zhou, X.; Liu, N.; Zang, X.; Tolbert, E.; Zhao, T.C.; Zhuang, S. Pharmacological targeting of BET proteins inhibits renal fibroblast activation and alleviates renal fibrosis. Oncotarget 2016, 7, 69291–69308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Díez, R.R.; Rodrigues-Diez, R.R.; Lavoz, C.; Carvajal, G.; Droguett, A.; Garcia-Redondo, A.B.; Rodriguez, I.; Ortiz, A.; Egido, J.; Mezzano, S.; et al. Gremlin Activates the Smad Pathway Linked to Epithelial Mesenchymal Transdifferentiation in Cultured Tubular Epithelial Cells. BioMed Res. Int. 2014, 2014, 1–11. [Google Scholar] [CrossRef]
- Christopoulos, P.F.; Gjølberg, T.T.; Krüger, S.; Haraldsen, G.; Andersen, J.T.; Sundlisæter, E. Targeting the Notch Signaling Pathway in Chronic Inflammatory Diseases. Front. Immunol. 2021, 12, 668207. [Google Scholar] [CrossRef] [PubMed]
- Piha-Paul, S.A.; Sachdev, J.C.; Barve, M.; Lorusso, P.; Szmulewitz, R.; Patel, S.P.; Lara, P.N., Jr.; Chen, X.; Hu, B.; Freise, K.J.; et al. First-in-Human Study of Mivebresib (ABBV-075), an Oral Pan-Inhibitor of Bromodomain and Extra Terminal Proteins, in Patients with Relapsed/Refractory Solid Tumors. Clin. Cancer Res. 2019, 25, 6309–6319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aggarwal, R.R.; Schweizer, M.T.; Nanus, D.M.; Pantuck, A.J.; Heath, E.I.; Campeau, E.; Attwell, S.; Norek, K.; Snyder, M.; Bauman, L.; et al. A Phase Ib/IIa Study of the Pan-BET Inhibitor ZEN-3694 in Combination with Enzalutamide in Patients with Metastatic Castration-resistant Prostate Cancer. Clin. Cancer Res. 2020, 26, 5338–5347. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Picaud, S.; Mangos, M.; Keates, T.; Lambert, J.-P.; Barsyte-Lovejoy, D.; Felletar, I.; Volkmer, R.; Müller, S.; Pawson, T.; et al. Histone Recognition and Large-Scale Structural Analysis of the Human Bromodomain Family. Cell 2012, 149, 214–231. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tejedor-Santamaria, L.; Morgado-Pascual, J.L.; Marquez-Exposito, L.; Suarez-Alvarez, B.; Rodrigues-Diez, R.R.; Tejera-Muñoz, A.; Marchant, V.; Mezzano, S.; Lopez-Larrea, C.; Sola, A.; et al. Epigenetic Modulation of Gremlin-1/NOTCH Pathway in Experimental Crescentic Immune-Mediated Glomerulonephritis. Pharmaceuticals 2022, 15, 121. https://0-doi-org.brum.beds.ac.uk/10.3390/ph15020121
Tejedor-Santamaria L, Morgado-Pascual JL, Marquez-Exposito L, Suarez-Alvarez B, Rodrigues-Diez RR, Tejera-Muñoz A, Marchant V, Mezzano S, Lopez-Larrea C, Sola A, et al. Epigenetic Modulation of Gremlin-1/NOTCH Pathway in Experimental Crescentic Immune-Mediated Glomerulonephritis. Pharmaceuticals. 2022; 15(2):121. https://0-doi-org.brum.beds.ac.uk/10.3390/ph15020121
Chicago/Turabian StyleTejedor-Santamaria, Lucia, Jose Luis Morgado-Pascual, Laura Marquez-Exposito, Beatriz Suarez-Alvarez, Raul R. Rodrigues-Diez, Antonio Tejera-Muñoz, Vanessa Marchant, Sergio Mezzano, Carlos Lopez-Larrea, Anna Sola, and et al. 2022. "Epigenetic Modulation of Gremlin-1/NOTCH Pathway in Experimental Crescentic Immune-Mediated Glomerulonephritis" Pharmaceuticals 15, no. 2: 121. https://0-doi-org.brum.beds.ac.uk/10.3390/ph15020121