
Elexacaftor-Tezacaftor-Ivacaftor as a Final Frontier in the Treatment of Cystic Fibrosis: Definition of the Clinical and Microbiological Implications in a Case-Control Study

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Clinical Results
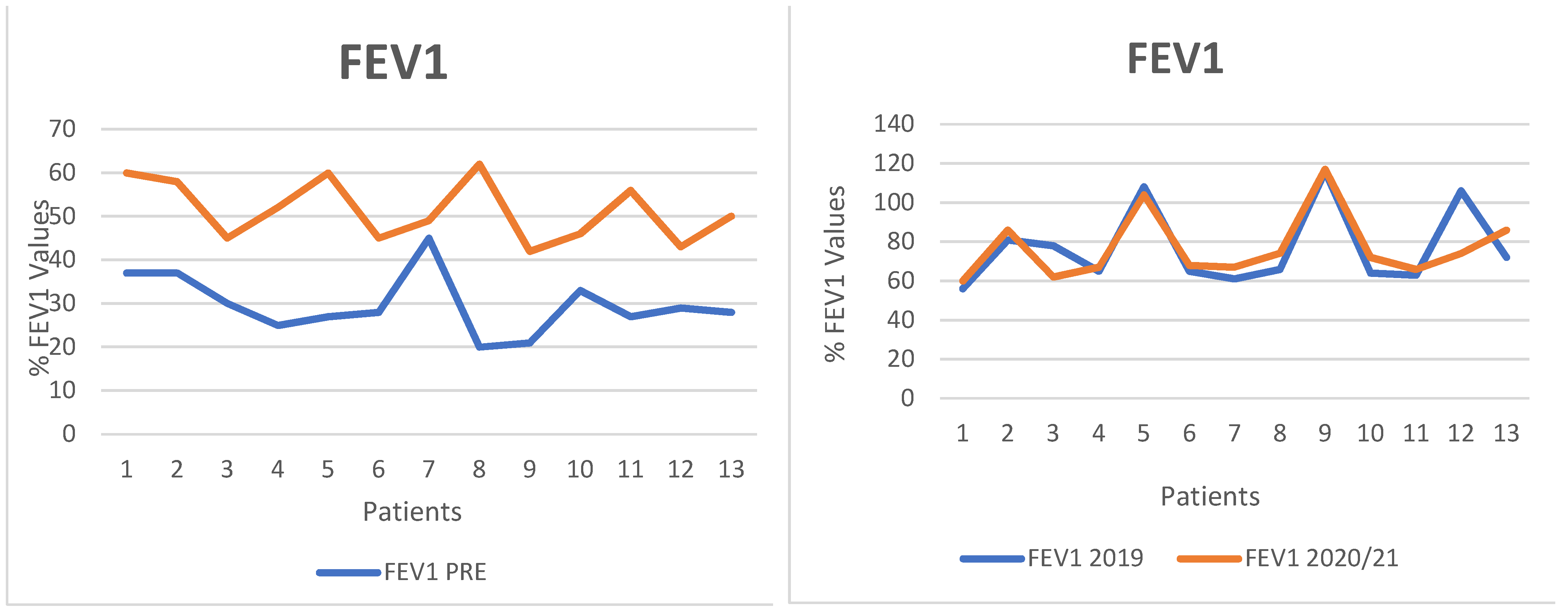
- FEV1
- Radiological Findings
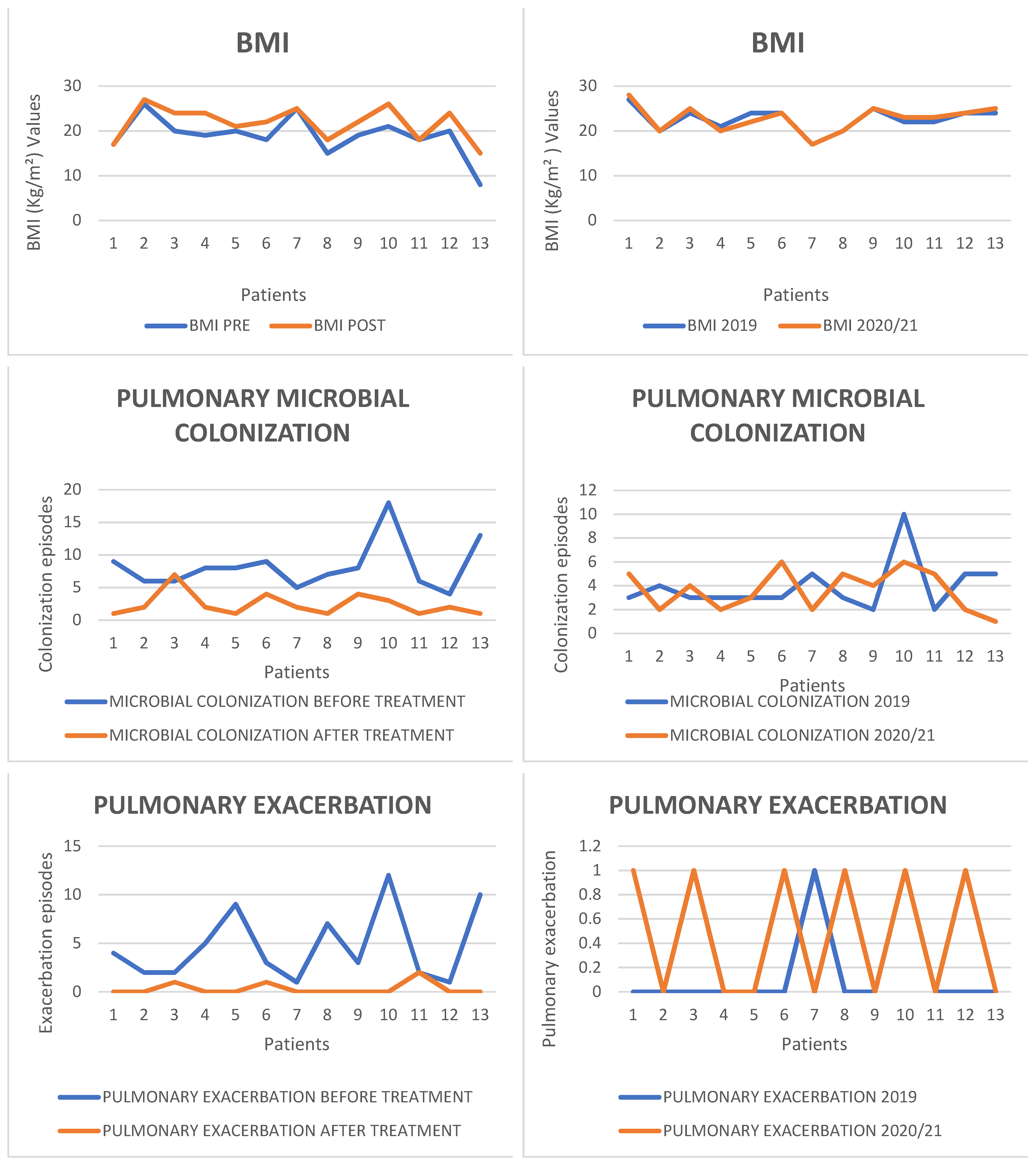
- Nutritional Status
- Sweat Chloride Values
- CFQ-R Questionnaire
2.2. Microbiological Results
2.3. Statistical Analysis
3. Discussion
4. Materials and Methods
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A


References
- European Cystic Fibrosis Society Patent Registry Annual Data Report; 2018.
- Patient Registry Annual Data Report; Cystic Fibrosis Foundation, 2019.
- Saint-Criq, V.; Gray, M.A. Role of CFTR in epithelial physiology. Cell Mol. Life Sci. 2017, 74, 93–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, E.; Sharma, S. Cystic Fibrosis. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Turcios, N.L. Cystic Fibrosis Lung Disease: An Overview. Respir. Care 2020, 65, 233–251. [Google Scholar] [CrossRef] [PubMed]
- Meoli, A.; Fainardi, V.; Deolmi, M.; Chiopris, G.; Marinelli, F.; Caminiti, C.; Esposito, S.; Pisi, G. State of the Art on Approved Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Modulators and Triple-Combination Therapy. Pharmaceuticals 2021, 14, 928. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Pacheco, M. CFTR Modulators: The Changing Face of Cystic Fibrosis in the Era of Precision Medicine. Front. Pharmacol. 2020, 10, 1662. [Google Scholar] [CrossRef] [Green Version]
- Bear, C.E. A Therapy for Most with Cystic Fibrosis. Cell 2020, 180, 211. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.aifa.gov.it/en/-/aifa-approva-nuovi-farmaci-per-il-trattamento-della-fibrosi-cistica (accessed on 10 January 2022).
- Montemayor, K.; Lechtzin, N. The PROSPECT Is Bright for CFTR Modulators. Ann. Am. Thorac. Soc. 2021, 18, 32–33. [Google Scholar] [CrossRef]
- Mayer-Hamblett, N.; Nichols, D.P.; Odem-Davis, K.; Riekert, K.A.; Sawicki, G.S.; Donaldson, S.H.; Ratjen, F.; Konstan, M.W.; Simon, N.; Rosenbluth, D.B.; et al. Evaluating the Impact of Stopping Chronic Therapies after Modulator Drug Therapy in Cystic Fibrosis: The SIMPLIFY Clinical Trial Study Design. Ann. Am. Thorac. Soc. 2021, 18, 1397–1405. [Google Scholar] [CrossRef]
- Zaher, A.; ElSaygh, J.; Elsori, D.; ElSaygh, H.; Sanni, A. A Review of Trikafta: Triple Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Modulator Therapy. Cureus 2021, 13, e16144. [Google Scholar] [CrossRef]
- Middleton, P.G.; Mall, M.A.; Dřevínek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef]
- Griese, M.; Costa, S.; Linnemann, R.W.; Mall, M.A.; McKone, E.F.; Polineni, D.; Quon, B.S.; Ringshausen, F.C.; Taylor-Cousar, J.L.; Withers, N.J.; et al. Safety and Efficacy of Elexacaftor/Tezacaftor/Ivacaftor for 24 Weeks or Longer in People with Cystic Fibrosis and One or More F508delAlleles: Interim Results of an Open-Label Phase 3 Clinical Trial. Am. J. Respir. Crit. Care Med. 2021, 203, 381–385. [Google Scholar] [CrossRef]
- Heijerman, H.G.M.; McKone, E.F.; Downey, D.G.; Van Braeckel, E.; Rowe, S.M.; Tullis, E.; Mall, M.A.; Welter, J.J.; Ramsey, B.W.; McKee, C.M.; et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: A double-blind, randomised, phase 3 trial. Lancet 2019, 394, 1940–1948, Erratum in Lancet 2020, 395, 1694. [Google Scholar] [CrossRef]
- Schäfer, J.; Griese, M.; Chandrasekaran, R.; Chotirmall, S.H.; Hartl, D. Pathogenesis, imaging and clinical characteristics of CF and non-CF bronchiectasis. BMC Pulm. Med. 2018, 18, 79. [Google Scholar] [CrossRef] [PubMed]
- Wielpütz, M.O.; Eichinger, M.; Biederer, J.; Wege, S.; Stahl, M.; Sommerburg, O.; Mall, M.A.; Kauczor, H.U.; Puderbach, M. Imaging of Cystic Fibrosis Lung Disease and Clinical Interpretation. Rofo 2016, 188, 834–845. (In English) [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuthbertson, L.; Walker, A.W.; Oliver, A.E.; Rogers, G.B.; Rivett, D.W.; Hampton, T.H.; Ashare, A.; Elborn, J.S.; De Soyza, A.; Carroll, M.P.; et al. Lung function and microbiota diversity in cystic fibrosis. Microbiome 2020, 8, 45. [Google Scholar] [CrossRef] [PubMed]
- Rogers, G.B.; Taylor, S.L.; Hoffman, L.R.; Burr, L.D. The impact of CFTR modulator therapies on CF airway microbiology. J. Cyst. Fibros. 2020, 19, 359–364. [Google Scholar] [CrossRef]
- Yi, B.; Dalpke, A.H.; Boutin, S. Changes in the Cystic Fibrosis Airway Microbiome in Response to CFTR Modulator Therapy. Front. Cell. Infect. Microbiol. 2021, 11, 548613. [Google Scholar] [CrossRef]
- Neerincx, A.H.; Whiteson, K.; Phan, J.L.; Brinkman, P.; Abdel-Aziz, M.I.; Weersink, E.J.M.; Altenburg, J.; Majoor, C.J.; Maitland-van der Zee, A.H.; Bos, L.D.J. Lumacaftor/ivacaftor changes the lung microbiome and metabolome in cystic fibrosis patients. ERJ Open Res. 2021, 7, 00731–02020. [Google Scholar] [CrossRef]
- Sosinski, L.M.; Christian Martin, H.; Neugebauer, K.A.; Ghuneim, L.J.; Guzior, D.V.; Castillo-Bahena, A.; Mielke, J.; Thomas, R.; McClelland, M.; Conrad, D.; et al. A restructuring of microbiome niche space is associated with Elexacaftor-Tezacaftor-Ivacaftor therapy in the cystic fibrosis lung. J. Cyst. Fibros. 2021; epub ahead of print. [Google Scholar] [CrossRef]
- Ratnayake, I.; Ahern, S.; Ruseckaite, R. A systematic review of patient-reported outcome measures (PROMs) in cystic fibrosis. BMJ Open 2020, 10, e033867. [Google Scholar] [CrossRef]
- Megalaa, R.; Gopalareddy, V.; Champion, E.; Goralski, J.L. Time for a gut check: Pancreatic sufficiency resulting from CFTR modulator use. Pediatr. Pulmonol. 2019, 54, E16–E18. [Google Scholar] [CrossRef]
- Munce, D.; Lim, M.; Akong, K. Persistent recovery of pancreatic function in patients with cystic fibrosis after ivacaftor. Pediatr. Pulmonol. 2020, 55, 3381–3383. [Google Scholar] [CrossRef]
- Ridley, K.; Condren, M. Elexacaftor-Tezacaftor-Ivacaftor: The First Triple-Combination Cystic Fibrosis Transmembrane Conductance Regulator Modulating Therapy. J. Pediatr. Pharmacol. Ther. 2020, 25, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.sifc.it/wp-content/uploads/2020/09/Raccomandazioni-Gruppo-Microbiologi-SIFC-2018.pdf (accessed on 10 January 2022).
- Shteinberg, M.; Haq, I.J.; Polineni, D.; Davies, J.C. Cystic fibrosis. Lancet 2021, 397, 2195–2211. [Google Scholar] [CrossRef]
- Daines, C.L.; Morgan, W.J. The Future of Highly Effective Modulator Therapy in Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2021, 203, 1453–1455. [Google Scholar] [CrossRef] [PubMed]
- Zemanick, E.T.; Taylor-Cousar, J.L.; Davies, J.; Gibson, R.L.; Mall, M.A.; McKone, E.F.; McNally, P.; Ramsey, B.W.; Rayment, J.H.; Rowe, S.M.; et al. A Phase 3 Open-Label Study of Elexacaftor/Tezacaftor/Ivacaftor in Children 6 through 11 Years of Age with Cystic Fibrosis and at Least One F508del Allele. Am. J. Respir. Crit. Care Med. 2021, 203, 1522–1532. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Patients | Best FEV1 | Best BMI | Number of Pulmonary Exacerbations | |||
|---|---|---|---|---|---|---|
| 2019/20 | 2020/21 | 2019/20 | 2020/21 | 2019/20 | 2020/21 | |
| 1 | 56 | 60 | 27.6 | 28.9 | 0 | 1 |
| 2 | 81 | 86 | 20.1 | 20.1 | 0 | 0 |
| 3 | 78 | 62 | 24.2 | 25.0 | 0 | 1 |
| 4 | 65 | 67 | 21.7 | 20.8 | 0 | 0 |
| 5 | 108 | 104 | 24.3 | 22.1 | 0 | 0 |
| 6 | 65 | 68 | 24.4 | 24.8 | 0 | 1 |
| 7 | 61 | 67 | 17.9 | 17.7 | 1 | 0 |
| 8 | 66 | 74 | 20.7 | 20.7 | 0 | 1 |
| 9 | 116 | 117 | 25.1 | 25.2 | 0 | 0 |
| 10 | 64 | 72 | 22.4 | 23.2 | 1 | 1 |
| 11 | 63 | 66 | 22.3 | 23.4 | 0 | 0 |
| 12 | 106 | 74 | 24.3 | 24.1 | 0 | 1 |
| 13 | 72 | 86 | 24.9 | 25.4 | 0 | 0 |
| Patients | Pre-Therapy FEV1 | Post-Therapy FEV1 | Pre-Therapy BMI | Post-Therapy BMI | Pre-Therapy Sweat Test | Post-Therapy Sweat Test | CFQR | Number of Pulmonary Exacerbations | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| T0 | T6 | T12 | T0 | T6 | T12 | T0 | T6 | T12 | T0 | T6 | T12 | T0 | T6 | T12 | T0 | T6 | T12 | Pre | Post | Pre | Post | |
| 1 | 28 | 35 | 37 | 49 | 57 | 60 | 17.2 | 17.5 | 17.4 | 17.1 | 17.1 | 17.5 | 82 | 84 | 78 | 86 | 84 | 78 | 33 | 100 | 4 | 0 |
| 2 | 29 | 37 | 24.6 | 32 | 45 | 58 | 26.2 | 26.2 | 26.9 | 27.1 | 27.2 | 27.2 | 109 | 90 | 86 | 86 | 44 | 35 | 34 | 100 | 2 | 0 |
| 3 | 30 | 28 | 30 | 32 | 45 | 44 | 20.2 | 19.4 | 20.1 | 19.2 | 24.1 | 24.8 | 119 | 100 | 90 | 88 | 66 | 65 | 36.6 | 100 | 2 | 1 |
| 4 | 23 | 25 | 21 | 18 | 51.7 | 52 | 19.1 | 19.6 | 19.4 | 21.6 | 24.1 | 23.0 | 109 | 105 | 98 | 103 | 46 | 36 | 22 | 100 | 5 | 0 |
| 5 | 22 | 27 | 22 | 26 | 48 | 60 | 19.3 | 20.1 | 19.5 | 19.5 | 21.5 | 22.3 | 67 | 75 | 72 | 67 | 36 | 30 | 72.2 | 100 | 9 | 0 |
| 6 | 27 | 25 | 28 | 29 | 38 | 45 | 17.6 | 18.0 | 17.7 | 20.3 | 22.9 | 22.1 | 98 | 95 | 96 | 98 | 74 | 100 | 49 | 100 | 3 | 1 |
| 7 | 45 | 37 | 45 | 37 | 49 | 45 | 25.1 | 25.3 | 25.2 | 25.3 | 25.2 | 25.4 | 60 | 68 | 65 | 107 | 60 | 38 | 33.3 | 100 | 1 | 0 |
| 8 | 20 | 18 | 15 | 18 | 24 | 45 | 15 | 15.1 | 14.8 | 14.8 | 18.2 | 19.5 | 109 | 119 | 98 | 104 | 73 | 36 | 44.4 | 100 | 7 | 0 |
| 9 | 21 | 15 | 17 | 17 | 36 | 42 | 19.1 | 19.2 | 18.2 | 20.7 | 22.4 | 22.7 | 78 | 86 | 90 | 95 | 50 | 48 | 61 | 100 | 3 | 0 |
| 10 | 33 | 31 | 33 | 25 | 42 | 46 | 21.4 | 19.7 | 20.8 | 21.4 | 25.4 | 26.6 | 109 | 98 | 109 | 109 | 67 | 38 | 77.8 | 100 | 12 | 0 |
| 11 | 25 | 27 | 25 | 27 | 45 | 56 | 17.7 | 18 | 17 | 18.2 | 18.5 | 18.6 | 93 | 82 | 82 | 93 | 34 | 37 | 78.9 | 100 | 2 | 2 |
| 12 | 25 | 27 | 29 | 22 | 38 | 43 | 19.2 | 20.0 | 19.4 | 20.1 | 24.2 | 23.1 | 130 | 118 | 101 | 124 | 37 | 38 | 44.4 | 100 | 1 | 0 |
| 13 | 22 | 25 | 28 | 30 | 45 | 50 | 7.3 | 7.8 | 8.1 | 7.3 | 14.5 | 15.4 | 128 | 108 | 115 | 138 | 46 | 48 | 22 | 100 | 10 | 0 |
| Treated Patients | Before Treatment | After Treatment | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Airway Colonization p < 0.05 | P. aeruginosa Dry Colony | P. aeruginosa Mucoid Colony | S. aureus | A. xylosoxidans | Other Microorganisms | Clinical Exacerbations p < 0.05 | Airway Colonization p < 0.05 | P. aeruginosa Dry Colony | P. aeruginosa Mucoid Colony | S. aureus | A. xylosoxidans | Other Microorganisms | Clinical Exacerbations p < 0.05 | |
| 1 | 9 | 11% | 100% | 67% | 4 | 1 | 100% | 0 | ||||||
| 2 | 6 | 100% | 33.3% | 2 | 2 | 100% | 50% | 0 | ||||||
| 3 | 6 | 100% | 17% | 2 | 7 | 100% | - | 1 | ||||||
| 4 | 8 | 25% | 87.5% | 12.5% | 12.5% | 5 | 2 | 100% | 50% | 0 | ||||
| 5 | 8 | 75% | 12.5% | 37.5% | 12.5% | 9 | 1 | 100% | 0 | |||||
| 6 | 9 | 22.2% | 100% | 44.4% | 3 | 4 | 75% | 50% | 1 | |||||
| 7 | 5 | 80% | 40% | 1 | 2 | 50% | 50% | 0 | ||||||
| 8 | 7 | 100% | 7 | 1 | 100% | 100% | 0 | |||||||
| 9 | 8 | 87.5% | 87.5% | 62.5% | 3 | 4 | 25% | 100% | 75% | 50% | 0 | |||
| 10 | 18 | 11.1% | 5.5% | 33.3% | 100% | 12 | 3 | 33.3% | 100% | 33.3% | 0 | |||
| 11 | 6 | 67% | 67% | 2 | 1 | 100% | 2 | |||||||
| 12 | 4 | 25% | 75% | 1 | 2 | 50% | 100% | 0 | ||||||
| 13 | 13 | 31% | 85% | 92.3% | 46% | 10 | 1 | 100% | 100% | 100% | 0 | |||
| Control Patients | Period of Observation: 2019–2020 | Period of Observation: 2020–2021 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Airway Colonization | P. aeruginosa Dry Colony | P. aeruginosa Mucoid Colony | S. aureus | A. xylosoxidans | Other Microorganisms | Clinical Exacerbations | Airway Colonization | P. aeruginosa Dry Colony | P. aeruginosa Mucoid Colony | S. aureus | A. xylosoxidans | Other Microorganisms | Clinical Exacerbations | |
| 1 | 3 | 100% | 33.3% | 33.3% | 0 | 5 | 80% | 40% | 40% | 1 | ||||
| 2 | 4 | 100% | 50% | 25% | 0 | 2 | 50% | 50% | 0 | |||||
| 3 | 3 | 33.3% | 100% | 33.3% | 0 | 4 | 100% | 1 | ||||||
| 4 | 3 | 100% | 33.3% | 0 | 2 | 100% | 0 | |||||||
| 5 | 3 | 100% | 0 | 3 | 100% | 33.3% | 0 | |||||||
| 6 | 3 | 100% | 100% | 0 | 6 | 17% | 100% | 1 | ||||||
| 7 | 5 | 40% | 100% | 20% | 1 | 2 | 50% | 100% | 0 | |||||
| 8 | 3 | 100% | 33.3% | 33.3% | 0 | 5 | 100% | 80% | 20% | 1 | ||||
| 9 | 2 | 50% | 50% | 0 | 4 | 25% | 75% | 0 | ||||||
| 10 | 10 | 80% | 100% | 60% | 10% | 1 | 6 | 100% | 83% | 83% | 1 | |||
| 11 | 2 | 100% | 50% | 0 | 5 | 20% | 80% | 80% | 20% | 0 | ||||
| 12 | 5 | 100% | 20% | 0 | 2 | 100% | 50% | 1 | ||||||
| 13 | 5 | 100% | 0 | 1 | 100% | 0 | ||||||||
| Patients Treated with Triple Combination Therapy | |||
|---|---|---|---|
| Patients | Age | Gender | Genotype |
| 1 | 25 | M | DF508/2183 AA > G |
| 2 | 50 | M | DF508/DF508 |
| 3 | 48 | M | DF508/del2 ins182 |
| 4 | 20 | M | DF508/G542X |
| 5 | 24 | F | DF508/DF508 |
| 6 | 28 | M | DF508/N1303K |
| 7 | 35 | M | DF508/2183 AA < G |
| 8 | 21 | F | DF508/DF508 |
| 9 | 23 | F | DF508/L102R |
| 10 | 23 | F | DF508/DF508 |
| 11 | 29 | F | DF508/DF508 |
| 12 | 43 | F | DF508/E585X |
| 13 | 18 | F | DF508/del ex2 |
| Control Group Patients | |||
| Patients | Age | Gender | Genotypes |
| 1 | 41 | M | DF508/2789 + G > A |
| 2 | 31 | F | DF508/DF508 |
| 3 | 19 | M | DF508/G542X |
| 4 | 44 | F | DF508/2183AA > G |
| 5 | 19 | F | DF508/D1152H |
| 6 | 43 | M | DF508/L558S |
| 7 | 18 | F | DF508/DF508 |
| 8 | 33 | M | DF508/R1158X |
| 9 | 30 | M | DF508/DF508 |
| 10 | 22 | F | DF508/G542X |
| 11 | 40 | M | DF508/2789 + 5G > A |
| 12 | 36 | F | DF508/2789 + 5G > A |
| 13 | 34 | M | DF508/G542X |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Migliorisi, G.; Collura, M.; Ficili, F.; Pensabene, T.; Bongiorno, D.; Collura, A.; Di Bernardo, F.; Stefani, S. Elexacaftor-Tezacaftor-Ivacaftor as a Final Frontier in the Treatment of Cystic Fibrosis: Definition of the Clinical and Microbiological Implications in a Case-Control Study. Pharmaceuticals 2022, 15, 606. https://0-doi-org.brum.beds.ac.uk/10.3390/ph15050606
Migliorisi G, Collura M, Ficili F, Pensabene T, Bongiorno D, Collura A, Di Bernardo F, Stefani S. Elexacaftor-Tezacaftor-Ivacaftor as a Final Frontier in the Treatment of Cystic Fibrosis: Definition of the Clinical and Microbiological Implications in a Case-Control Study. Pharmaceuticals. 2022; 15(5):606. https://0-doi-org.brum.beds.ac.uk/10.3390/ph15050606
Chicago/Turabian StyleMigliorisi, Giuseppe, Mirella Collura, Francesca Ficili, Tiziana Pensabene, Dafne Bongiorno, Antonina Collura, Francesca Di Bernardo, and Stefania Stefani. 2022. "Elexacaftor-Tezacaftor-Ivacaftor as a Final Frontier in the Treatment of Cystic Fibrosis: Definition of the Clinical and Microbiological Implications in a Case-Control Study" Pharmaceuticals 15, no. 5: 606. https://0-doi-org.brum.beds.ac.uk/10.3390/ph15050606