
Role of Brain Modulators in Neurodevelopment: Focus on Autism Spectrum Disorder and Associated Comorbidities



Abstract
:1. Introduction
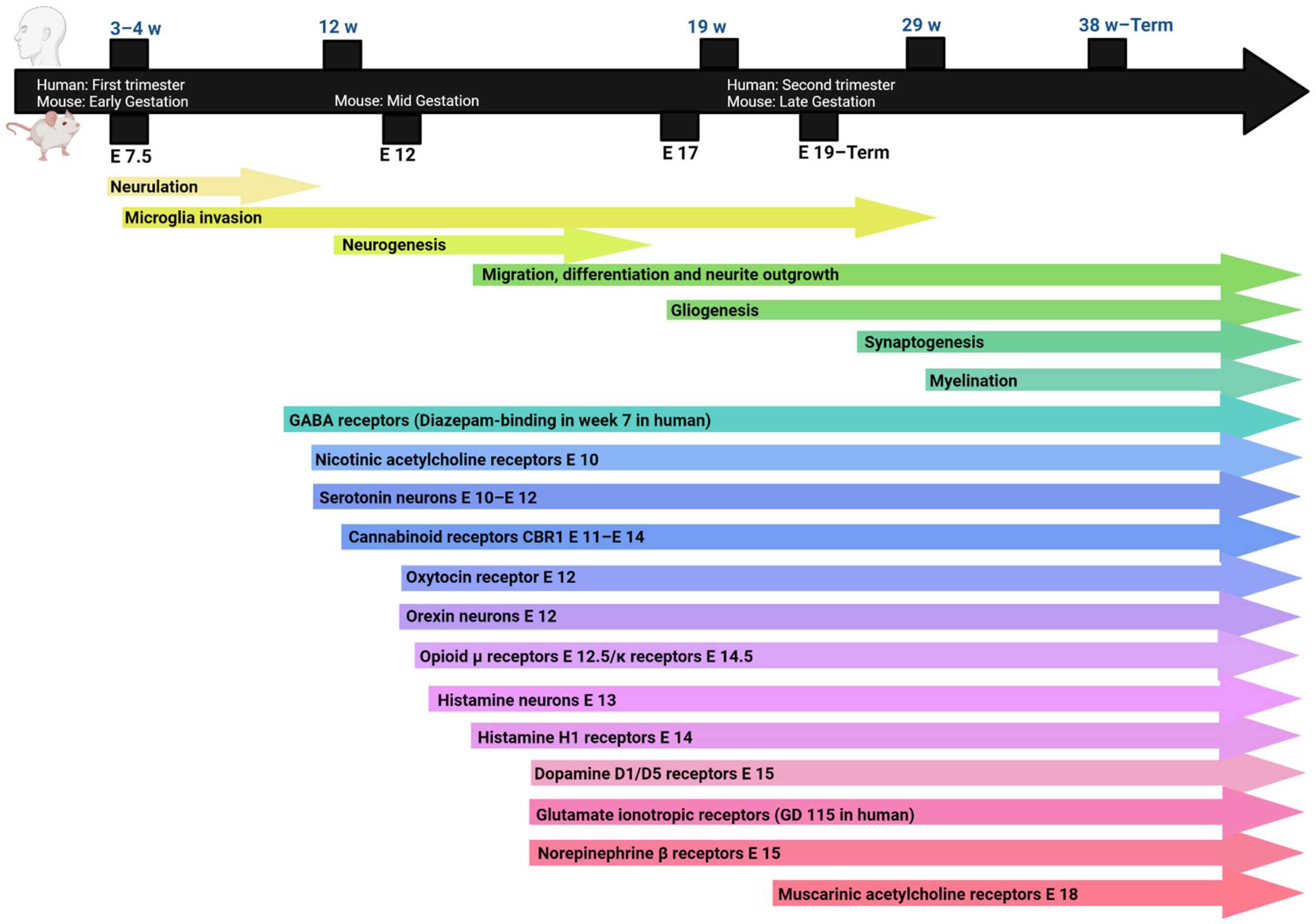
2. Neurodevelopment
2.1. Neurogenesis and Synaptogenesis
2.2. Synaptic Plasticity
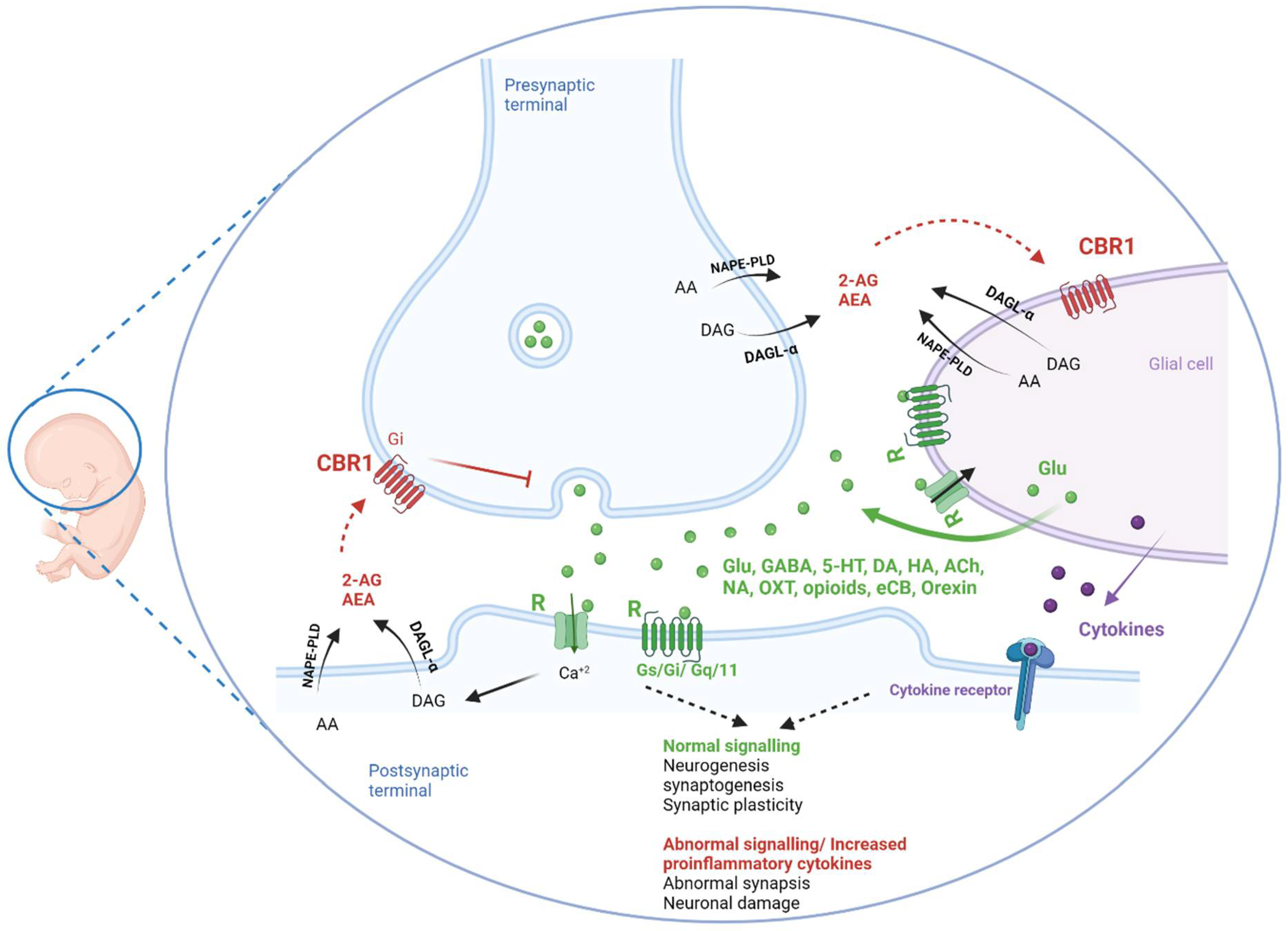
2.3. Neuroinflammation
2.4. Growth Factors, Synaptic Proteins and Intracellular Calcium
3. Endogenous Neuromodulators and Pharmacological Agents in Brain Development
3.1. Neurotransmitters
3.1.1. Excitatory/Inhibitory Balance (Glutamate/GABA)
3.1.2. Dopamine
3.1.3. Serotonin
3.1.4. Acetylcholine
3.1.5. Norepinephrine
3.1.6. Histamine
3.2. Neuropeptides in Brain Development
3.2.1. Oxytocin
3.2.2. Opioids
3.2.3. Cannabinoids
3.2.4. Orexin
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 2-AG | 2-Arachidonoylglycerol |
| 5-HT | Serotonin |
| ADHD | Attention deficit hyperactivity disorder |
| AEA | Anandamide |
| Akt | Ak strain transforming also known as protein kinase B |
| AMPA | α-Amino-3-hydroxy-5-methyl-4-isoxazole propionic acid |
| ASD BDNF | Autism spectrum disorder Brain-derived neurotrophic factor |
| BTBR | Black and Tan Brachyury |
| CBR | Cannabinoid receptor |
| CSF | Cerebrospinal fluid |
| DSM DAG | Diagnostic and statistical manual of mental disorders Diaceylglycerol |
| eCB | Endogenous cannabinoids |
| E or ED | Embryonic day |
| EMA | European Medicine Agency |
| EPSP | Excitatory postsynaptic potential |
| ERK | Extracellular signal-regulated kinase |
| FDA | Food and Drug Administration in USA |
| GABA | Gamma aminobutyric acid |
| GDNF | Glial cell line-derived neurotrophic factor |
| Gi | Inhibitory G-protein |
| Glu | Glutamate |
| GPC | Glial progenitor cell |
| Gs IGF-1 | Stimulatory G protein Insulin growth factor 1 |
| IL-6 IL-8 IL-β IL-10 IPSCs LC | Interleukin-6 Interleukin-8 Interleukin-beta Interleukin-10 Inhibitory postsynaptic currents Locus coeruleus |
| LTD | Long-term depression |
| LTP | Long-term potentiation |
| mGluR MCP-1 | Metabotropic glutamate receptor Monocyte chemoattractant protein 1, |
| MIF | Macrophage migration inhibitory factor |
| NAPE-PLD | N-acyl phosphatidylethanolamine-specific phospholipase D |
| NMDA | N-methyl-D-aspartate |
| NPC NT OXR2 | Neuronal progenitor cells Neurotensin Orexin receptor 2 |
| PI3Ks | Phosphoinositide 3-kinases |
| SCH | Schizophrenia |
| SHANK | SH3 and multiple ankyrin repeat domains protein |
| SSRIs | Selective serotonin reuptake inhibitors |
| TGF-β | Transforming growth factor beta |
| VTA | Ventral tegmental area |
References
- ASA. Neurodevelopmental Disorders: DSM-5® Selections; American Psychiatric Publishing: Washington, DC, USA, 2015. [Google Scholar]
- Posar, A.; Visconti, P. Neurodevelopmental disorders between past and future. J. Pediatr. Neurosci. 2017, 12, 301–302. [Google Scholar] [CrossRef] [PubMed]
- Taheri, A.; Perry, A.; Factor, D.C. Brief report: A further examination of the DSM. 5 autism spectrum disorder criteria in practice. J. Dev. Disabil. 2014, 20, 116. [Google Scholar]
- Hwang, Y.I.; Srasuebkul, P.; Foley, K.R.; Arnold, S.; Trollor, J.N. Mortality and cause of death of Australians on the autism spectrum. Autism Res. 2019, 12, 806–815. [Google Scholar] [CrossRef] [PubMed]
- Ivanović, I. Psychiatric comorbidities in children with ASD: Autism Centre experience. Front. Psychiatry 2021, 12, 673169. [Google Scholar] [CrossRef] [PubMed]
- Ingersoll, B.; Wainer, A. The Broader Autism Phenotype. In Handbook of Autism and Pervasive Developmental Disorders, 4th ed.; Volume Diagnosis, Development, and Brain Mechanisms; Volkmar, F.R., Paul, R., Rogers, S.J., Pelphrey, K.A., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2014. [Google Scholar]
- Hull, L.; Petrides, K.; Mandy, W. The female autism phenotype and camouflaging: A narrative review. Rev. J. Autism Dev. Disord. 2020, 7, 306–317. [Google Scholar] [CrossRef] [Green Version]
- McLennan, J.D.; Lord, C.; Schopler, E. Sex differences in higher functioning people with autism. J. Autism Dev. Disord. 1993, 23, 217–227. [Google Scholar] [CrossRef]
- Zafeiriou, D.I.; Ververi, A.; Dafoulis, V.; Kalyva, E.; Vargiami, E. Autism spectrum disorders: The quest for genetic syndromes. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2013, 162, 327–366. [Google Scholar] [CrossRef]
- Grabrucker, A.M. Environmental factors in autism. Front. Psychiatry 2013, 3, 118. [Google Scholar] [CrossRef] [Green Version]
- Urbán, N.; Guillemot, F. Neurogenesis in the embryonic and adult brain: Same regulators, different roles. Front. Cell. Neurosci. 2014, 8, 396. [Google Scholar] [CrossRef] [Green Version]
- Kriegstein, A.R.; Noctor, S.C. Patterns of neuronal migration in the embryonic cortex. Trends Neurosci. 2004, 27, 392–399. [Google Scholar] [CrossRef]
- Miller, K.E.; Suter, D.M. An integrated cytoskeletal model of neurite outgrowth. Front. Cell. Neurosci. 2018, 12, 447. [Google Scholar] [CrossRef] [PubMed]
- McAllister, A.K. Dynamic Aspects of CNS Synapse Formation. Annu. Rev. Neurosci. 2007, 30, 425–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshihara, Y.; De Roo, M.; Muller, D. Dendritic spine formation and stabilization. Curr. Opin. Neurobiol. 2009, 19, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; van Praag, H. Steps towards standardized quantification of adult neurogenesis. Nat. Commun. 2020, 11, 4275. [Google Scholar] [CrossRef]
- Atluri, P.; Fleck, M.; Shen, Q.; Mah, S.; Stadfelt, D.; Barnes, W.; Goderie, S.; Temple, S.; Schneider, A. Functional nicotinic acetylcholine receptor expression in stem and progenitor cells of the early embryonic mouse cerebral cortex. Dev. Biol. 2001, 240, 143–156. [Google Scholar] [CrossRef]
- Höhmann, C.F.; Pert, C.C.; Ebner, F.F. Development of cholinergic markers in mouse forebrain. II. Muscarinic receptor binding in cortex. Dev. Brain Res. 1985, 23, 243–253. [Google Scholar] [CrossRef]
- Saboory, E.; Ghasemi, M.; Mehranfard, N. Norepinephrine, neurodevelopment and behavior. Neurochem. Int. 2020, 135, 104706. [Google Scholar] [CrossRef]
- Popolo, M.; McCarthy, D.M.; Bhide, P.G. Influence of dopamine on precursor cell proliferation and differentiation in the embryonic mouse telencephalon. Dev. Neurosci. 2004, 26, 229–244. [Google Scholar] [CrossRef] [Green Version]
- Herlenius, E.; Lagercrantz, H. Development of neurotransmitter systems during critical periods. Exp. Neurol. 2004, 190, 8–21. [Google Scholar] [CrossRef]
- Ritter, L.M.; Unis, A.S.; Meador-Woodruff, J.H. Ontogeny of ionotropic glutamate receptor expression in human fetal brain. Dev. Brain Res. 2001, 127, 123–133. [Google Scholar] [CrossRef]
- Hebebrand, J.; Hofmann, D.; Reichelt, R.; Schnarr, S.; Knapp, M.; Propping, P.; Födisch, H. Early ontogeny of the central benzodiazepine receptor in human embryos and fetuses. Life Sci. 1988, 43, 2127–2136. [Google Scholar] [CrossRef]
- Kinnunen, A.; Lintunen, M.; Karlstedt, K.; Fukui, H.; Panula, P. In situ detection of H1-receptor mRNA and absence of apoptosis in the transient histamine system of the embryonic rat brain. J. Comp. Neurol. 1998, 394, 127–137. [Google Scholar] [CrossRef]
- Tribollet, E.; Charpak, S.; Schmidt, A.; Dubois-Dauphin, M.; Dreifuss, J. Appearance and transient expression of oxytocin receptors in fetal, infant, and peripubertal rat brain studied by autoradiography and electrophysiology. J. Neurosci. 1989, 9, 1764–1773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rius, R.A.; Barg, J.; Bem, W.T.; Coscia, C.J.; Loh, Y.P. The prenatal developmental profile of expression of opioid peptides and receptors in the mouse brain. Dev. Brain Res. 1991, 58, 237–241. [Google Scholar] [CrossRef] [Green Version]
- Steininger, T.L.; Kilduff, T.S.; Behan, M.; Benca, R.M.; Landry, C.F. Comparison of hypocretin/orexin and melanin-concentrating hormone neurons and axonal projections in the embryonic and postnatal rat brain. J. Chem. Neuroanat. 2004, 27, 165–181. [Google Scholar] [CrossRef]
- Ogawa, S.; Iriguchi, M.; Lee, Y.-A.; Yoshikawa, S.; Goto, Y. Atypical social rank recognition in autism spectrum disorder. Sci. Rep. 2019, 9, 15657. [Google Scholar] [CrossRef] [PubMed]
- Maffei, A. Long-term potentiation and long-term depression. In Oxford Research Encyclopedia of Neuroscience; Oxford University Press: Oxford, UK, 2018. [Google Scholar]
- Luscher, C.; Malenka, R.C. NMDA Receptor-Dependent Long-Term Potentiation and Long-Term Depression (LTP/LTD). Cold Spring Harb. Perspect. Biol. 2012, 4, a005710. [Google Scholar] [CrossRef] [Green Version]
- Ransohoff, R.M.; Schafer, D.; Vincent, A.; Blachère, N.E.; Bar-Or, A. Neuroinflammation: Ways in which the immune system affects the brain. Neurotherapeutics 2015, 12, 896–909. [Google Scholar] [CrossRef] [Green Version]
- Hendriksen, E.; van Bergeijk, D.; Oosting, R.S.; Redegeld, F.A. Mast cells in neuroinflammation and brain disorders. Neurosci. Biobehav. Rev. 2017, 79, 119–133. [Google Scholar] [CrossRef]
- DiSabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The devil is in the details. J. Neurochem. 2016, 139, 136–153. [Google Scholar] [CrossRef] [Green Version]
- Meyer, U. Developmental neuroinflammation and schizophrenia. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2013, 42, 20–34. [Google Scholar] [CrossRef] [PubMed]
- Slezak, M.; Pfrieger, F.W. New roles for astrocytes: Regulation of CNS synaptogenesis. Trends Neurosci. 2003, 26, 531–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franzen, R.; Bouhy, D.; Schoenen, J. Nervous system injury: Focus on the inflammatory cytokine ‘granulocyte-macrophage colony stimulating factor’. Neurosci. Lett. 2004, 361, 76–78. [Google Scholar] [CrossRef] [PubMed]
- Theoharides, T.C.; Zhang, B. Neuro-inflammation, blood-brain barrier, seizures and autism. J. Neuroinflamm. 2011, 8, 168. [Google Scholar] [CrossRef] [Green Version]
- Vargas, D.L.; Nascimbene, C.; Krishnan, C.; Zimmerman, A.W.; Pardo, C.A. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 2005, 57, 67–81. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Chiba, K.J. Involvement of neuroinflammation during brain development in social cognitive deficits in autism spectrum disorder and schizophrenia. J. Pharmacol. Exp. Ther. 2016, 358, 504–515. [Google Scholar] [CrossRef] [Green Version]
- Nadeem, A.; Ahmad, S.F.; Al-Harbi, N.O.; Al-Ayadhi, L.Y.; Sarawi, W.; Attia, S.M.; Bakheet, S.A.; Alqarni, S.A.; Ali, N.; AsSobeai, H.M. Imbalance in pro-inflammatory and anti-inflammatory cytokines milieu in B cells of children with autism. Mol. Immunol. 2022, 141, 297–304. [Google Scholar] [CrossRef]
- Tsilioni, I.; Pantazopoulos, H.; Conti, P.; Leeman, S.E.; Theoharides, T.C. IL-38 inhibits microglial inflammatory mediators and is decreased in amygdala of children with autism spectrum disorder. Proc. Natl. Acad. Sci. USA 2020, 117, 16475–16480. [Google Scholar] [CrossRef]
- Zhao, H.; Zhang, H.; Liu, S.; Luo, W.; Jiang, Y.; Gao, J. Association of peripheral blood levels of cytokines with autism spectrum disorder: A meta-analysis. Front. Psychiatry 2021, 12, 1006. [Google Scholar] [CrossRef]
- Giacobbo, B.L.; Doorduin, J.; Klein, H.C.; Dierckx, R.A.; Bromberg, E.; de Vries, E.F. Brain-derived neurotrophic factor in brain disorders: Focus on neuroinflammation. Mol. Neurobiol. 2019, 56, 3295–3312. [Google Scholar] [CrossRef] [Green Version]
- Rea, K.; Dinan, T.G.; Cryan, J.F. The microbiome: A key regulator of stress and neuroinflammation. Neurobiol. Stress 2016, 4, 23–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowiański, P.; Lietzau, G.; Czuba, E.; Waśkow, M.; Steliga, A.; Moryś, J. BDNF: A key factor with multipotent impact on brain signaling and synaptic plasticity. Cell. Mol. Neurobiol. 2018, 38, 579–593. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, P.; Feng, G. SHANK proteins: Roles at the synapse and in autism spectrum disorder. Nat. Rev. Neurosci. 2017, 18, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Koshimizu, H.; Adachi, N.; Matsuoka, H.; Fushimi, S.; Ono, J.; Ohta, K.-I.; Miki, T. Functional interaction between BDNF and mGluR II in vitro: BDNF down-regulated mGluR II gene expression and an mGluR II agonist enhanced BDNF-induced BDNF gene expression in rat cerebral cortical neurons. Peptides 2017, 89, 42–49. [Google Scholar] [CrossRef]
- Jaggar, M.; Vaidya, V.A. 5-HT 2A receptors and BDNF regulation: Implications for psychopathology. In 5-HT2A Receptors in the Central Nervous System; Springer: Berlin/Heidelberg, Germany, 2018; pp. 395–438. [Google Scholar]
- Trobiani, L.; Meringolo, M.; Diamanti, T.; Bourne, Y.; Marchot, P.; Martella, G.; Dini, L.; Pisani, A.; De Jaco, A.; Bonsi, P. The neuroligins and the synaptic pathway in Autism Spectrum Disorder. Neurosci. Biobehav. Rev. 2020, 119, 37–51. [Google Scholar] [CrossRef]
- Glaser, T.; Sampaio, V.F.A.; Lameu, C.; Ulrich, H. Calcium signalling: A common target in neurological disorders and neurogenesis. Semin. Cell Dev. Biol. 2019, 95, 25–33. [Google Scholar] [CrossRef]
- Konur, S.; Ghosh, A. Calcium signaling and the control of dendritic development. Neuron 2005, 46, 401–405. [Google Scholar] [CrossRef] [Green Version]
- Lohmann, C. Calcium signaling and the development of specific neuronal connections. Prog. Brain Res. 2009, 175, 443–452. [Google Scholar] [CrossRef]
- Brigadski, T.; Leßmann, V. The physiology of regulated BDNF release. Cell Tissue Res. 2020, 382, 15–45. [Google Scholar] [CrossRef]
- Nguyen, L.; Rigo, J.-M.; Rocher, V.; Belachew, S.; Malgrange, B.; Rogister, B.; Leprince, P.; Moonen, G. Neurotransmitters as early signals for central nervous system development. Cell Tissue Res. 2001, 305, 187–202. [Google Scholar] [CrossRef]
- Herlenius, E.; Lagercrantz, H. Neurotransmitters and neuromodulators during early human development. Early Hum. Dev. 2001, 65, 21–37. [Google Scholar] [CrossRef]
- Medvedeva, V.P.; Pierani, A. How do electric fields coordinate neuronal migration and maturation in the developing cortex? Front. Cell Dev. Biol. 2020, 8, 580657. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, P.R.; Lahiri, S.; Rajamma, U. Glutamate mediated signaling in the pathophysiology of autism spectrum disorders. Pharmacol. Biochem. Behav. 2012, 100, 841–849. [Google Scholar] [CrossRef] [PubMed]
- Peça, J.; Feliciano, C.; Ting, J.T.; Wang, W.; Wells, M.F.; Venkatraman, T.N.; Lascola, C.D.; Fu, Z.; Feng, G. Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature 2011, 472, 437–442. [Google Scholar] [CrossRef] [Green Version]
- Bariselli, S.; Tzanoulinou, S.; Glangetas, C.; Prévost-Solié, C.; Pucci, L.; Viguié, J.; Bezzi, P.; O’Connor, E.C.; Georges, F.; Lüscher, C. SHANK3 controls maturation of social reward circuits in the VTA. Nat. Neurosci. 2016, 19, 926–934. [Google Scholar] [CrossRef] [Green Version]
- Herbert, M.R. SHANK3, the synapse, and autism. N. Engl. J. Med. 2011, 365, 173–175. [Google Scholar] [CrossRef] [Green Version]
- Zimmerman, R.; Smith, A.; Fech, T.; Mansour, Y.; Kulesza, R.J. In utero exposure to valproic acid disrupts ascending projections to the central nucleus of the inferior colliculus from the auditory brainstem. Exp. Brain Res. 2020, 238, 551–563. [Google Scholar] [CrossRef]
- Lenart, J.; Augustyniak, J.; Lazarewicz, J.W.; Zieminska, E. Altered expression of glutamatergic and GABAergic genes in the valproic acid-induced rat model of autism: A screening test. Toxicology 2020, 440, 152500. [Google Scholar] [CrossRef]
- Silvestrin, R.B.; Bambini-Junior, V.; Galland, F.; Bobermim, L.D.; Quincozes-Santos, A.; Abib, R.T.; Zanotto, C.; Batassini, C.; Brolese, G.; Gonçalves, C.-A. Animal model of autism induced by prenatal exposure to valproate: Altered glutamate metabolism in the hippocampus. Brain Res. 2013, 1495, 52–60. [Google Scholar] [CrossRef] [Green Version]
- Miyazaki, K.; Narita, N.; Narita, M. Maternal administration of thalidomide or valproic acid causes abnormal serotonergic neurons in the offspring: Implication for pathogenesis of autism. Int. J. Dev. Neurosci. 2005, 23, 287–297. [Google Scholar] [CrossRef]
- Dufour-Rainfray, D.; Vourc’h, P.; Le Guisquet, A.-M.; Garreau, L.; Ternant, D.; Bodard, S.; Jaumain, E.; Gulhan, Z.; Belzung, C.; Andres, C.R. Behavior and serotonergic disorders in rats exposed prenatally to valproate: A model for autism. Neurosci. Lett. 2010, 470, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.-C.; Zhang, H.-F.; Schön, M.; Böckers, T.M.; Han, S.-P.; Han, J.-S.; Zhang, R. Neonatal oxytocin treatment ameliorates autistic-like behaviors and oxytocin deficiency in valproic acid-induced rat model of autism. Front. Cell. Neurosci. 2018, 12, 355. [Google Scholar] [CrossRef] [PubMed]
- Kellogg, C.K.; Primus, R.J.; Bitran, D.J. Sexually dimorphic influence of prenatal exposure to diazepam on behavioral responses to environmental challenge and on gamma-aminobutyric acid (GABA)-stimulated chloride uptake in the brain. J. Pharmacol. Exp. Ther. 1991, 256, 259–265. [Google Scholar] [PubMed]
- Abekawa, T.; Ito, K.; Nakagawa, S.; Koyama, T. Prenatal exposure to an NMDA receptor antagonist, MK-801 reduces density of parvalbumin-immunoreactive GABAergic neurons in the medial prefrontal cortex and enhances phencyclidine-induced hyperlocomotion but not behavioral sensitization to methamphetamine in postpubertal rats. Psychopharmacology 2007, 192, 303–316. [Google Scholar] [CrossRef]
- Alonazi, M.A.; El Gezeery, A.; El-Ansary, A.; Bhat, R.S. Neurochemical changes in the brain after fetal exposure to fluoxetine, a serotonin reuptake inhibitor (SRI) in rodents. Curr. Proteom. 2021, 18, 499–504. [Google Scholar] [CrossRef]
- Lawrence, R.H.; Palumbo, M.C.; Freeman, S.M.; Guoynes, C.D.; Bales, K.L. Developmental fluoxetine exposure alters behavior and neuropeptide receptors in the prairie vole. Front. Behav. Neurosci. 2020, 14, 215. [Google Scholar] [CrossRef]
- Rosengarten, H.; Quartermain, D. Effect of prenatal administration of haloperidol, risperidone, quetiapine and olanzapine on spatial learning and retention in adult rats. Pharmacol. Biochem. Behav. 2002, 72, 575–579. [Google Scholar] [CrossRef]
- Previc, F.H. Prenatal influences on brain dopamine and their relevance to the rising incidence of autism. Med. Hypotheses 2007, 68, 46–60. [Google Scholar] [CrossRef]
- Zhao, T.; Chen, Y.; Sun, Z.; Shi, Z.; Qin, J.; Lu, J.; Li, C.; Ma, D.; Zhou, L.; Song, X. Prenatal sevoflurane exposure causes neuronal excitatory/inhibitory imbalance in the prefrontal cortex and neurofunctional abnormality in rats. Neurobiol. Dis. 2020, 146, 105121. [Google Scholar] [CrossRef]
- Borges, S.; Coimbra, B.; Soares-Cunha, C.; Pêgo, J.M.; Sousa, N.; Rodrigues, A.J. Dopaminergic modulation of affective and social deficits induced by prenatal glucocorticoid exposure. Neuropsychopharmacology 2013, 38, 2068–2079. [Google Scholar] [CrossRef] [Green Version]
- Alkam, T.; Mamiya, T.; Kimura, N.; Yoshida, A.; Kihara, D.; Tsunoda, Y.; Aoyama, Y.; Hiramatsu, M.; Kim, H.-C.; Nabeshima, T.J.P. Prenatal nicotine exposure decreases the release of dopamine in the medial frontal cortex and induces atomoxetine-responsive neurobehavioral deficits in mice. Psychopharmacology 2017, 234, 1853–1869. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, J.B.; Cardenas, A.; Franke, R.M.; Chen, Y.; Bai, Y.; Belluzzi, J.D.; Lotfipour, S.; Leslie, F.M. Prenatal nicotine sex-dependently alters adolescent dopamine system development. Transl. Psychiatry 2019, 9, 304. [Google Scholar] [CrossRef] [PubMed]
- Morgan, A.J.; Harrod, S.B.; Lacy, R.T.; Stanley, E.M.; Fadel, J.R. Intravenous prenatal nicotine exposure increases orexin expression in the lateral hypothalamus and orexin innervation of the ventral tegmental area in adult male rats. Drug Alcohol Depend. 2013, 132, 562–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanwood, G.; Washington, R.; Shumsky, J.; Levitt, P. Prenatal cocaine exposure produces consistent developmental alterations in dopamine-rich regions of the cerebral cortex. Neuroscience 2001, 106, 5–14. [Google Scholar] [CrossRef]
- Chai, L.; Choi, W.S.; Rönnekleiv, O.K. Maternal cocaine treatment alters dynorphin and enkephalin mRNA expression in brains of fetal rhesus macaques. J. Neurosci. Res. 1997, 17, 1112. [Google Scholar] [CrossRef]
- Chai, L.; Bosch, M.A.; Moore, J.M.; Rønnekleiv, O.K. Chronic prenatal cocaine treatment down-regulates μ-opioid receptor mRNA expression in the brain of fetal Rhesus Macaque. Neurosci. Lett. 1999, 261, 45–48. [Google Scholar] [CrossRef]
- Kim, P.; Park, J.H.; Choi, C.S.; Choi, I.; Joo, S.H.; Kim, M.K.; Kim, S.Y.; Kim, K.C.; Park, S.H.; Kwon, K.J. Effects of ethanol exposure during early pregnancy in hyperactive, inattentive and impulsive behaviors and MeCP2 expression in rodent offspring. Neurochem. Res. 2013, 38, 620–631. [Google Scholar] [CrossRef]
- Carneiro, L.M.; Diógenes, J.P.L.; Vasconcelos, S.M.; Aragão, G.F.; Noronha, E.C.; Gomes, P.B.; Viana, G.S.J. Behavioral and neurochemical effects on rat offspring after prenatal exposure to ethanol. Neurotoxicology Teratol. 2005, 27, 585–592. [Google Scholar] [CrossRef]
- Facciol, A.; Bailleul, C.; Nguyen, S.; Chatterjee, D.; Gerlai, R. Developmental stage-dependent deficits induced by embryonic ethanol exposure in zebrafish: A neurochemical analysis. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2020, 99, 109859. [Google Scholar] [CrossRef]
- Waddell, J.; Mooney, S.M. Choline and working memory training improve cognitive deficits caused by prenatal exposure to ethanol. Nutrients 2017, 9, 1080. [Google Scholar] [CrossRef]
- Smiley, J.F.; Bleiwas, C.; Canals-Baker, S.; Williams, S.Z.; Sears, R.; Teixeira, C.M.; Wilson, D.A.; Saito, M. Neonatal ethanol causes profound reduction of cholinergic cell number in the basal forebrain of adult animals. Alcohol 2021, 97, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Holman, P.J.; Ellis, L.; Morgan, E.; Weinberg, J. Prenatal alcohol exposure disrupts male adolescent social behavior and oxytocin receptor binding in rodents. Horm. Behav. 2018, 105, 115–127. [Google Scholar] [CrossRef] [PubMed]
- He, F.-Q.; Zhang, J.; Guo, X. Prenatal ethanol exposure increases depressive-like behavior and central estrogen receptor α and oxytocin expressions in adult female Mandarin Voles. Zool. Stud. 2012, 51, 1–11. [Google Scholar]
- Arias, C.; Chotro, M.G. Increased palatability of ethanol after prenatal ethanol exposure is mediated by the opioid system. Pharmacol. Biochem. Behav. 2005, 82, 434–442. [Google Scholar] [CrossRef] [PubMed]
- Collier, A.D.; Halkina, V.; Min, S.S.; Roberts, M.Y.; Campbell, S.D.; Camidge, K.; Leibowitz, S.F. Embryonic ethanol exposure affects the early development, migration, and location of hypocretin/orexin neurons in zebrafish. Alcohol. Clin. Exp. Res. 2019, 43, 1702–1713. [Google Scholar] [CrossRef] [PubMed]
- Collier, A.D.; Yasmin, N.; Khalizova, N.; Campbell, S.; Onoichenco, A.; Fam, M.; Albeg, A.S.; Leibowitz, S.F. Sexually dimorphic and asymmetric effects of embryonic ethanol exposure on hypocretin/orexin neurons as related to behavioral changes in zebrafish. Sci. Rep. 2021, 11, 16078. [Google Scholar] [CrossRef]
- Di Cristo, G. Development of cortical GABAergic circuits and its implications for neurodevelopmental disorders. Clin. Genet. 2007, 72, 1–8. [Google Scholar] [CrossRef]
- De Caro, C.; Iannone, L.F.; Citraro, R.; Striano, P.; De Sarro, G.; Constanti, A.; Cryan, J.F.; Russo, E. Can we ‘seize’ the gut microbiota to treat epilepsy? Neurosci. Biobehav. Rev. 2019, 107, 750–764. [Google Scholar] [CrossRef]
- Gatto, C.L.; Broadie, K. Genetic controls balancing excitatory and inhibitory synaptogenesis in neurodevelopmental disorder models. Front. Synaptic Neurosci. 2010, 2, 4. [Google Scholar] [CrossRef] [Green Version]
- Miranda-Contrerasa, P.B.-D.L.; Mendoza-Briceñoa, R.V.; Palacios-Prüa, Z.P.-C.E. Prenatal and postnatal contents of amino acid neurotransmitters in mouse parietal cortex. Dev. Neurosci. 2003, 25, 366–374. [Google Scholar]
- Luján, R.; Shigemoto, R.; López-Bendito, G. Glutamate and GABA receptor signalling in the developing brain. Neuroscience 2005, 130, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.; Maussion, G.; Jefri, M.; Peng, H.; Theroux, J.-F.; Silveira, H.; Soubannier, V.; Wu, H.; Hu, P.; Galat, E. Disruption of GRIN2B impairs differentiation in human neurons. Stem Cell Rep. 2018, 11, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Roullet, F.I.; Lai, J.K.; Foster, J.A. In utero exposure to valproic acid and autism—a current review of clinical and animal studies. Neurotoxicol. Teratol. 2013, 36, 47–56. [Google Scholar] [CrossRef] [PubMed]
- van Gelder, M.M.; Bos, J.H.; Roeleveld, N.; de Jong-van den Berg, L.T.W. Drugs associated with teratogenic mechanisms. Part I: Dispensing rates among pregnant women in the Netherlands, 1998–2009. Hum. Reprod. 2014, 29, 161–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandhi, R.; Luk, K.C.; Rymar, V.V.; Sadikot, A.F. Group I mGluR5 metabotropic glutamate receptors regulate proliferation of neuronal progenitors in specific forebrain developmental domains. J. Neurochem. 2008, 104, 155–172. [Google Scholar] [CrossRef]
- Gogolla, N.; LeBlanc, J.J.; Quast, K.B.; Südhof, T.C.; Fagiolini, M.; Hensch, T.K. Common circuit defect of excitatory-inhibitory balance in mouse models of autism. J. Neurodev. Disord. 2009, 1, 172–181. [Google Scholar] [CrossRef] [Green Version]
- Olloquequi, J.; Cornejo-Córdova, E.; Verdaguer, E.; Soriano, F.X.; Binvignat, O.; Auladell, C.; Camins, A. Excitotoxicity in the pathogenesis of neurological and psychiatric disorders: Therapeutic implications. J. Psychopharmacol. 2018, 32, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Essa, M.; Braidy, N.; Vijayan, K.; Subash, S.; Guillemin, G.J. Excitotoxicity in the pathogenesis of autism. Neurotox. Res. 2013, 23, 393–400. [Google Scholar] [CrossRef]
- Viviani, B.; Boraso, M.; Marchetti, N.; Marinovich, M. Perspectives on neuroinflammation and excitotoxicity: A neurotoxic conspiracy? Neurotoxicology 2014, 43, 10–20. [Google Scholar] [CrossRef]
- Nakamoto, M.; Nalavadi, V.; Epstein, M.P.; Narayanan, U.; Bassell, G.J.; Warren, S.T. Fragile X mental retardation protein deficiency leads to excessive mGluR5-dependent internalization of AMPA receptors. Proc. Natl. Acad. Sci. USA 2007, 104, 15537–15542. [Google Scholar] [CrossRef] [Green Version]
- Bovetti, S.; Gribaudo, S.; Puche, A.C.; De Marchis, S.; Fasolo, A. From progenitors to integrated neurons: Role of neurotransmitters in adult olfactory neurogenesis. J. Chem. Neuroanat. 2011, 42, 304–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, H.G.S.; Manzoni, O. Late onset deficits in synaptic plasticity in the valproic acid rat model of autism. Front. Cell. Neurosci. 2014, 8, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, J.R.; Mukherjee, B.; Rogers, B.C.; Nafar, F.; Gosse, M.; Parsons, M.P. The relationship between glutamate dynamics and activity-dependent synaptic plasticity. J. Neurosci. 2020, 40, 2793–2807. [Google Scholar] [CrossRef]
- Kehrer, C.; Maziashvili, N.; Dugladze, T.; Gloveli, T. Altered excitatory-inhibitory balance in the NMDA-hypofunction model of schizophrenia. Front. Mol. Neurosci. 2008, 1, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lahti, A.C.; Weiler, M.A.; Michaelidis, B.T.; Parwani, A.; Tamminga, C.A. Effects of ketamine in normal and schizophrenic volunteers. Neuropsychopharmacology 2001, 25, 455–467. [Google Scholar] [CrossRef] [Green Version]
- Jacob, T.C.; Wan, Q.; Vithlani, M.; Saliba, R.S.; Succol, F.; Pangalos, M.N.; Moss, S.J. GABAA receptor membrane trafficking regulates spine maturity. Proc. Natl. Acad. Sci. USA 2009, 106, 12500–12505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajamani, K.T.; Wagner, S.; Grinevich, V.; Harony-Nicolas, H. Oxytocin as a Modulator of Synaptic Plasticity: Implications for Neurodevelopmental Disorders. Front. Synaptic Neurosci. 2018, 10, 17. [Google Scholar] [CrossRef] [Green Version]
- Arion, D.; Lewis, D.A. Altered expression of regulators of the cortical chloride transporters NKCC1 and KCC2 in Schizophrenia. Arch. Gen. Psychiatry 2011, 68, 21. [Google Scholar] [CrossRef]
- Hyde, T.M.; Lipska, B.K.; Ali, T.; Mathew, S.V.; Law, A.J.; Metitiri, O.E.; Straub, R.E.; Ye, T.; Colantuoni, C.; Herman, M.M.; et al. Expression of GABA signaling molecules KCC2, NKCC1, and GAD1 in cortical development and schizophrenia. J. Neurosci. 2011, 31, 11088–11095. [Google Scholar] [CrossRef] [Green Version]
- Mabunga, D.F.N.; Gonzales, E.L.T.; Kim, J.-W.; Kim, K.C.; Shin, C.Y. Exploring the validity of valproic acid animal model of autism. Exp. Neurobiol. 2015, 24, 285. [Google Scholar] [CrossRef] [Green Version]
- Plioplys, A.V. Autism: Electroencephalogram abnormalities and clinical improvement with valproic acid. Arch. Pediatrics Adolesc. Med. 1994, 148, 220–222. [Google Scholar] [CrossRef] [PubMed]
- Money, K.; Stanwood, G. Developmental origins of brain disorders: Roles for dopamine. Front. Cell. Neurosci. 2013, 7, 260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozawa, K.; Hashimoto, K.; Kishimoto, T.; Shimizu, E.; Ishikura, H.; Iyo, M. Immune activation during pregnancy in mice leads to dopaminergic hyperfunction and cognitive impairment in the offspring: A neurodevelopmental animal model of schizophrenia. Biol. Psychiatry 2006, 59, 546–554. [Google Scholar] [CrossRef]
- Vuillermot, S.; Weber, L.; Feldon, J.; Meyer, U. A longitudinal examination of the neurodevelopmental impact of prenatal immune activation in mice reveals primary defects in dopaminergic development relevant to schizophrenia. J. Neurosci. Methods 2010, 30, 1270–1287. [Google Scholar] [CrossRef] [PubMed]
- Spencer, G.; Klumperman, J.; Syed, N. Neurotransmitters and neurodevelopment. Role of dopamine in neurite outgrowth, target selection and specific synapse formation. Perspect. Dev. Neurobiol. 1998, 5, 451–467. [Google Scholar]
- Areal, L.B.; Blakely, R.D. Neurobehavioral changes arising from early life dopamine signaling perturbations. Neurochem. Int. 2020, 137, 104747. [Google Scholar] [CrossRef] [PubMed]
- Azmitia, E. Serotonin neurons, neuroplasticity, and homeostasis of neural tissue. Neuropsychopharmacology 1999, 21, S33–S45. [Google Scholar] [CrossRef]
- Lesch, K.-P.; Waider, J. Serotonin in the modulation of neural plasticity and networks: Implications for neurodevelopmental disorders. Neuron 2012, 76, 175–191. [Google Scholar] [CrossRef] [Green Version]
- Kepser, L.-J.; Homberg, J.R. The neurodevelopmental effects of serotonin: A behavioural perspective. Behav. Brain Res. 2015, 277, 3–13. [Google Scholar] [CrossRef]
- Chugani, D.C. Role of altered brain serotonin mechanisms in autism. Mol. Psychiatry 2002, 7, S16–S17. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.W.; Huel, G.; Campagna, D.; Hellier, G.; Boissinot, C.; Blot, P. Neurodevelopmental evaluation of 9-month-old infants exposed to low levels of lead in utero: Involvement of monoamine neurotransmitters. J. Appl. Toxicol. 1999, 19, 167–172. [Google Scholar] [CrossRef]
- Eissa, N.; Al-Houqani, M.; Sadeq, A.; Ojha, S.K.; Sasse, A.; Sadek, B. Current enlightenment about etiology and pharmacological treatment of autism spectrum disorder. Front. Neurosci. 2018, 12, 304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eissa, N.; Jayaprakash, P.; Azimullah, S.; Ojha, S.K.; Al-Houqani, M.; Jalal, F.Y.; Łażewska, D.; Kieć-Kononowicz, K.; Sadek, B. The histamine H3R antagonist DL77 attenuates autistic behaviors in a prenatal valproic acid-induced mouse model of autism. Sci. Rep. 2018, 8, 13077. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Avramets, D.; Jeon, B.; Choo, H. Modulation of serotonin receptors in neurodevelopmental disorders: Focus on 5-HT7 receptor. Molecules 2021, 26, 3348. [Google Scholar] [CrossRef]
- Court, J.; Perry, E.; Spurden, D.; Griffiths, M.; Kerwin, J.; Morris, C.; Johnson, M.; Oakley, A.; Birdsall, N.; Clementi, F. The role of the cholinergic system in the development of the human cerebellum. Dev. Brain Res. 1995, 90, 159–167. [Google Scholar] [CrossRef]
- Abreu-Villaça, Y.; Filgueiras, C.C.; Manhães, A.C. Developmental aspects of the cholinergic system. Behav. Brain Res. 2011, 221, 367–378. [Google Scholar] [CrossRef]
- Bruel-Jungerman, E.; Lucassen, P.J.; Francis, F. Cholinergic influences on cortical development and adult neurogenesis. Behav. Brain Res. 2011, 221, 379–388. [Google Scholar] [CrossRef]
- Deutsch, S.I.; Burket, J.A. An evolving therapeutic rationale for targeting the α 7 nicotinic acetylcholine receptor in autism spectrum disorder. In Behavioral Pharmacology of the Cholinergic System; Springer: Berlin/Heidelberg, Germany, 2020; pp. 167–208. [Google Scholar]
- Poppi, L.A.; Ho-Nguyen, K.T.; Shi, A.; Daut, C.T.; Tischfield, M.A. Recurrent implication of striatal cholinergic interneurons in a range of neurodevelopmental, neurodegenerative, and neuropsychiatric disorders. Cells 2021, 10, 907. [Google Scholar] [CrossRef]
- Adams, C.E.; Stevens, K.E. Evidence for a role of nicotinic acetylcholine receptors in schizophrenia. Front. Biosci. 2007, 12, 55–4772. [Google Scholar] [CrossRef] [Green Version]
- Artoni, P.; Piffer, A.; Vinci, V.; Leblanc, J.; Nelson, C.A.; Hensch, T.K.; Fagiolini, M. Deep learning of spontaneous arousal fluctuations detects early cholinergic defects across neurodevelopmental mouse models and patients. Proc. Natl. Acad. Sci. USA 2020, 117, 23298–23303. [Google Scholar] [CrossRef] [Green Version]
- Mostafalou, S.; Abdollahi, M. The link of organophosphorus pesticides with neurodegenerative and neurodevelopmental diseases based on evidence and mechanisms. Toxicology 2018, 409, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Ni, L.; Acevedo, G.; Muralidharan, B.; Padala, N.; To, J.; Jonakait, G.M. Toll-like receptor ligands and CD154 stimulate microglia to produce a factor(s) that promotes excess cholinergic differentiation in the developing rat basal forebrain: Implications for neurodevelopmental disorders. Pediatric Res. 2007, 61, 15–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vivanti, G.; Nuske, H.J. Autism, attachment, and social learning: Three challenges and a way forward. Behav. Brain Res. 2017, 325, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Stowell, R.D.; Sipe, G.O.; Dawes, R.P.; Batchelor, H.N.; Lordy, K.A.; Whitelaw, B.S.; Stoessel, M.B.; Bidlack, J.M.; Brown, E.; Sur, M. Noradrenergic signaling in the wakeful state inhibits microglial surveillance and synaptic plasticity in the mouse visual cortex. Nat. Neurosci. 2019, 22, 1782–1792. [Google Scholar] [CrossRef] [PubMed]
- Eissa, N.; Sadeq, A.; Sasse, A.; Sadek, B. Role of Neuroinflammation in Autism Spectrum Disorder and the Emergence of Brain Histaminergic System. Lessons Also for BPSD? Front. Pharmacol. 2020, 11, 886. [Google Scholar] [CrossRef]
- Haas, H.L.; Sergeeva, O.A.; Selbach, O. Histamine in the nervous system. Physiol. Rev. 2008, 88, 1183–1241. [Google Scholar] [CrossRef]
- Carthy, E.; Ellender, T. Histamine, neuroinflammation and neurodevelopment: A review. Front. Neurosci. 2021, 15, 680214. [Google Scholar] [CrossRef]
- Baronio, D.; Puttonen, H.A.; Sundvik, M.; Semenova, S.; Lehtonen, E.; Panula, P. Embryonic exposure to valproic acid affects the histaminergic system and the social behaviour of adult zebrafish (Danio rerio). Br. J. Pharmacol. 2018, 175, 797–809. [Google Scholar] [CrossRef] [Green Version]
- Mahmood, D. Histamine H3 receptors and its antagonism as a novel mechanism for antipsychotic effect: A current preclinical & clinical perspective. Int. J. Health Sci. 2016, 10, 564. [Google Scholar]
- Eissa, N.; Venkatachalam, K.; Jayaprakash, P.; Falkenstein, M.; Dubiel, M.; Frank, A.; Reiner-Link, D.; Stark, H.; Sadek, B. The multi-targeting ligand ST-2223 with histamine H3 receptor and dopamine D2/D3 receptor antagonist properties mitigates autism-like repetitive behaviors and brain oxidative stress in mice. Int. J. Mol. Sci. 2021, 22, 1947. [Google Scholar] [CrossRef]
- Sadek, B.; Saad, A.; Subramanian, D.; Shafiullah, M.; Lazewska, D.; Kiec-Kononowiczc, K. Anticonvulsant and procognitive properties of the non-imidazole histamine H3 receptor antagonist DL77 in male adult rats. Neuropharmacology 2016, 106, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.W.; Whitehead, C.A.; Basso, A.M.; Rueter, L.E.; Zhang, M. Preclinical evaluation of non-imidazole histamine H3 receptor antagonists in comparison to atypical antipsychotics for the treatment of cognitive deficits associated with schizophrenia. Int. J. Neuropsychopharmacol. 2013, 16, 889–904. [Google Scholar] [CrossRef] [PubMed]
- Ellenbroek, B.A.; Ghiabi, B. The other side of the histamine H3 receptor. Trends Neurosci. 2014, 37, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Bhowmik, M.; Khanam, R.; Vohora, D. Histamine H3receptor antagonists in relation to epilepsy and neurodegeneration: A systemic consideration of recent progress and perspectives. Br. J. Pharmacol. 2012, 167, 1398–1414. [Google Scholar] [CrossRef] [Green Version]
- Cowan, M.; Petri, W.A., Jr. Microglia: Immune regulators of neurodevelopment. Front. Immunol. 2018, 9, 2576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Zhang, X.; Zhang, Y.; Qu, C.; Zhou, X.; Zhang, S. Histamine induces microglia activation and the release of proinflammatory mediators in rat brain via H1R or H4R. J. Neuroimmune Pharmacol. 2020, 15, 280–291. [Google Scholar] [CrossRef]
- Ahmad, S.F.; Nadeem, A.; Ansari, M.A.; Bakheet, S.A.; Al-Mazroua, H.A.; Khan, M.R.; Alasmari, A.F.; Alanazi, W.A.; Sobeai, H.M.A.; Attia, S.M. The histamine-4 receptor antagonist JNJ7777120 prevents immune abnormalities by inhibiting RORγt/T-bet transcription factor signaling pathways in BTBR T+ Itpr3tf/J mice exposed to gamma rays. Mol. Immunol. 2019, 114, 561–570. [Google Scholar] [CrossRef]
- Monks, D.; Palanisamy, A. Oxytocin: At birth and beyond. A systematic review of the long-term effects of peripartum oxytocin. Anaesthesia 2021, 76, 1526–1537. [Google Scholar] [CrossRef]
- Tamborski, S.; Mintz, E.; Caldwell, H. Sex differences in the embryonic development of the central oxytocin system in mice. J. Neuroendocrinol. 2016, 28, 1–7. [Google Scholar] [CrossRef]
- Wu, Z.; Xie, C.; Kuang, H.; Wu, J.; Chen, X.; Liu, H.; Liu, T. Oxytocin mediates neuroprotection against hypoxic-ischemic injury in hippocampal CA1 neuron of neonatal rats. Neuropharmacology 2021, 187, 108488. [Google Scholar] [CrossRef]
- El Falougy, H.; Filova, B.; Ostatnikova, D.; Bacova, Z.; Bakos, J. Neuronal morphology alterations in autism and possible role of oxytocin. Endocr. Regul. 2019, 53, 46–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filova, B.; Reichova, A.; Zatkova, M.; Srancikova, A.; Bukatova, S.; Bacova, Z.; Bakos, J. Expression of synaptic proteins in the hippocampus is modulated by neonatal oxytocin treatment. Neurosci. Lett. 2020, 725, 134912. [Google Scholar] [CrossRef] [PubMed]
- Mairesse, J.; Zinni, M.; Pansiot, J.; Hassan-Abdi, R.; Demene, C.; Colella, M.; Charriaut-Marlangue, C.; Novais, A.R.B.; Tanter, M.; Maccari, S. Oxytocin receptor agonist reduces perinatal brain damage by targeting microglia. Glia 2019, 67, 345–359. [Google Scholar] [CrossRef] [PubMed]
- Pekarek, B.T.; Hunt, P.J.; Arenkiel, B.R. Oxytocin and sensory network plasticity. Front. Neurosci. 2020, 14, 30. [Google Scholar] [CrossRef] [PubMed]
- Aulino, E.A.; Caldwell, H.K. Pharmacological manipulation of oxytocin receptor signaling during mouse embryonic development results in sex-specific behavioral effects in adulthood. Horm. Behav. 2021, 135, 105026. [Google Scholar] [CrossRef]
- Nunes, A.R.; Gliksberg, M.; Varela, S.A.; Teles, M.; Wircer, E.; Blechman, J.; Petri, G.; Levkowitz, G.; Oliveira, R.F. Developmental effects of oxytocin neurons on social affiliation and processing of social information. J. Neurosci. 2021, 41, 8742–8760. [Google Scholar] [CrossRef]
- Williams, A.V.; Duque-Wilckens, N.; Ramos-Maciel, S.; Campi, K.L.; Bhela, S.K.; Xu, C.K.; Jackson, K.; Chini, B.; Pesavento, P.A.; Trainor, B.C. Social approach and social vigilance are differentially regulated by oxytocin receptors in the nucleus accumbens. Neuropsychopharmacology 2020, 45, 1423–1430. [Google Scholar] [CrossRef]
- Fineberg, S.K.; Ross, D.A. Oxytocin and the social brain. Biol. Psychiatry 2017, 81, e19. [Google Scholar] [CrossRef] [Green Version]
- Siu, M.T.; Goodman, S.J.; Yellan, I.; Butcher, D.T.; Jangjoo, M.; Grafodatskaya, D.; Rajendram, R.; Lou, Y.; Zhang, R.; Zhao, C. DNA methylation of the oxytocin receptor across neurodevelopmental disorders. J. Autism Dev. Disord. 2021, 51, 3610–3623. [Google Scholar] [CrossRef]
- Moerkerke, M.; Bonte, M.-L.; Daniels, N.; Chubar, V.; Alaerts, K.; Steyaert, J.; Boets, B. Oxytocin receptor gene (OXTR) DNA methylation is associated with autism and related social traits–A systematic review. Res. Autism Spectr. Disord. 2021, 85, 101785. [Google Scholar] [CrossRef]
- Boggess, T.; Risher, W.C. Clinical and basic research investigations into the long-term effects of prenatal opioid exposure on brain development. J. Neurosci. Res. 2020, 100, 396–409. [Google Scholar] [CrossRef] [PubMed]
- Méndez, M.; Hernández-Fonseca, K.; Abate, P. Prenatal ethanol exposure and enkephalinergic neurotransmission. In Opioid Hormones; Litwack, G., Ed.; Elsevier Science: Amsterdam, The Netherlands, 2019; Volume 111, pp. 313–337. [Google Scholar]
- Nummenmaa, L.; Tuominen, L. Opioid system and human emotions. Br. J. Pharmacol. 2018, 175, 2737–2749. [Google Scholar] [CrossRef] [PubMed]
- Georges, F.; Normand, E.; Bloch, B.; Le Moine, C. Opioid receptor gene expression in the rat brain during ontogeny, with special reference to the mesostriatal system: An in situ hybridization study. Dev. Brain Res. 1998, 109, 187–199. [Google Scholar] [CrossRef]
- Pellissier, L.P.; Gandía, J.; Laboute, T.; Becker, J.A.; Le Merrer, J. μ opioid receptor, social behaviour and autism spectrum disorder: Reward matters. Br. J. Pharmacol. 2018, 175, 2750–2769. [Google Scholar] [CrossRef]
- Tarnowska, K.; Gruczyńska–Sękowska, E.; Kowalska, D.; Majewska, E.; Kozłowska, M.; Winkler, R. The opioid excess theory in autism spectrum disorders-is it worth investigating further? Crit. Rev. Food Sci. Nutr. 2021, 1–14. [Google Scholar] [CrossRef]
- Anugu, V.; Ringhisen, J.; Johnson, B. Case Report: Cause and treatment of “high opioid tone” autism. Front. Psychol. 2021, 12, 657952. [Google Scholar] [CrossRef]
- Roy, A.; Roy, M.; Deb, S.; Unwin, G.; Roy, A. Are opioid antagonists effective in attenuating the core symptoms of autism spectrum conditions in children: A systematic review. J. Intellect. Disabil. Res. 2015, 59, 293–306. [Google Scholar] [CrossRef]
- Lutz, P.-E.; Kieffer, B.L. Opioid receptors: Distinct roles in mood disorders. Trends Neurosci. 2013, 36, 195–206. [Google Scholar] [CrossRef] [Green Version]
- Monnelly, V.J.; Hamilton, R.; Chappell, F.M.; Mactier, H.; Boardman, J.P. Childhood neurodevelopment after prescription of maintenance methadone for opioid dependency in pregnancy: A systematic review and meta-analysis. Dev. Med. Child Neurol. 2019, 61, 750–760. [Google Scholar] [CrossRef] [Green Version]
- Conradt, E.; Crowell, S.E.; Lester, B.M. Early life stress and environmental influences on the neurodevelopment of children with prenatal opioid exposure. Neurobiol. Stress 2018, 9, 48–54. [Google Scholar] [CrossRef]
- Eschenroeder, A.C.; Vestal-Laborde, A.A.; Sanchez, E.S.; Robinson, S.E.; Sato-Bigbee, C. Oligodendrocyte responses to buprenorphine uncover novel and opposing roles of μ-opioid-and nociceptin/orphanin FQ receptors in cell development: Implications for drug addiction treatment during pregnancy. Glia 2012, 60, 125–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauser, K.F.; Houdi, A.A.; Turbek, C.S.; Elde, R.P.; Iii, W.M. Opioids intrinsically inhibit the genesis of mouse cerebellar granule neuron precursors in vitro: Differential impact of μ and δ receptor activation on proliferation and neurite elongation. Eur. J. Neurosci. 2000, 12, 1281–1293. [Google Scholar] [CrossRef] [PubMed]
- Narita, M.; Kuzumaki, N.; Miyatake, M.; Sato, F.; Wachi, H.; Seyama, Y.; Suzuki, T. Role of δ-opioid receptor function in neurogenesis and neuroprotection. J. Neurochem. 2006, 97, 1494–1505. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, P.J.; Tobias, S.W.; Lang, C.M.; Zagon, I.S. Chronic exposure to the opioid antagonist naltrexone during pregnancy: Maternal and offspring effects. Physiol. Behav. 1997, 62, 501–508. [Google Scholar] [CrossRef]
- Wille-Bille, A.; Bellia, F.; García, A.M.J.; Miranda-Morales, R.S.; D’Addario, C.; Pautassi, R.M. Early exposure to environmental enrichment modulates the effects of prenatal ethanol exposure upon opioid gene expression and adolescent ethanol intake. Neuropharmacology 2020, 165, 107917. [Google Scholar] [CrossRef] [PubMed]
- Zagon, I.S.; Tobias, S.W.; McLaughlin, P.J. Endogenous opioids and prenatal determinants of neuroplasticity. In Brain Plasticity; Filogamo, G., Vernadakis, A., Gremo, F., Privat, A.M., Timiras, P.S., Eds.; Springer: Boston, MA, USA, 1997; Volume 429, pp. 289–303. [Google Scholar]
- Gomes, T.M.; da Silva, D.D.; Carmo, H.; Carvalho, F.; Silva, J.P. Epigenetics and the endocannabinoid system signaling: An intricate interplay modulating neurodevelopment. Pharmacol. Res. 2020, 162, 105237. [Google Scholar] [CrossRef] [PubMed]
- Walter, L.; Stella, N. Cannabinoids and neuroinflammation. Br. J. Pharmacol. 2004, 141, 775–785. [Google Scholar] [CrossRef] [Green Version]
- Zamberletti, E.; Rubino, T.; Parolaro, D. Therapeutic potential of cannabidivarin for epilepsy and autism spectrum disorder. Pharmacol. Ther. 2021, 226, 107878. [Google Scholar] [CrossRef]
- Nashed, M.G.; Hardy, D.B.; Laviolette, S.R. Prenatal Cannabinoid Exposure: Emerging Evidence of Physiological and Neuropsychiatric Abnormalities. Front. Psychiatry 2021, 11, 1577. [Google Scholar] [CrossRef]
- Mato, S.; Del Olmo, E.; Pazos, A. Ontogenetic development of cannabinoid receptor expression and signal transduction functionality in the human brain. Eur. J. Neurosci. 2003, 17, 1747–1754. [Google Scholar] [CrossRef]
- Alexandre, J.; Carmo, H.; Carvalho, F.; Silva, J.P. Synthetic cannabinoids and their impact on neurodevelopmental processes. Addict. Biol. 2020, 25, e12824. [Google Scholar] [CrossRef] [PubMed]
- Carbone, E.; Manduca, A.; Cacchione, C.; Vicari, S.; Trezza, V. Healing autism spectrum disorder with cannabinoids: A neuroinflammatory story. Neurosci. Biobehav. Rev. 2020, 121, 128–143. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Ruiz, J.; De Fonseca, F.R.; Navarro, M.; Ramos, J. Maternal cannabinoid exposure and brain development: Changes in the ontogeny of dopaminergic neurons. In Marijuana/Cannabinoids: Neurobiology and Neurophysiology, 1st ed.; Murphy, L., Bartke, A., Eds.; CRC Press: Boca Raton, FL, USA, 2019; pp. 119–164. [Google Scholar]
- Fernández-Ruiz, J.; Berrendero, F.; Hernández, M.L.; Ramos, J.A. The endogenous cannabinoid system and brain development. Trends Neurosci. 2000, 23, 14–20. [Google Scholar] [CrossRef]
- Lisboa, S.F.; Niraula, A.; Resstel, L.B.; Guimaraes, F.S.; Godbout, J.P.; Sheridan, J.F. Repeated social defeat-induced neuroinflammation, anxiety-like behavior and resistance to fear extinction were attenuated by the cannabinoid receptor agonist WIN55, 212–2. Neuropsychopharmacology 2018, 43, 1924–1933. [Google Scholar] [CrossRef] [PubMed]
- García-Baos, A.; Alegre-Zurano, L.; Cantacorps, L.; Martín-Sánchez, A.; Valverde, O. Role of cannabinoids in alcohol-induced neuroinflammation. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2021, 104, 110054. [Google Scholar] [CrossRef]
- Nezgovorova, V.; Ferretti, C.; Taylor, B.; Shanahan, E.; Uzunova, G.; Hong, K.; Devinsky, O.; Hollander, E. Potential of cannabinoids as treatments for autism spectrum disorders. J. Psychiatr. Res. 2021, 137, 194–201. [Google Scholar] [CrossRef]
- Fusar-Poli, L.; Cavone, V.; Tinacci, S.; Concas, I.; Petralia, A.; Signorelli, M.S.; Díaz-Caneja, C.M.; Aguglia, E. Cannabinoids for people with ASD: A systematic review of published and ongoing studies. Brain Sci. 2020, 10, 572. [Google Scholar] [CrossRef]
- Chieffi, S.; Carotenuto, M.; Monda, V.; Valenzano, A.; Villano, I.; Precenzano, F.; Tafuri, D.; Salerno, M.; Filippi, N.; Nuccio, F.; et al. Orexin system: The key for a healthy life. Front. Physiol. 2017, 8, 357. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, Y.; Kanda, T.; Vogt, K.; Yanagisawa, M. Anatomical and electrophysiological development of the hypothalamic orexin neurons from embryos to neonates. J. Comp. Neurol. 2017, 525, 3809–3820. [Google Scholar] [CrossRef]
- Aran, A.; Shors, I.; Lin, L.; Mignot, E.; Schimmel, M.S.J.S. CSF levels of hypocretin-1 (orexin-A) peak during early infancy in humans. Sleep 2012, 35, 187–191. [Google Scholar] [CrossRef] [Green Version]
- Dickinson, H.; Walker, D.W.; Castillo-Melendez, M. Onset of feeding at birth—Perinatal development of the hypothalamic mechanisms that induce appetite and feeding in the newborn. Neurosci. Lett. 2008, 436, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Sokołowska, P.; Urbańska, A.; Biegańska, K.; Wagner, W.; Ciszewski, W.; Namiecińska, M.; Zawilska, J.B. Orexins protect neuronal cell cultures against hypoxic stress: An involvement of Akt signaling. J. Mol. Neurosci. 2014, 52, 48–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokołowska, P.; Urbańska, A.; Namiecińska, M.; Biegańska, K.; Zawilska, J.B. Orexins promote survival of rat cortical neurons. Neurosci. Lett. 2012, 506, 303–306. [Google Scholar] [CrossRef] [PubMed]
- Baykal, S.; Albayrak, Y.; Durankuş, F.; Güzel, S.; Abbak, Ö.; Potas, N.; Beyazyüz, M.; Karabekiroğlu, K.; Donma, M.M. Decreased serum orexin A levels in drug-naive children with attention deficit and hyperactivity disorder. Neurol. Sci. 2019, 40, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Omokawa, M.; Ayabe, T.; Nagai, T.; Imanishi, A.; Omokawa, A.; Nishino, S.; Sagawa, Y.; Shimizu, T.; Kanbayashi, T. Decline of CSF orexin (hypocretin) levels in Prader–Willi syndrome. Am. J. Med. Genet. Part A 2016, 170, 1181–1186. [Google Scholar] [CrossRef] [PubMed]
- Van Den Pol, A.N.; Patrylo, P.R.; Ghosh, P.K.; Gao, X.B. Lateral hypothalamus: Early developmental expression and response to hypocretin (orexin). J. Comp. Neurol. 2001, 433, 349–363. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Drug | Affected System | Preclinical Outcomes | References |
|---|---|---|---|
| Valproate | GABA Glutamate Serotonin Oxytocin | Auditory dysfunction relevant to ASD Autism-like behaviour Autism-like behaviour Autism-like behaviour Autism-like behaviour | [61] [62,63] [64,65] [66] |
| Diazepam | GABA | Anxiety Sex-specific changes in social interaction | [67] |
| Ketamine | GABA | Model of treatment-resistant schizophrenia | [68] |
| Selective serotonin reuptake inhibitors (SSRIs) | Serotonin Dopamine Noradrenaline Oxytocin | Neurodevelopmental changes, warning against intake during pregnancy elevated anxiety + low sociability | [69] [70] |
| Antipsychotics | Dopamine | Reduced spatial learning and reduced postnatal activity of dopaminergic neurons | [71,72] |
| Sevoflurane | Glutamate/GABA balance | Impaired learning Depression-like behaviour | [73] |
| Glucocorticoids | Dopamine | Altered sociability Anhedonia Depression-like behaviour | [74] |
| Nicotine | Dopamine Orexin | Disturbed dopaminergic activity ADHD-like behaviour Increased drug abuse | [75,76] [77] |
| Amphetamine | Dopamine | Altered activity of dopaminergic neurons | [72] |
| Cocaine | Dopamine Opioids | Disrupted development and activity of dopaminergic neurons Disrupted opioid expression in dopaminergic neurons | [72,78] [79,80] |
| Ethanol | Dopamine, Serotonin Acetylcholine Oxytocin Opioids Orexin | ADHD-like behaviour Depressive behaviour Cognitive deficit Impaired sociability Impairment in working memory and reversal learning Impaired attentional set shifting Deficit in social interaction and depression-like behaviour Increased likability of ethanol taste Ethanol abuse | [81,82,83] [84,85] [86,87] [88] [89,90] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saad, A.K.; Akour, A.; Mahboob, A.; AbuRuz, S.; Sadek, B. Role of Brain Modulators in Neurodevelopment: Focus on Autism Spectrum Disorder and Associated Comorbidities. Pharmaceuticals 2022, 15, 612. https://0-doi-org.brum.beds.ac.uk/10.3390/ph15050612
Saad AK, Akour A, Mahboob A, AbuRuz S, Sadek B. Role of Brain Modulators in Neurodevelopment: Focus on Autism Spectrum Disorder and Associated Comorbidities. Pharmaceuticals. 2022; 15(5):612. https://0-doi-org.brum.beds.ac.uk/10.3390/ph15050612
Chicago/Turabian StyleSaad, Ali K., Amal Akour, Abdulla Mahboob, Salahdein AbuRuz, and Bassem Sadek. 2022. "Role of Brain Modulators in Neurodevelopment: Focus on Autism Spectrum Disorder and Associated Comorbidities" Pharmaceuticals 15, no. 5: 612. https://0-doi-org.brum.beds.ac.uk/10.3390/ph15050612