
Artonin F Induces the Ubiquitin-Proteasomal Degradation of c-Met and Decreases Akt-mTOR Signaling

 , and
, and
Abstract
:
1. Introduction
2. Results
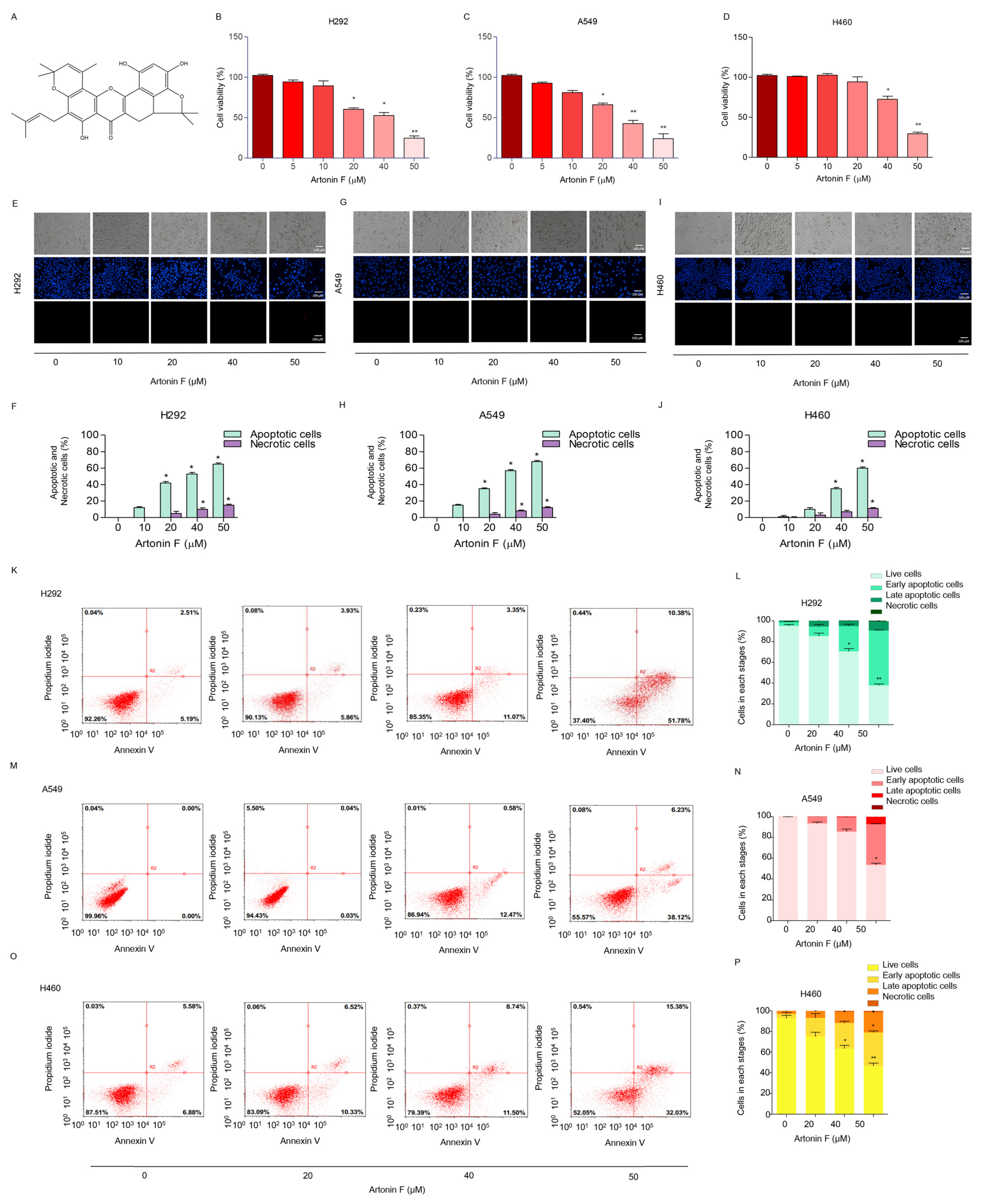
2.1. Artonin F Induces Apoptosis in Human Lung Cancer Cells
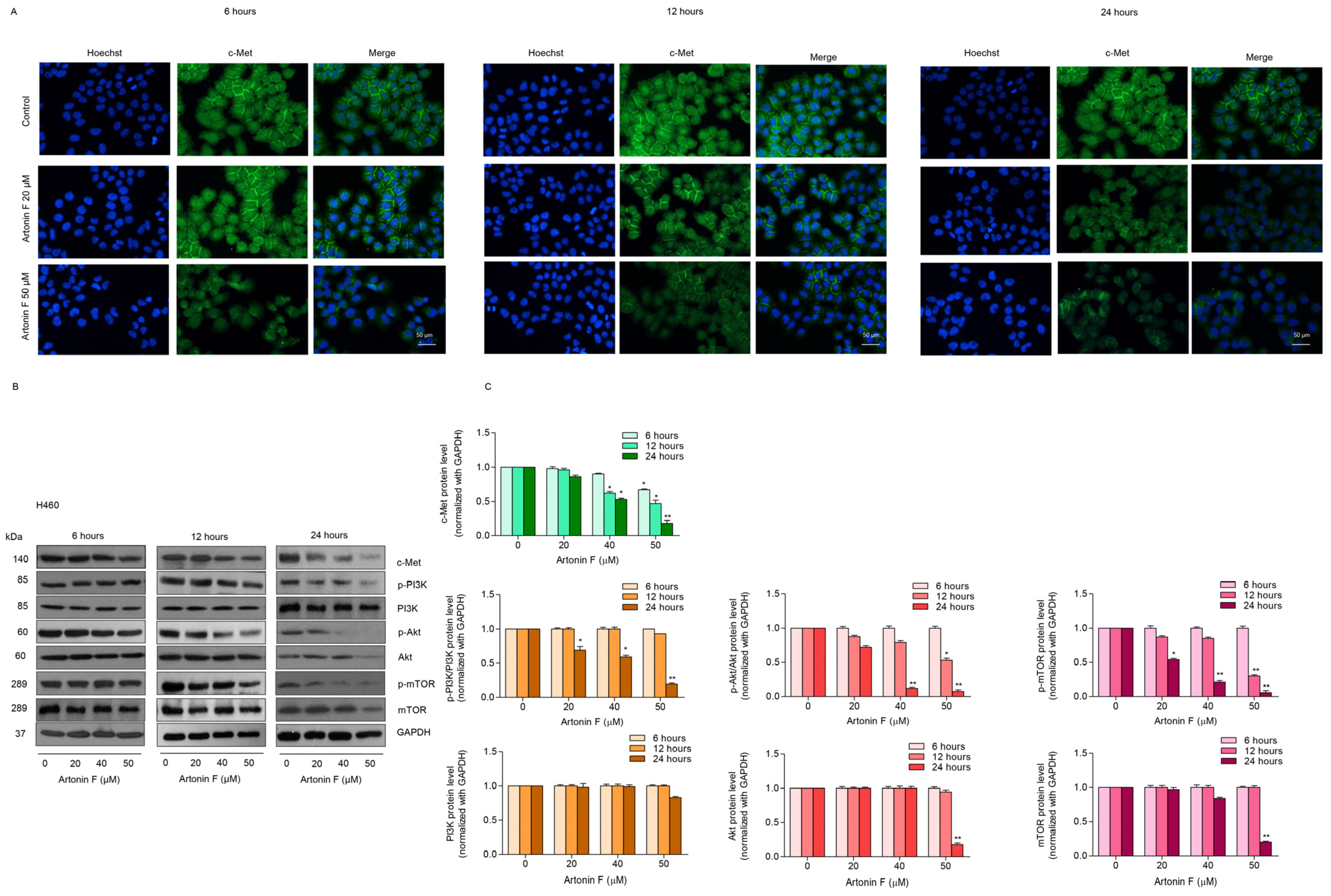
2.2. Artonin F Triggers Apoptosis Cascade through Mechanisms That Involve c-Met Downregulation
2.3. Artonin F Decreased the Levels of c-Met and p-PI3K
2.4. Artonin F Decreases c-Met Levels through the Induction of c-Met Proteasomal Degradation
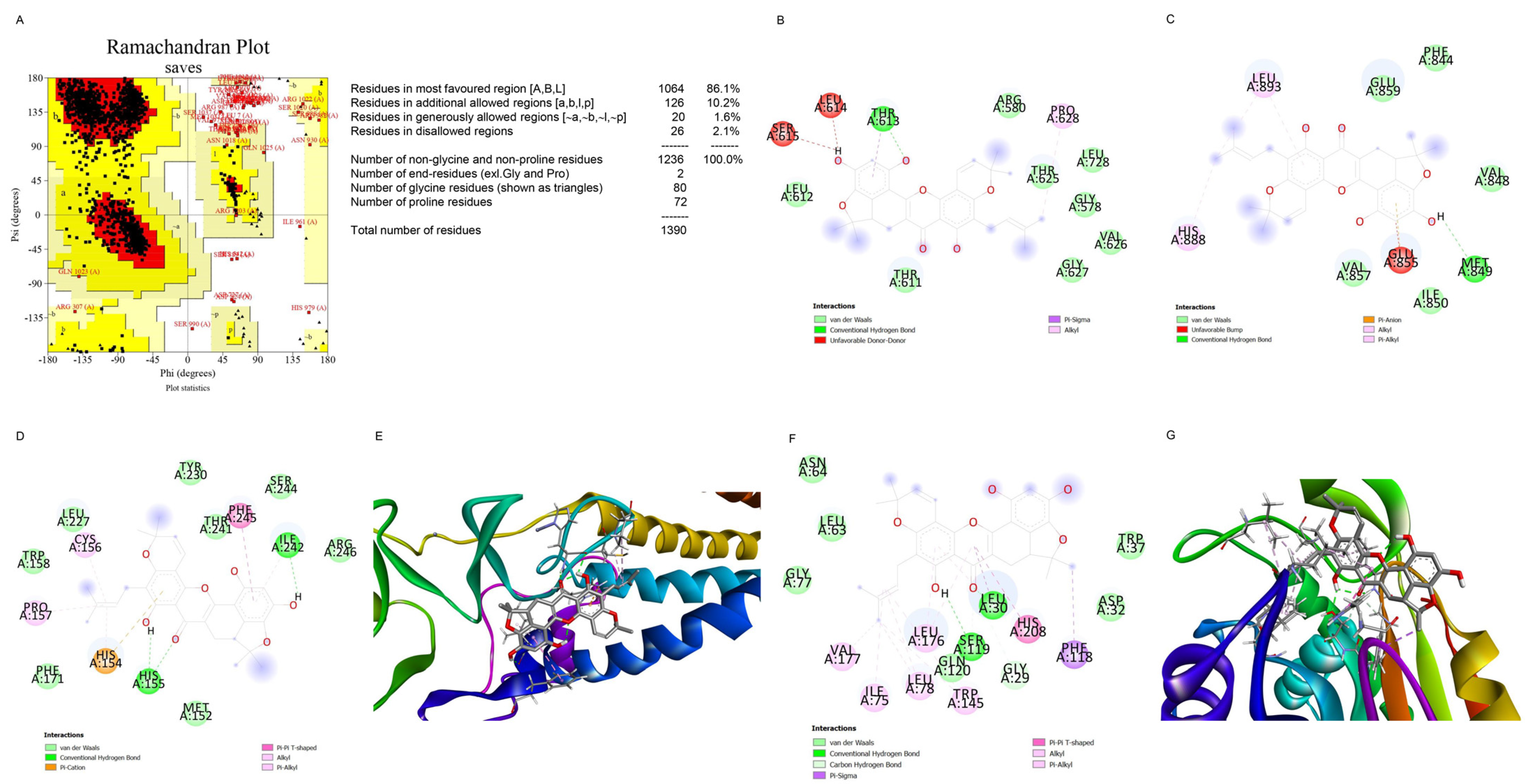
2.5. Computational Modeling Analysis of the Binding of Artonin F to c-Met
2.6. Computational Modeling Analysis of the Binding of Artonin F to USP8
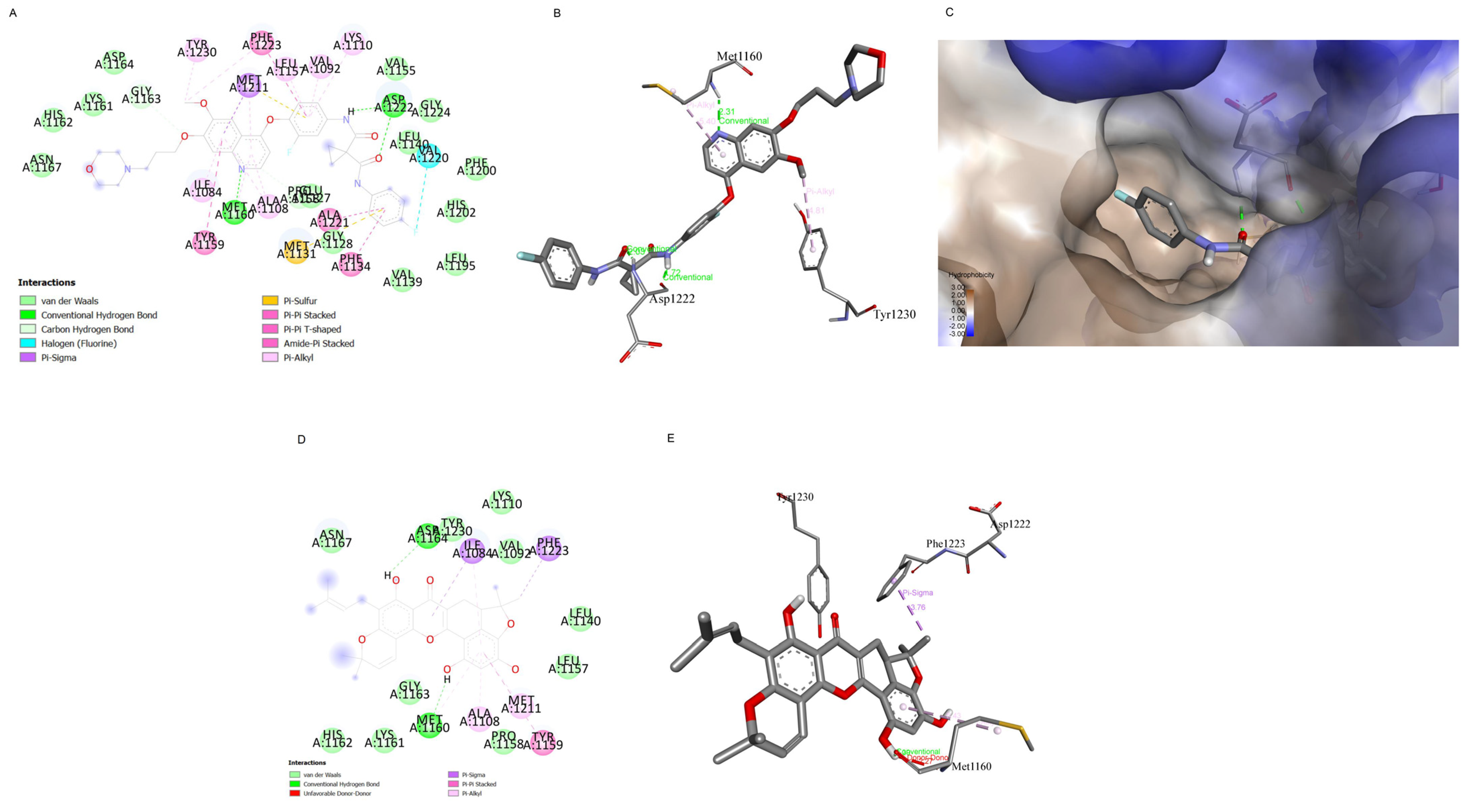
2.7. Computational Modelling Analysis of the Artonin F Interaction with c-Met Compared to Foretinib Interaction with c-Met
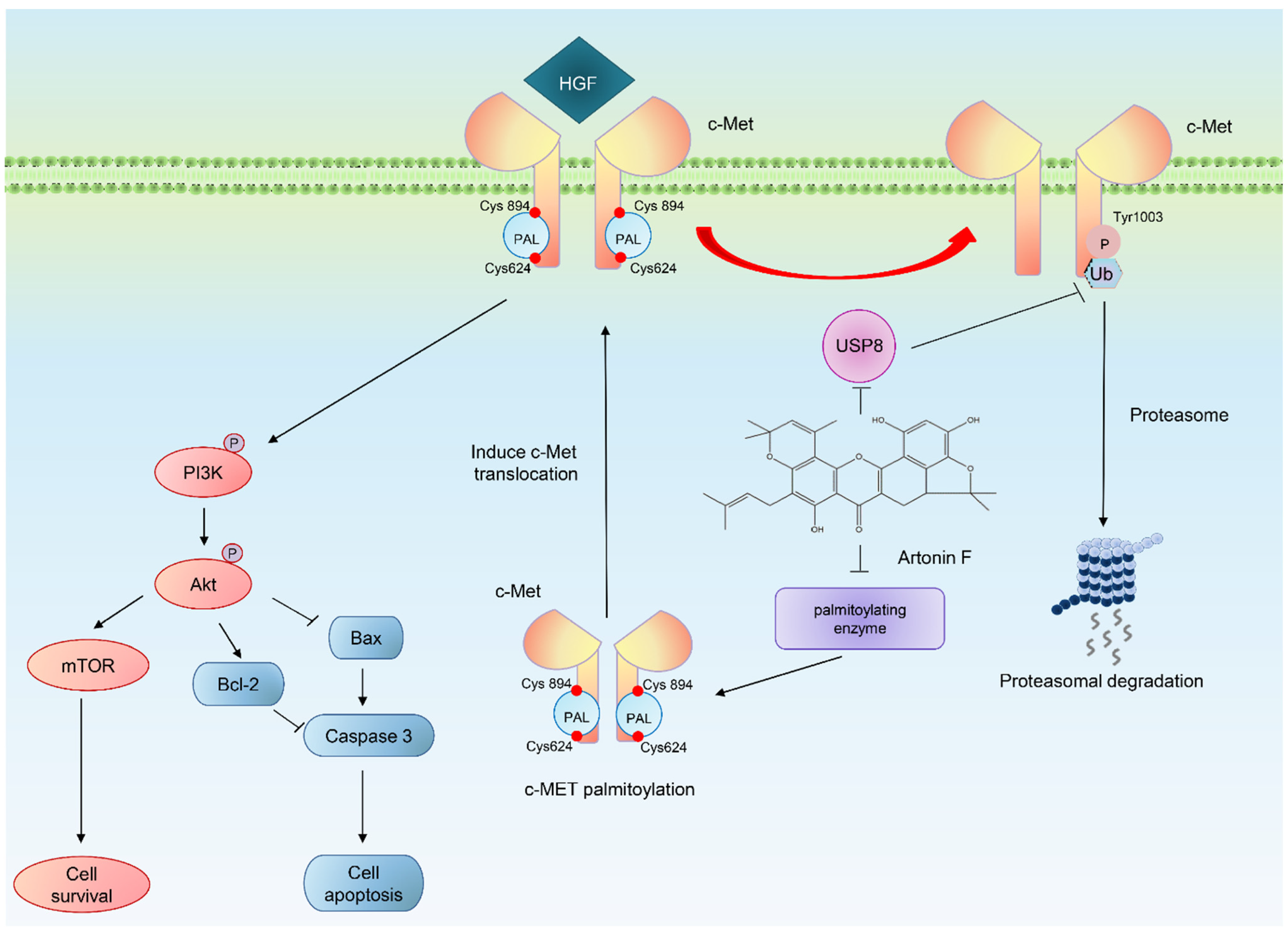
3. Discussion
4. Materials and Methods
4.1. Isolation of Artonin F
4.2. Cell Culture and Reagents
4.3. Cytotoxicity Assay
4.4. Nuclear Staining Assay
4.5. Annexin V-FITC/PI Flow Cytometry
4.6. Western Blot Analysis
4.7. Immunofluorescence
4.8. Co-Immunoprecipitation (Co-IP)
4.9. Computational Method
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lemjabbar-Alaoui, H.; Hassan, O.U.; Yang, Y.-W.; Buchanan, P. Lung cancer: Biology and treatment options. Biochim. Biophys. Acta Rev. Cancer 2015, 1856, 189–210. [Google Scholar] [CrossRef] [Green Version]
- Bhullar, K.S.; Lagarón, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-targeted cancer therapies: Progress, challenges and future directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [CrossRef] [PubMed]
- Sudhesh Dev, S.; Zainal Abidin, S.A.; Farghadani, R.; Othman, I.; Naidu, R. Receptor Tyrosine Kinases and Their Signaling Pathways as Therapeutic Targets of Curcumin in Cancer. Front. Pharmacol. 2021, 12, 772510. [Google Scholar] [CrossRef] [PubMed]
- Organ, S.L.; Tsao, M.-S. An overview of the c-MET signaling pathway. Ther. Adv. Med. Oncol. 2011, 3, 7–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landi, L.; Minuti, G.; D’Incecco, A.; Salvini, J.; Cappuzzo, F. MET overexpression and gene amplification in NSCLC: A clinical perspective. Lung Cancer 2013, 4, 15–25. [Google Scholar]
- Zhang, Y.; Xia, M.; Jin, K.; Wang, S.; Wei, H.; Fan, C.; Wu, Y.; Li, X.; Li, X.; Li, G.; et al. Function of the c-Met receptor tyrosine kinase in carcinogenesis and associated therapeutic opportunities. Mol. Cancer 2018, 17, 45. [Google Scholar] [CrossRef]
- Venepalli, N.K.; Goff, L. Targeting the HGF-cMET Axis in Hepatocellular Carcinoma. Int. J. Hepatol. 2013, 2013, 341636. [Google Scholar] [CrossRef] [Green Version]
- Liang, H.; Wang, M. MET Oncogene in Non-Small Cell Lung Cancer: Mechanism of MET Dysregulation and Agents Targeting the HGF/c-Met Axis. Onco Targets Ther. 2020, 13, 2491–2510. [Google Scholar] [CrossRef] [Green Version]
- Jung, H.-Y.; Joo, H.-J.; Park, J.K.; Kim, Y.H. The Blocking of c-Met Signaling Induces Apoptosis through the Increase of p53 Protein in Lung Cancer. Cancer Res. Treat. 2012, 44, 251–261. [Google Scholar] [CrossRef]
- Abrami, L.; Leppla, S.H.; van der Goot, F.G. Receptor palmitoylation and ubiquitination regulate anthrax toxin endocytosis. J. Cell Biol. 2006, 172, 309–320. [Google Scholar] [CrossRef]
- Coleman, D.T.; Cardelli, J.A. Abstract A83: c-Met requires palmitoylation for proper stability and trafficking in cancer cells. Cancer Res. 2014, 73, 73–83. [Google Scholar]
- Coleman, D.T.; Gray, A.L.; Kridel, S.J.; Cardelli, J.A. Palmitoylation regulates the intracellular trafficking and stability of c-Met. Oncotarget 2016, 7, 32664–32677. [Google Scholar] [CrossRef] [PubMed]
- Jeffers, M.; Taylor, G.A.; Weidner, K.M.; Omura, S.; Vande Woude, G.F. Degradation of the Met tyrosine kinase receptor by the ubiquitin-proteasome pathway. Mol. Cell. Biol. 1997, 17, 799–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Critchley, W.R.; Pellet-Many, C.; Ringham-Terry, B.; Harrison, M.A.; Zachary, I.C.; Ponnambalam, S. Receptor Tyrosine Kinase Ubiquitination and De-Ubiquitination in Signal Transduction and Receptor Trafficking. Cells 2018, 7, 22. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.T.; Chen, F.; Chen, H. The oncogenic role of ubiquitin specific peptidase (USP8) and its signaling pathways targeting for cancer therapeutics. Arch. Biochem. Biophys. 2021, 701, 108811–108821. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y.M.; Lee, S.B.; Choi, J.; Suh, H.-Y.; Shim, S.; Song, Y.-J.; Kim, B.; Lee, J.M.; Oh, S.J.; Jeong, Y.; et al. USP8 modulates ubiquitination of LRIG1 for Met degradation. Sci. Rep. 2014, 4, 4980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salgia, R. Role of c-Met in cancer: Emphasis on lung cancer. Semin. Oncol. 2009, 36, 52–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prashanth, S.J.; Suresh, D.; Potty, V.H.; Sadananda Maiya, P. In Vitro Anticancer and Hepatoprotective Activities of Artocarpus gomezianus. Int. J. Med. Sci. 2014, 7, 18–23. [Google Scholar] [CrossRef]
- Losuwannarak, N.; Sritularak, B.; Chanvorachote, P. Cycloartobiloxanthone Induces Human Lung Cancer Cell Apoptosis via Mitochondria-dependent Apoptotic Pathway. In Vivo 2018, 32, 71–78. [Google Scholar]
- Bailly, C. Anticancer mechanism of artonin E and related prenylated flavonoids from the medicinal plant Artocarpus elasticus. Asian J. Nat. Prod. Biochem. 2021, 19, 44–56. [Google Scholar] [CrossRef]
- Ren, Y.; de Blanco, E.J.C.; Fuchs, J.R.; Soejarto, D.D.; Burdette, J.E.; Swanson, S.M.; Kinghorn, A.D. Potential Anticancer Agents Characterized from Selected Tropical Plants. J. Nat. Prod. 2019, 82, 657–679. [Google Scholar] [CrossRef]
- França, F.; Silva, P.M.A.; Soares, J.X.; Henriques, A.C.; Loureiro, D.R.P.; Azevedo, C.M.G.; Afonso, C.M.M.; Bousbaa, H. A Pyranoxanthone as a Potent Antimitotic and Sensitizer of Cancer Cells to Low Doses of Paclitaxel. Molecules 2020, 25, 5845. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.A.; Ramli, F.; Karimian, H.; Dehghan, F.; Nordin, N.; Ali, H.M.; Mohan, S.; Hashim, N.M. Artonin E Induces Apoptosis via Mitochondrial Dysregulation in SKOV-3 Ovarian Cancer Cells. PLoS ONE 2016, 11, 0151466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yangnok, K.; Innajak, S.; Sawasjirakij, R.; Mahabusarakam, W.; Watanapokasin, R. Effects of Artonin E on Cell Growth Inhibition and Apoptosis Induction in Colon Cancer LoVo and HCT116 Cells. Molecules 2022, 27, 2095. [Google Scholar] [CrossRef] [PubMed]
- Plaibua, K.; Pongrakhananon, V.; Chunhacha, P.; Sritularak, B.; Chanvorachote, P. Effects of Artonin E on Migration and Invasion Capabilities of Human Lung Cancer Cells. Anticancer Res. 2013, 33, 3079–3088. [Google Scholar]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef]
- Park, K.C.; Richardson, D.R. The c-MET oncoprotein: Function, mechanisms of degradation and its targeting by novel anti-cancer agents. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2020, 1864, 129650. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Avvakumov, G.V.; Walker, J.R.; Xue, S.; Finerty, P.J., Jr.; Mackenzie, F.; Newman, E.M.; Dhe-Paganon, S. Amino-terminal dimerization, NRDP1-rhodanese interaction, and inhibited catalytic domain conformation of the ubiquitin-specific protease 8 (USP8). J. Biol. Chem. 2006, 281, 38061–38070. [Google Scholar] [CrossRef] [Green Version]
- Bukowski, K.; Kciuk, M.; Kontek, R. Mechanisms of Multidrug Resistance in Cancer Chemotherapy. Int. J. Mol. Sci. 2020, 21, 3233. [Google Scholar] [CrossRef] [PubMed]
- Pistritto, G.; Trisciuoglio, D.; Ceci, C.; Garufi, A.; D’Orazi, G. Apoptosis as anticancer mechanism: Function and dysfunction of its modulators and targeted therapeutic strategies. Aging 2016, 8, 603–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Li, N.; Wang, T.-M.; Di, L. Natural Products with Activity against Lung Cancer: A Review Focusing on the Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 10827. [Google Scholar] [CrossRef] [PubMed]
- Wlodkowic, D.; Telford, W.; Skommer, J.; Darzynkiewicz, Z. Apoptosis and beyond: Cytometry in studies of programmed cell death. Methods Cell Biol. 2011, 103, 55–98. [Google Scholar]
- Pfeffer, C.M.; Singh, A.T.K. Apoptosis: A Target for Anticancer Therapy. Int. J. Mol. Sci. 2018, 19, 448. [Google Scholar] [CrossRef] [Green Version]
- MacKenzie, S.H.; Clark, A.C. Targeting cell death in tumors by activating caspases. Curr. Cancer Drug Targets 2008, 8, 98–109. [Google Scholar]
- Lam, B.Q.; Dai, L.; Qin, Z. The role of HGF/c-MET signaling pathway in lymphoma. J. Hematol. Oncol. 2016, 9, 135. [Google Scholar] [CrossRef] [Green Version]
- Baldanzi, G.; Graziani, A. Physiological Signaling and Structure of the HGF Receptor MET. Biomedicines 2015, 3, 1–31. [Google Scholar] [CrossRef] [Green Version]
- Usatyuk, P.V.; Fu, P.; Mohan, V.; Epshtein, Y.; Jacobson, J.R.; Gomez-Cambronero, J.; Wary, K.K.; Bindokas, V.; Dudek, S.M.; Salgia, R.; et al. Role of c-Met/Phosphatidylinositol 3-Kinase (PI3k)/Akt Signaling in Hepatocyte Growth Factor (HGF)-mediated Lamellipodia Formation, Reactive Oxygen Species (ROS) Generation, and Motility of Lung Endothelial Cells. J. Biol. Chem. 2014, 289, 13476–13491. [Google Scholar] [CrossRef] [Green Version]
- Damghani, T.; Moosavi, F.; Khoshneviszadeh, M.; Mortazavi, M.; Pirhadi, S.; Kayani, Z.; Saso, L.; Edraki, N.; Firuzi, O. Imidazopyridine hydrazone derivatives exert antiproliferative effect on lung and pancreatic cancer cells and potentially inhibit receptor tyrosine kinases including c-Met. Sci. Rep. 2021, 11, 3644. [Google Scholar] [CrossRef]
- Tang, X.-l.; Yan, L.; Zhu, L.; Jiao, D.-m.; Chen, J.; Chen, Q.-y. Salvianolic acid A reverses cisplatin resistance in lung cancer A549 cells by targeting c-met and attenuating Akt/mTOR pathway. J. Pharmacol. Sci. 2017, 135, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Theodore, F.L. Foretinib (XL880): c-MET inhibitor with activity in papillary renal cell cancer. Curr. Oncol. Rep. 2013, 2, 83–90. [Google Scholar]
- Petpiroon, N.; Sritularak, B.; Chanvorachote, P. Phoyunnanin E inhibits migration of non-small cell lung cancer cells via suppression of epithelial-to-mesenchymal transition and integrin αv and integrin β3. BMC Complement. Altern. Med. 2017, 17, 553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krieger, E.; Joo, K.; Lee, J.; Lee, J.; Raman, S.; Thompson, J.; Tyka, M.; Baker, D.; Karplus, K. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins 2009, 77, 114–122. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Collie, G.W.; Koh, C.M.; O’Neill, D.J.; Stubbs, C.J.; Khurana, P.; Eddershaw, A.; Snijder, A.; Mauritzson, F.; Barlind, L.; Dale, I.L.; et al. Structural and Molecular Insight into Resistance Mechanisms of First Generation cMET Inhibitors. ACS Med. Chem. Lett. 2019, 9, 1322–1327. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Affinity (kcal/mol) | Estimated Ki (μM) | Type of Interaction | Interacting Residue(s) |
|---|---|---|---|---|
| hDHHC20 palmitoyl transferase | −8.03 | 1.30 | Hydrogen bond | His155, Ile242 |
| Pi Cation | His154 | |||
| Pi–Pi T-Shaped | Phe245 | |||
| Alkyl and Pi–Alkyl | Pro157, Cys156, His154, Ile242 | |||
| Van der Waals | Trp158, Leu227, Tyr230, Thr241, Ser244, Arg246, Met152, Phe171 | |||
| Acyl-protein thioesterase 1 | −9.27 | 0.16 | Hydrogen bond | Leu30, Ser119 |
| Pi–Sigma | Phe118, Leu176, Leu30 | |||
| Pi–Pi T-Shaped | His208 | |||
| Alkyl | Ile75, Val177, Leu78, Trp145, Leu176 | |||
| Van der Waals | Leu63, Asn64, Gly77, Gln120, Gly29, Trp37, Asp32, Ser209 |
| Binding Affinity (kcal/mol) | Interacting Residues |
|---|---|
| −6.8 | Conventional Hydrogen Bond: Asp1084 [bond length = 1.93 Å] Pi interaction: Tyr1060, His1067, Asp1084, Asp1085 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soonnarong, R.; Putra, I.D.; Sriratanasak, N.; Sritularak, B.; Chanvorachote, P. Artonin F Induces the Ubiquitin-Proteasomal Degradation of c-Met and Decreases Akt-mTOR Signaling. Pharmaceuticals 2022, 15, 633. https://0-doi-org.brum.beds.ac.uk/10.3390/ph15050633
Soonnarong R, Putra ID, Sriratanasak N, Sritularak B, Chanvorachote P. Artonin F Induces the Ubiquitin-Proteasomal Degradation of c-Met and Decreases Akt-mTOR Signaling. Pharmaceuticals. 2022; 15(5):633. https://0-doi-org.brum.beds.ac.uk/10.3390/ph15050633
Chicago/Turabian StyleSoonnarong, Rapeepun, Ismail Dwi Putra, Nicharat Sriratanasak, Boonchoo Sritularak, and Pithi Chanvorachote. 2022. "Artonin F Induces the Ubiquitin-Proteasomal Degradation of c-Met and Decreases Akt-mTOR Signaling" Pharmaceuticals 15, no. 5: 633. https://0-doi-org.brum.beds.ac.uk/10.3390/ph15050633