
DNA-DAPI Interaction-Based Method for Cell Proliferation Rate Evaluation in 3D Structures

 , ,
, ,
Abstract
:1. Introduction
2. Methods
2.1. Materials
2.2. Cell Culture
2.3. Scaffolds Printing
2.4. Assessment of the Background Fluorescence of Lysis Buffers
2.5. Fluorescence Emission Sweep
2.6. Immunofluorescence Staining
2.7. Calibration Curve
2.8. Assessment of the Cell Number in 3D Surroundings
2.9. Statistical Analysis
3. Results
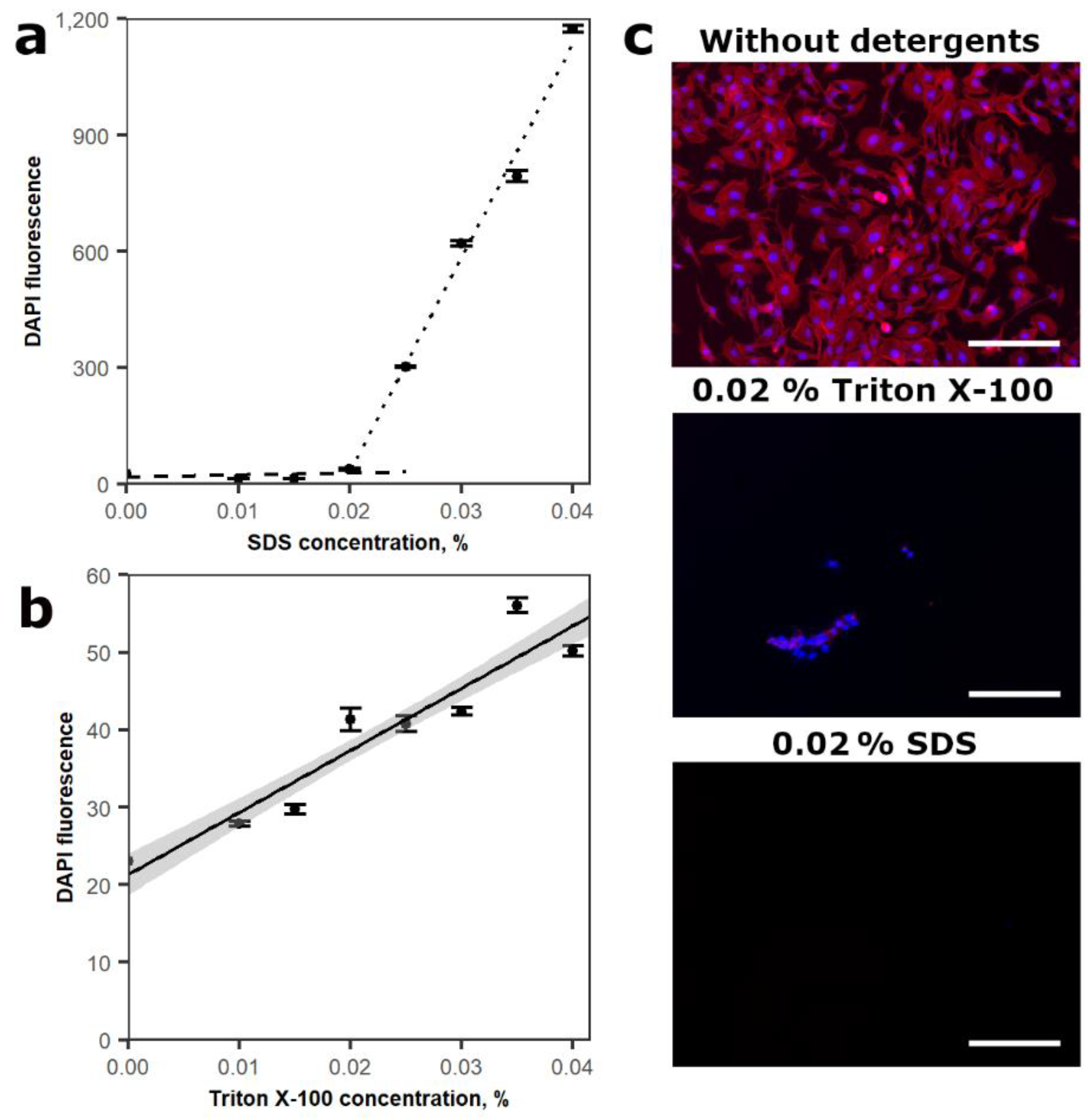
3.1. Assessment of Lysis Buffer Composition
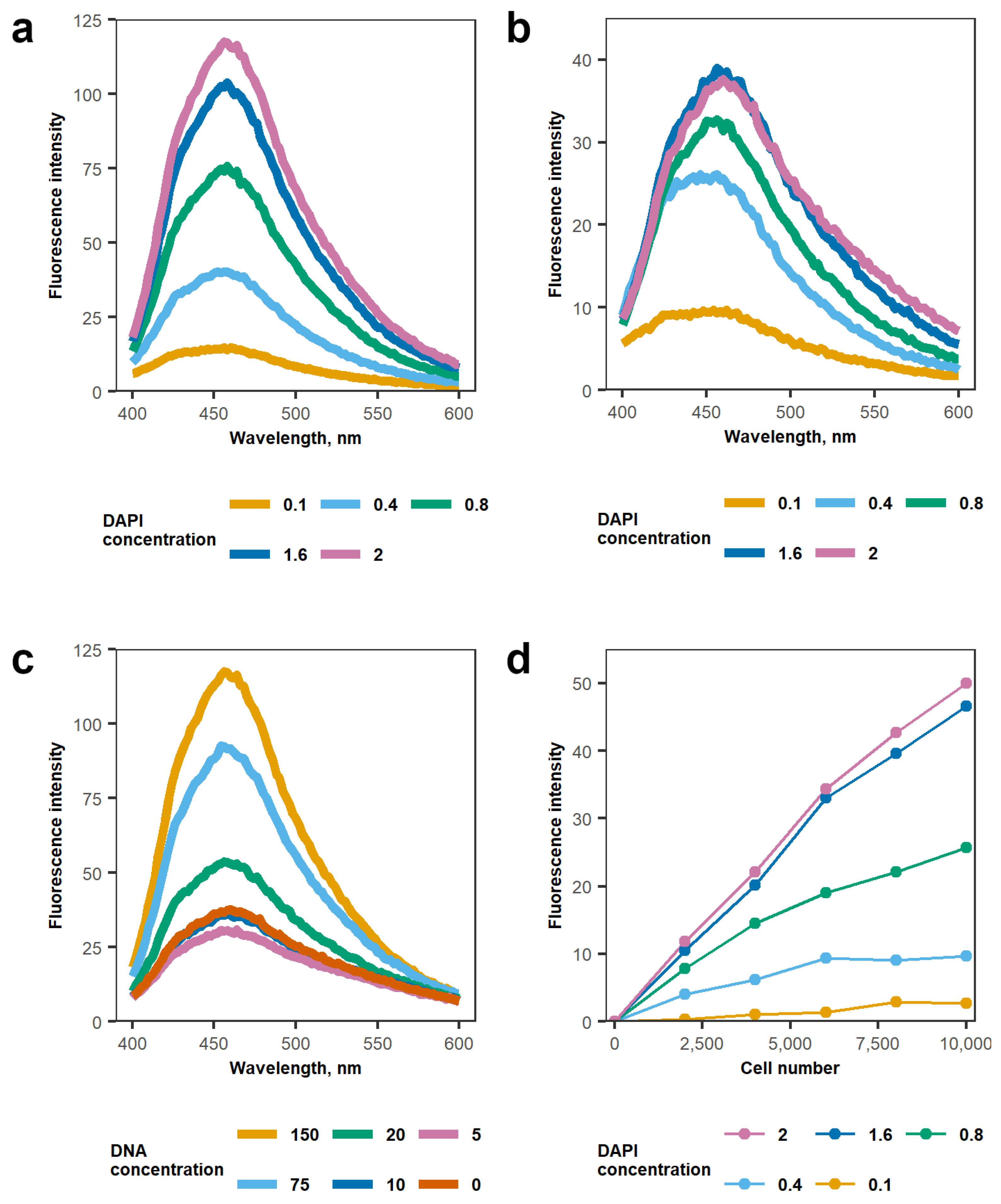
3.2. Determination of DAPI Working Concentration
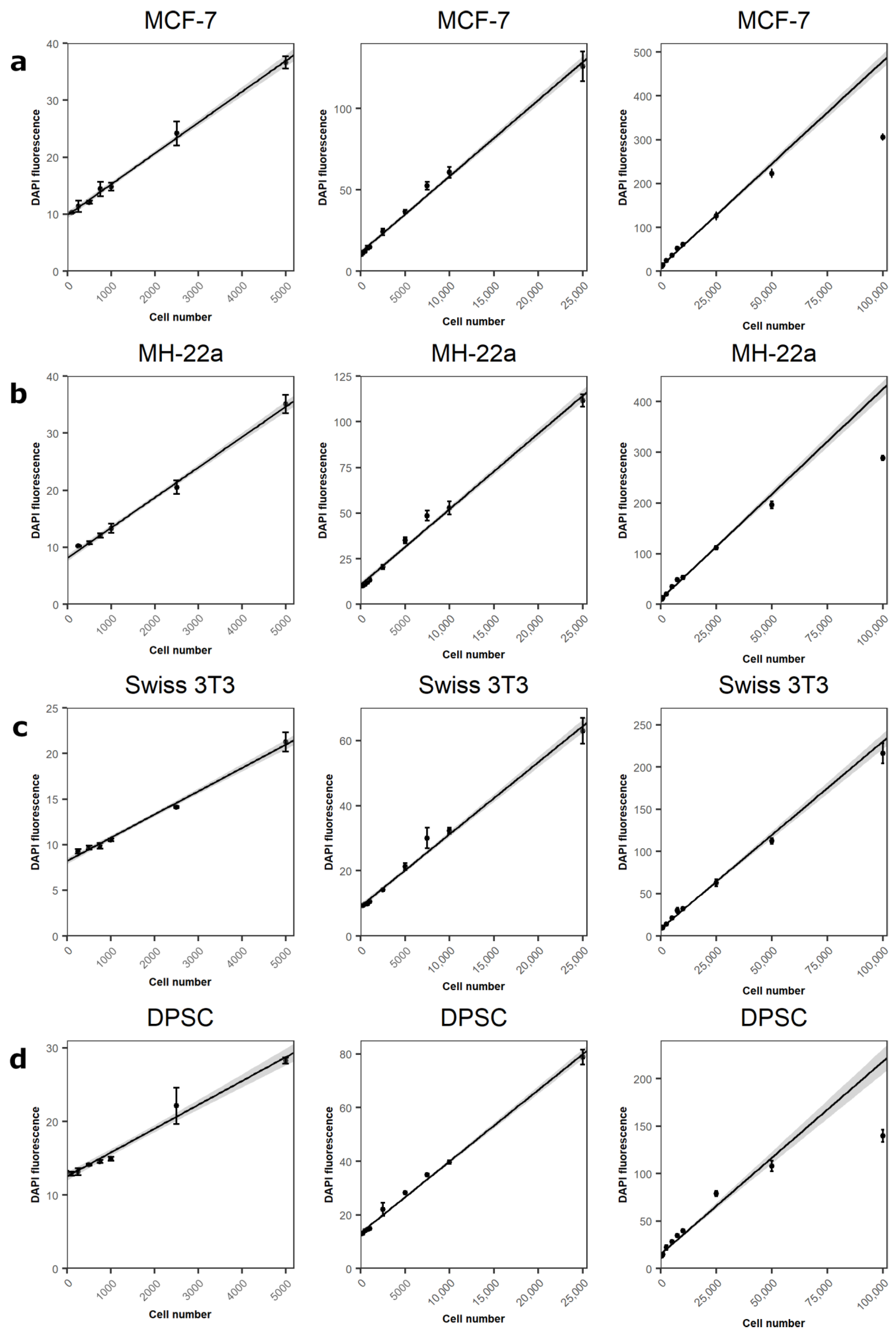
3.3. Assessment of the Method with Various Cell Lines
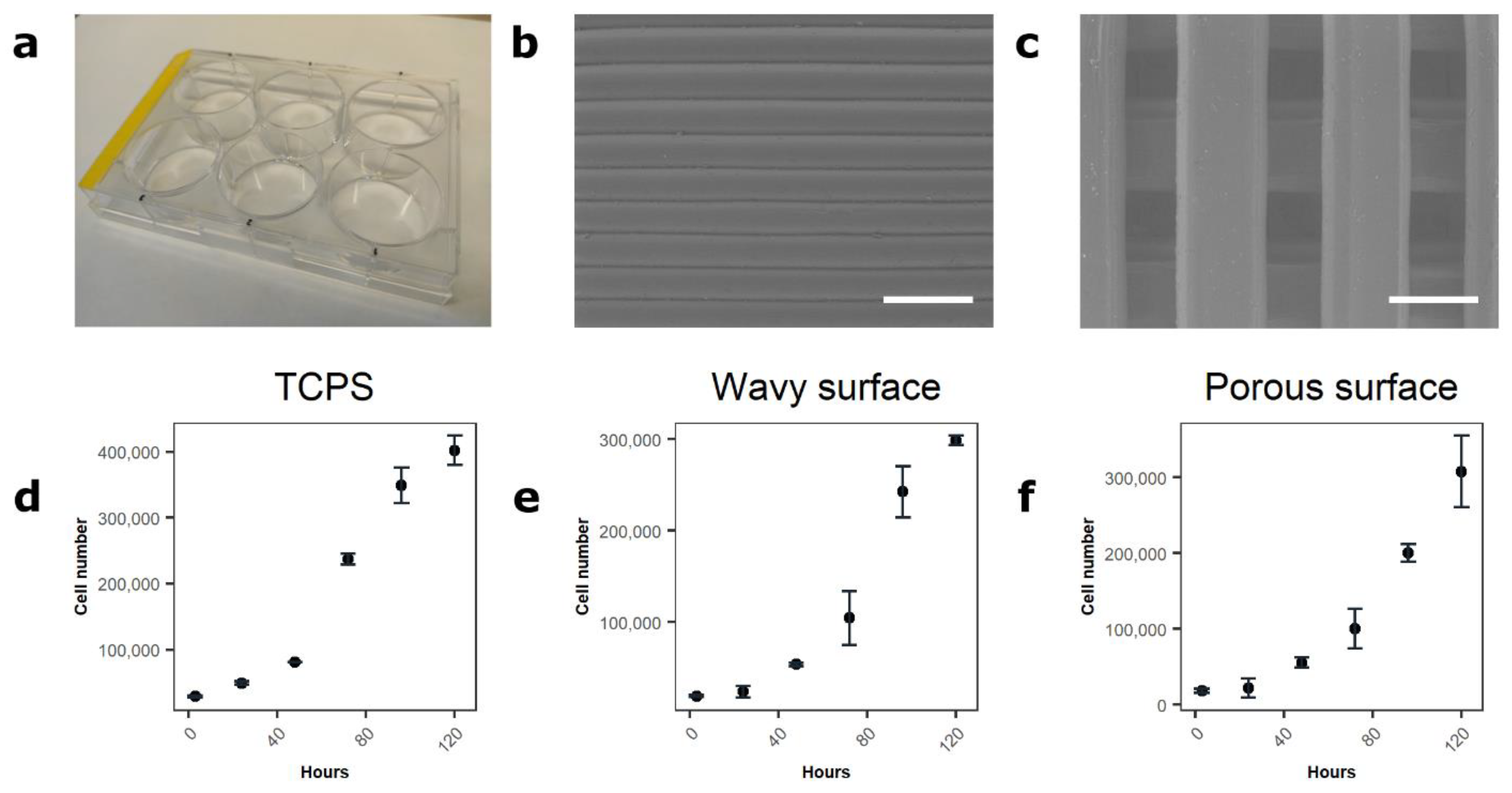
3.4. Assessment of the Method in 2D, 2.5D, and 3D Environments
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Knapp, B.; Rebhan, I.; Kumar, A.; Matula, P.; Kiani, N.A.; Binder, M.; Erfle, H.; Rohr, K.; Eils, R.; Bartenschlager, R.; et al. Normalizing for individual cell population context in the analysis of high-content cellular screens. BMC Bioinform. 2011, 12, 485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Choung, S.; Sun, R.X.; Ung, N.; Hashemi, N.; Fong, E.J.; Lau, R.; Spiller, E.; Gasho, J.; Foo, J.; et al. Comparison of Cell and Organoid-Level Analysis of Patient-Derived 3D Organoids to Evaluate Tumor Cell Growth Dynamics and Drug Response. SLAS Discov. 2020, 25, 744–754. [Google Scholar] [CrossRef] [PubMed]
- Śliwka, L.; Wiktorska, K.; Suchocki, P.; Milczarek, M.; Mielczarek, S.; Lubelska, K.; Cierpiał, T.; Łyżwa, P.; Kiełbasiński, P.; Jaromin, A.; et al. The Comparison of MTT and CVS Assays for the Assessment of Anticancer Agent Interactions. PLoS ONE 2016, 11, e0155772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghali, O.; Broux, O.; Falgayrac, G.; Haren, N.; Van Leeuwen, J.; Penel, G.; Hardouin, P.; Chauveau, C. Dexamethasone in osteogenic medium strongly induces adipocyte differentiation of mouse bone marrow stromal cells and increases osteoblast differentiation. BMC Cell Biol. 2015, 16, 9. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Kim, B.-Y.; Choi, S. On-Chip Cell Staining and Counting Platform for the Rapid Detection of Blood Cells in Cerebrospinal Fluid. Sensors 2018, 18, 1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Zhou, J.; Xu, Y. Study of the in vitro cytotoxicity testing of medical devices (Review). Biomed. Rep. 2015, 3, 617–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romar, G.A.; Kupper, T.S.; DiVito, S.J. Research Techniques Made Simple: Techniques to Assess Cell Proliferation. J. Investig. Dermatol. 2016, 136, e1–e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loh, Q.L.; Choong, C. Three-Dimensional Scaffolds for Tissue Engineering Applications: Role of Porosity and Pore Size. Tissue Eng. Part B Rev. 2013, 19, 485–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neto, A.S.; Ferreira, J.M.F. Synthetic and Marine-Derived Porous Scaffolds for Bone Tissue Engineering. Materials 2018, 11, 1702. [Google Scholar] [CrossRef] [Green Version]
- Abbasi, N.; Hamlet, S.; Love, R.M.; Nguyen, N.-T. Porous scaffolds for bone regeneration. J. Sci. Adv. Mater. Devices 2020, 5, 1–9. [Google Scholar] [CrossRef]
- Chen, Z.; Yan, X.; Yin, S.; Liu, L.; Liu, X.; Zhao, G.; Ma, W.; Qi, W.; Ren, Z.; Liao, H.; et al. Influence of the pore size and porosity of selective laser melted Ti6Al4V ELI porous scaffold on cell proliferation, osteogenesis and bone ingrowth. Mater. Sci. Eng. C 2020, 106, 110289. [Google Scholar] [CrossRef] [PubMed]
- Alksne, M.; Simoliunas, E.; Kalvaityte, M.; Skliutas, E.; Rinkunaite, I.; Gendviliene, I.; Baltriukiene, D.; Rutkunas, V.; Bukelskiene, V. The effect of larger than cell diameter polylactic acid surface patterns on osteogenic differentiation of rat dental pulp stem cells. J. Biomed. Mater. Res. Part A 2019, 107A, 174–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gantenbein-Ritter, B.; Potier, E.; Zeiter, S.; Van Der Werf, M.; Sprecher, C.M.; Ito, K. Accuracy of Three Techniques to Determine Cell Viability in 3D Tissues or Scaffolds. Tissue Eng. Part C Methods 2008, 14, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Hu, N.; Wei, X.; Gong, L.; Zhang, B.; Wan, H.; Wang, P. 3D cell-based biosensor for cell viability and drug assessment by 3D electric cell/matrigel-substrate impedance sensing. Biosens. Bioelectron. 2019, 130, 344–351. [Google Scholar] [CrossRef]
- Divieto, C.; Sassi, M.P. A first approach to evaluate the cell dose in highly porous scaffolds by using a nondestructive metabolic method. Futur. Sci. OA 2015, 1, fso.15.58. [Google Scholar] [CrossRef]
- Riss, T.L.; Moravec, R.A.; Niles, A.L.; Duellman, S.; Benink, H.A.; Worzella, T.J.; Minor, L. Cell Viability Assays. Assay Guidance Manual; Eli Lilly & Company: Indianapolis, IN, USA; National Center for Advancing Translational Sciences: Bethesda, MD, USA, 2016. [Google Scholar]
- Qi, L.; Knapton, E.K.; Zhang, X.; Zhang, T.; Gu, C.; Zhao, Y. Pre-culture Sudan Black B treatment suppresses autofluorescence signals emitted from polymer tissue scaffolds. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Gao, Z.; Chen, Y.; Guan, M.-X. The role of mitochondria in osteogenic, adipogenic and chondrogenic differentiation of mesenchymal stem cells. Protein Cell 2017, 8, 439–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, P.; Nagarajan, A.; Uchil, P.D. Analysis of Cell Viability by the MTT Assay. Cold Spring Harb. Protoc. 2018, 2018, 469–471. [Google Scholar] [CrossRef]
- Stepanenko, A.A.; Dmitrenko, V.V. Pitfalls of the MTT assay: Direct and off-target effects of inhibitors can result in over/underestimation of cell viability. Gene 2015, 574, 193–203. [Google Scholar] [CrossRef]
- Forsey, R.W.; Chaudhuri, J.B. Validity of DNA analysis to determine cell numbers in tissue engineering scaffolds. Biotechnol. Lett. 2009, 31, 819–823. [Google Scholar] [CrossRef]
- Silva, L.P.; Lorenzi, P.L.; Purwaha, P.; Yong, V.; Hawke, D.H.; Weinstein, J.N. Measurement of DNA Concentration as a Normalization Strategy for Metabolomic Data from Adherent Cell Lines. Anal. Chem. 2013, 85, 9536–9542. [Google Scholar] [CrossRef] [Green Version]
- Hayton, S.; Maker, G.L.; Mullaney, I.; Trengove, R.D. Experimental design and reporting standards for metabolomics studies of mammalian cell lines. Cell. Mol. Life Sci. 2017, 74, 4421–4441. [Google Scholar] [CrossRef]
- Ng, K.W.; Leong, D.T.; Hutmacher, D.W. The Challenge to Measure Cell Proliferation in Two and Three Dimensions. Tissue Eng. 2005, 11, 182–191. [Google Scholar] [CrossRef]
- Orellana, E.A.; Kasinski, A.L. Sulforhodamine B (SRB) Assay in Cell Culture to Investigate Cell Proliferation. Bio-Protocol 2016, 6, e1984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, A.; Romero, R.; Petty, H.R. An enzymatic colorimetric assay for glucose-6-phosphate. Anal. Biochem. 2011, 419, 266–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaja, S.; Payne, A.J.; Naumchuk, Y.; Koulen, P. Quantification of Lactate Dehydrogenase for Cell Viability Testing Using Cell Lines and Primary Cultured Astrocytes. Curr. Protoc. Toxicol. 2017, 72, 2.26.1–2.26.10. [Google Scholar] [CrossRef] [Green Version]
- Gomes, C.J.; Harman, M.W.; Centuori, S.M.; Wolgemuth, C.W.; Martinez, J.D. Measuring DNA content in live cells by fluorescence microscopy. Cell Div. 2018, 13, 6. [Google Scholar] [CrossRef] [PubMed]
- Ruoß, M.; Kieber, V.; Rebholz, S.; Linnemann, C.; Rinderknecht, H.; Häussling, V.; Häcker, M.; Damink, L.H.H.O.; Ehnert, S.; Nussler, A.K. Cell-Type-Specific Quantification of a Scaffold-Based 3D Liver Co-Culture. Methods Protoc. 2019, 3, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivero, R.E.; Capella, V.; Liaudat, A.C.; Bosch, P.; Barbero, C.A.; Rodríguez, N.; Rivarola, C.R. Mechanical and physicochemical behavior of a 3D hydrogel scaffold during cell growth and proliferation. RSC Adv. 2020, 10, 5827–5837. [Google Scholar] [CrossRef]
- Quent, V.M.; Loessner, D.L.; Friis, T.E.; Reichert, J.C.; Hutmacher, D.W. Discrepancies between metabolic activity and DNA content as tool to assess cell proliferation in cancer research. J. Cell. Mol. Med. 2010, 14, 1003–1013. [Google Scholar] [CrossRef] [Green Version]
- Nava, M.M.; Miroshnikova, Y.A.; Biggs, L.C.; Whitefield, D.B.; Metge, F.; Boucas, J.; Vihinen, H.; Jokitalo, E.; Li, X.; Arcos, J.M.G.; et al. Heterochromatin-Driven Nuclear Softening Protects the Genome against Mechanical Stress-Induced Damage. Cell 2020, 181, 800–817.e22. [Google Scholar] [CrossRef]
- Ligasová, A.; Koberna, K. Quantification of fixed adherent cells using a strong enhancer of the fluorescence of DNA dyes. Sci. Rep. 2019, 9, 1–12. [Google Scholar]
- Malinauskas, M.; Rekštytė, S.; Lukoševičius, L.; Butkus, S.; Balčiūnas, E.; Pečiukaitytė, M.; Baltriukienė, D.; Bukelskienė, V.; Butkevičius, A.; Kucevičius, P.; et al. 3D Microporous Scaffolds Manufactured via Combination of Fused Filament Fabrication and Direct Laser Writing Ablation. Micromachines 2014, 5, 839–858. [Google Scholar] [CrossRef] [Green Version]
- Gillooly, J.F.; Hein, A.; Damiani, R. Nuclear DNA Content Varies with Cell Size across Human Cell Types. Cold Spring Harb. Perspect. Biol. 2015, 7, a019091. [Google Scholar] [CrossRef] [Green Version]
- Mirzayans, R.; Andrais, B.; Murray, D. Roles of Polyploid/Multinucleated Giant Cancer Cells in Metastasis and Disease Relapse Following Anticancer Treatment. Cancers 2018, 10, 118. [Google Scholar] [CrossRef] [Green Version]
- Bhaisare, M.L.; Pandey, S.; Khan, M.S.; Talib, A.; Wu, H.-F. Fluorophotometric determination of critical micelle concentration (CMC) of ionic and non-ionic surfactants with carbon dots via Stokes shift. Talanta 2015, 132, 572–578. [Google Scholar] [CrossRef] [PubMed]
- Favilla, R.; Parisoli, A.; Mazzini, A. Alkaline denaturation and partial refolding of pepsin investigated with DAPI as an extrinsic probe. Biophys. Chem. 1997, 67, 75–83. [Google Scholar] [CrossRef]
- Shlien, A.; Malkin, D. Copy number variations and cancer. Genome Med. 2009, 1, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emes, R.D.; Goodstadt, L.; Winter, E.E.; Ponting, C.P. Comparison of the genomes of human and mouse lays the foundation of genome zoology. Hum. Mol. Genet. 2003, 12, 701–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, K.; Zhang, J.; Zhu, Y.; Chen, L.; Ye, J. Zinc doping induced differences in the surface composition, surface morphology and osteogenesis performance of the calcium phosphate cement hydration products. Mater. Sci. Eng. C 2019, 105, 110065. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell line | k | b | R2 |
|---|---|---|---|
| MCF-7 | 0.0047 | 11.6 | 0.988 |
| MH-22a | 0.0041 | 10.9 | 0.988 |
| Swiss 3T3 | 0.0021 | 9.1 | 0.986 |
| DPSC | 0.0026 | 13.6 | 0.996 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Šimoliūnas, E.; Kantakevičius, P.; Kalvaitytė, M.; Bagdzevičiūtė, L.; Alksnė, M.; Baltriukienė, D. DNA-DAPI Interaction-Based Method for Cell Proliferation Rate Evaluation in 3D Structures. Curr. Issues Mol. Biol. 2021, 43, 251-263. https://0-doi-org.brum.beds.ac.uk/10.3390/cimb43010021
Šimoliūnas E, Kantakevičius P, Kalvaitytė M, Bagdzevičiūtė L, Alksnė M, Baltriukienė D. DNA-DAPI Interaction-Based Method for Cell Proliferation Rate Evaluation in 3D Structures. Current Issues in Molecular Biology. 2021; 43(1):251-263. https://0-doi-org.brum.beds.ac.uk/10.3390/cimb43010021
Chicago/Turabian StyleŠimoliūnas, Egidijus, Paulius Kantakevičius, Miglė Kalvaitytė, Lina Bagdzevičiūtė, Milda Alksnė, and Daiva Baltriukienė. 2021. "DNA-DAPI Interaction-Based Method for Cell Proliferation Rate Evaluation in 3D Structures" Current Issues in Molecular Biology 43, no. 1: 251-263. https://0-doi-org.brum.beds.ac.uk/10.3390/cimb43010021