
Evaluating the Effect of 3′-UTR Variants in DICER1 and DROSHA on Their Tissue-Specific Expression by miRNA Target Prediction


Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Collection
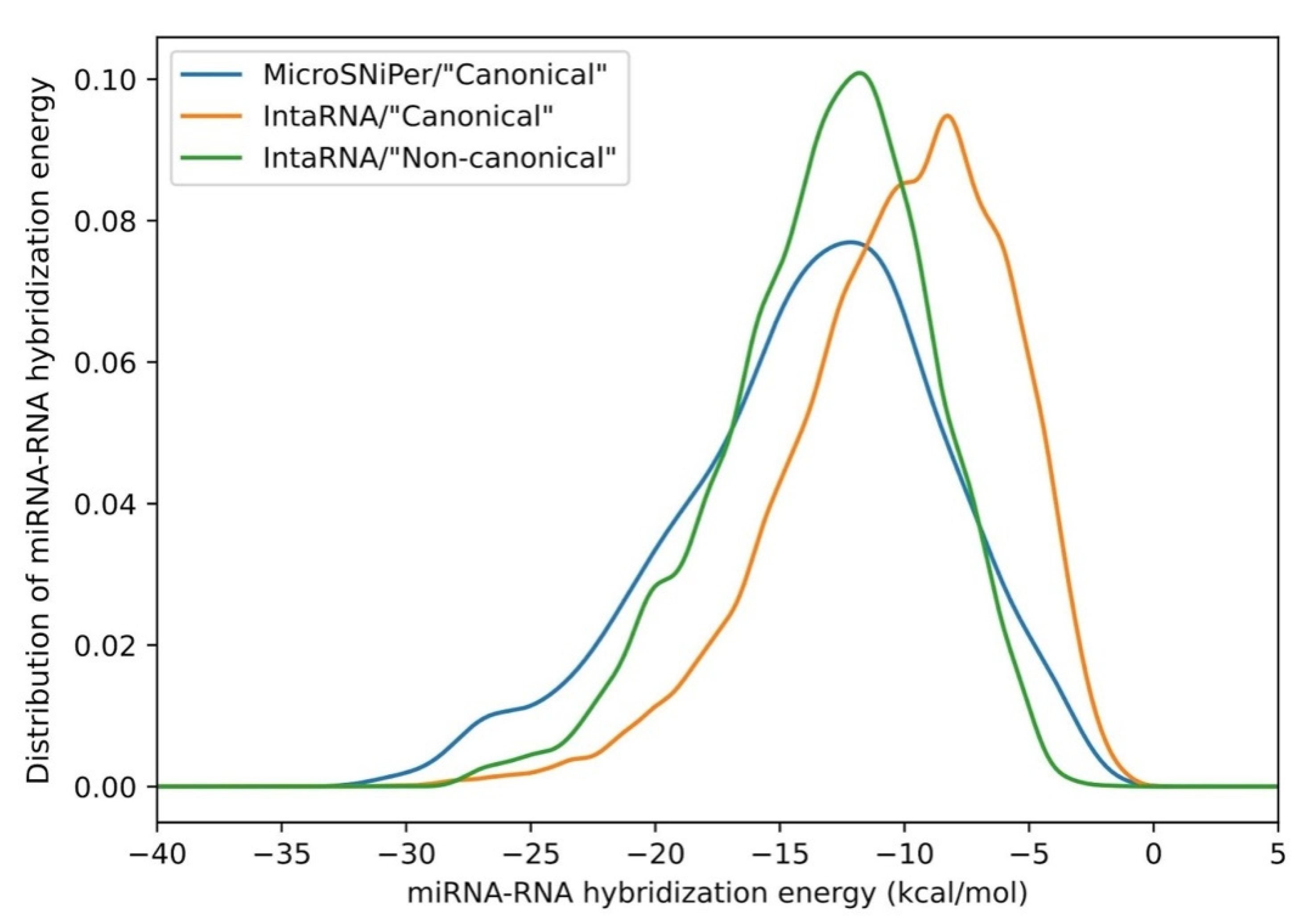
2.2. ”Canonical” Binding Analysis
2.3. Energy Evaluation of mRNA-miRNA Duplex
2.4. Tissue-Specific miRNA Profiling
2.5. Resulting Genes Variant Selection
3. Results
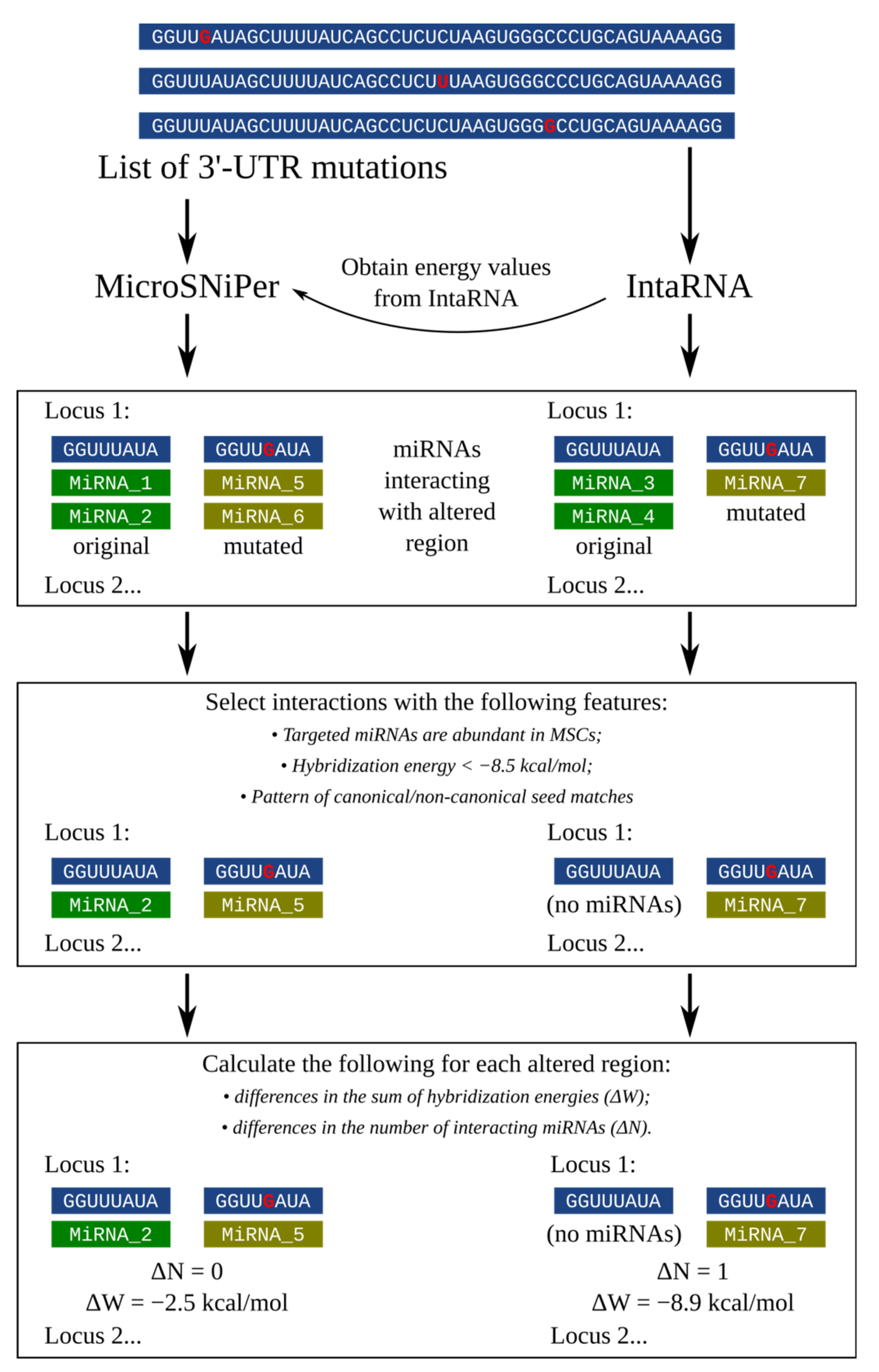
3.1. Building the Pipeline for 3′-UTR SNPs Analysis Considering Tissue-Specific miRNA Expression
3.2. Variants in DROSHA 3′-UTR Predicted to Downregulate Gene Expression
3.3. Variants in DICER1 3′-UTR Predicted to Downregulate Gene Expression
4. Discussion
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Taylor, J.C.; Martin, H.C.; Lise, S.; Broxholme, J.; Cazier, J.-B.; Rimmer, A.; Kanapin, A.; Lunter, G.; Fiddy, S.; Allan, C.; et al. Factors influencing success of clinical genome sequencing across a broad spectrum of disorders. Nat. Genet. 2015, 47, 717–726. [Google Scholar] [CrossRef]
- Lionel, A.C.; Costain, G.; Monfared, N.; Walker, S.; Reuter, M.S.; Hosseini, S.M.; Thiruvahindrapuram, B.; Merico, D.; Jobling, R.; Nalpathamkalam, T.; et al. Improved diagnostic yield compared with targeted gene sequencing panels suggests a role for whole-genome sequencing as a first-tier genetic test. Genet. Med. 2018, 20, 435–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yépez, V.A.; Mertes, C.; Müller, M.F.; Klaproth-Andrade, D.; Wachutka, L.; Frésard, L.; Gusic, M.; Scheller, I.F.; Goldberg, P.F.; Prokisch, H.; et al. Detection of aberrant gene expression events in RNA sequencing data. Nat. Protoc. 2021, 16, 1276–1296. [Google Scholar] [CrossRef]
- Hindorff, L.A.; Sethupathy, P.; Junkins, H.A.; Ramos, E.M.; Mehta, J.P.; Collins, F.S.; Manolio, T.A. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl. Acad. Sci. USA 2009, 106, 9362–9367. [Google Scholar] [CrossRef] [Green Version]
- Schaub, M.A.; Boyle, A.P.; Kundaje, A.; Batzoglou, S.; Snyder, M. Linking disease associations with regulatory information in the human genome. Genome Res. 2012, 22, 1748–1759. [Google Scholar] [CrossRef] [Green Version]
- Khurana, E.; Fu, Y.; Chakravarty, D.; Demichelis, F.; Rubin, M.A.; Gerstein, M. Role of non-coding sequence variants in cancer. Nat. Rev. Genet. 2016, 17, 93–108. [Google Scholar] [CrossRef] [PubMed]
- Moszyńska, A.; Gebert, M.; Collawn, J.F.; Bartoszewski, R. SNPs in microRNA target sites and their potential role in human disease. Open Biol. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved Seed Pairing, Often Flanked by Adenosines, Indicates that Thousands of Human Genes are MicroRNA Targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef]
- Hrdlickova, B.; de Almeida, R.C.; Borek, Z.; Withoff, S. Genetic variation in the non-coding genome: Involvement of micro-RNAs and long non-coding RNAs in disease. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2014, 1842, 1910–1922. [Google Scholar] [CrossRef] [Green Version]
- Friedman, R.C.; Farh, K.K.-H.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartel, D.P. MicroRNAs: Target Recognition and Regulatory Functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [Green Version]
- Bartel, D.P. Metazoan MicroRNAs. Cell 2018, 173, 20–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chipman, L.B.; Pasquinelli, A.E. miRNA Targeting: Growing beyond the Seed. Trends Genet. 2019, 35, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Stavast, C.J.; Erkeland, S.J. The Non-Canonical Aspects of MicroRNAs: Many Roads to Gene Regulation. Cells 2019, 8, 1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hafner, M.; Landthaler, M.; Burger, L.; Khorshid, M.; Hausser, J.; Berninger, P.; Rothballer, A.; Ascano, M.; Jungkamp, A.-C.; Munschauer, M.; et al. Transcriptome-wide Identification of RNA-Binding Protein and MicroRNA Target Sites by PAR-CLIP. Cell 2010, 141, 129–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X. Composition of seed sequence is a major determinant of microRNA targeting patterns. Bioinformatics 2014, 30, 1377–1383. [Google Scholar] [CrossRef] [Green Version]
- Doench, J.G.; Sharp, P.A. Specificity of microRNA target selection in translational repression. Genes Dev. 2004, 18, 504–511. [Google Scholar] [CrossRef] [Green Version]
- Brennecke, J.; Stark, A.; Russell, R.B.; Cohen, S.M. Principles of MicroRNA–Target Recognition. PLoS Biol. 2005, 3, e85. [Google Scholar] [CrossRef]
- Shin, C.; Nam, J.-W.; Farh, K.K.-H.; Chiang, H.R.; Shkumatava, A.; Bartel, D.P. Expanding the MicroRNA Targeting Code: Functional Sites with Centered Pairing. Mol. Cell 2010, 38, 789–802. [Google Scholar] [CrossRef] [Green Version]
- Cho, S.H.; Ko, J.J.; Kim, J.O.; Jeon, Y.J.; Yoo, J.K.; Oh, J.; Oh, D.; Kim, J.W.; Kim, N.K. 3′-UTR Polymorphisms in the MiRNA Machinery Genes DROSHA, DICER1, RAN, and XPO5 Are Associated with Colorectal Cancer Risk in a Korean Population. PLoS ONE 2015, 10, e0131125. [Google Scholar] [CrossRef] [Green Version]
- Steensma, D.P.; Bejar, R.; Jaiswal, S.; Lindsley, R.C.; Sekeres, M.A.; Hasserjian, R.P.; Ebert, B.L. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015, 126, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Meggendorfer, M.; Haferlach, C.; Kern, W.; Haferlach, T. Molecular analysis of myelodysplastic syndrome with isolated deletion of the long arm of chromosome 5 reveals a specific spectrum of molecular mutations with prognostic impact: A study on 123 patients and 27 genes. Haematologica 2017, 102, 1502–1510. [Google Scholar] [CrossRef] [PubMed]
- Raaijmakers, M.H.G.P.; Mukherjee, S.; Guo, S.; Zhang, S.; Kobayashi, T.; Schoonmaker, J.A.; Ebert, B.L.; Al-Shahrour, F.; Hasserjian, R.P.; Scadden, E.O.; et al. Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature 2010, 464, 852–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santamaria, C.; Muntion, S.; Roson, B.; Blanco, B.; Lopez-Villar, O.; Carrancio, S.; Sanchez-Guijo, F.M.; Diez-Campelo, M.; Alvarez-Fernandez, S.; Sarasquete, M.E.; et al. Impaired expression of DICER, DROSHA, SBDS and some microRNAs in mesenchymal stromal cells from myelodysplastic syndrome patients. Haematologica 2012, 97, 1218–1224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moiseev, I.S.; Tcvetkov, N.Y.; Barkhatov, I.M.; Barabanshikova, M.V.; Bug, D.S.; Petuhova, N.V.; Tishkov, A.V.; Bakin, E.A.; Izmailova, E.A.; Shakirova, A.I.; et al. High mutation burden in the checkpoint and micro-RNA processing genes in myelodysplastic syndrome. PLoS ONE 2021, 16, e0248430. [Google Scholar] [CrossRef] [PubMed]
- Barenboim, M.; Zoltick, B.J.; Guo, Y.; Weinberger, D.R. MicroSNiPer: A web tool for prediction of SNP effects on putative microRNA targets. Hum. Mutat. 2010, 31, 1223–1232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearson, W.R.; Lipman, D.J. Improved tools for biological sequence comparison. Proc. Natl. Acad. Sci. USA 1988, 85, 2444–2448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffiths-Jones, S.; Grocock, R.J.; Van Dongen, S.; Bateman, A.; Enright, A.J. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006, 34, D140–D144. [Google Scholar] [CrossRef]
- Mann, M.; Wright, P.R.; Backofen, R. IntaRNA 2.0: Enhanced and customizable prediction of RNA–RNA interactions. Nucleic Acids Res. 2017, 45, W435–W439. [Google Scholar] [CrossRef]
- Lorenz, R.; Bernhart, S.H.; Höner Zu Siederdissen, C.; Tafer, H.; Flamm, C.; Stadler, P.F.; Hofacker, I.L. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011, 6, 26. [Google Scholar] [CrossRef] [PubMed]
- Ragan, C.; Zuker, M.; Ragan, M.A. Quantitative Prediction of miRNA-mRNA Interaction Based on Equilibrium Concentrations. PLoS Comput. Biol. 2011, 7, e1001090. [Google Scholar] [CrossRef] [Green Version]
- Clark, E.A.; Kalomoiris, S.; Nolta, J.A.; Fierro, F.A. Concise Review: MicroRNA Function in Multipotent Mesenchymal Stromal Cells. Stem Cells 2014, 32, 1074–1082. [Google Scholar] [CrossRef] [PubMed]
- Bellayr, I.H.; Kumar, A.; Puri, R.K. MicroRNA expression in bone marrow-derived human multipotent Stromal cells. BMC Genom. 2017, 18, 605. [Google Scholar] [CrossRef] [Green Version]
- Baglio, S.R.; Rooijers, K.; Koppers-Lalic, D.; Verweij, F.J.; Pérez Lanzón, M.; Zini, N.; Naaijkens, B.; Perut, F.; Niessen, H.W.M.; Baldini, N.; et al. Human bone marrow- and adipose-mesenchymal stem cells secrete exosomes enriched in distinctive miRNA and tRNA species. Stem Cell Res. Ther. 2015, 6, 127. [Google Scholar] [CrossRef] [Green Version]
- Shao, L.; Zhang, Y.; Lan, B.; Wang, J.; Zhang, Z.; Zhang, L.; Xiao, P.; Meng, Q.; Geng, Y.-J.; Yu, X.-Y.; et al. MiRNA-Sequence Indicates That Mesenchymal Stem Cells and Exosomes Have Similar Mechanism to Enhance Cardiac Repair. BioMed Res. Int. 2017, 2017, 4150705. [Google Scholar] [CrossRef]
- Luther, K.M.; Haar, L.; McGuinness, M.; Wang, Y.; Lynch Iv, T.L.; Phan, A.; Song, Y.; Shen, Z.; Gardner, G.; Kuffel, G.; et al. Exosomal miR-21a-5p mediates cardioprotection by mesenchymal stem cells. J. Mol. Cell. Cardiol. 2018, 119, 125–137. [Google Scholar] [CrossRef]
- Collino, F.; Deregibus, M.C.; Bruno, S.; Sterpone, L.; Aghemo, G.; Viltono, L.; Tetta, C.; Camussi, G. Microvesicles Derived from Adult Human Bone Marrow and Tissue Specific Mesenchymal Stem Cells Shuttle Selected Pattern of miRNAs. PLoS ONE 2010, 5, e11803. [Google Scholar] [CrossRef]
- Ferguson, S.W.; Wang, J.; Lee, C.J.; Liu, M.; Neelamegham, S.; Canty, J.M.; Nguyen, J. The microRNA regulatory landscape of MSC-derived exosomes: A systems view. Sci. Rep. 2018, 8, 1419. [Google Scholar] [CrossRef]
- Furuta, T.; Miyaki, S.; Ishitobi, H.; Ogura, T.; Kato, Y.; Kamei, N.; Miyado, K.; Higashi, Y.; Ochi, M. Mesenchymal Stem Cell-Derived Exosomes Promote Fracture Healing in a Mouse Model. STEM CELLS Transl. Med. 2016, 5, 1620–1630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rehmsmeier, M.; Steffen, P.; Höchsmann, M.; Giegerich, R. Fast and effective prediction of microRNA/target duplexes. RNA 2004, 10, 1507–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tafer, H.; Hofacker, I.L. RNAplex: A fast tool for RNA–RNA interaction search. Bioinformatics 2008, 24, 2657–2663. [Google Scholar] [CrossRef]
- Maragkakis, M.; Alexiou, P.; Papadopoulos, G.L.; Reczko, M.; Dalamagas, T.; Giannopoulos, G.; Goumas, G.; Koukis, E.; Kourtis, K.; Simossis, V.A.; et al. Accurate microRNA target prediction correlates with protein repression levels. BMC Bioinform. 2009, 10, 295. [Google Scholar] [CrossRef] [Green Version]
- Gaidatzis, D.; Van Nimwegen, E.; Hausser, J.; Zavolan, M. Inference of miRNA targets using evolutionary conservation and pathway analysis. BMC Bioinform. 2007, 8, 69. [Google Scholar] [CrossRef] [Green Version]
- Kertesz, M.; Iovino, N.; Unnerstall, U.; Gaul, U.; Segal, E. The role of site accessibility in microRNA target recognition. Nat. Genet. 2007, 39, 1278–1284. [Google Scholar] [CrossRef]
- Busch, A.; Richter, A.S.; Backofen, R. IntaRNA: Efficient prediction of bacterial sRNA targets incorporating target site accessibility and seed regions. Bioinformatics 2008, 24, 2849–2856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharya, A.; Ziebarth, J.D.; Cui, Y. PolymiRTS Database 3.0: Linking polymorphisms in microRNAs and their target sites with human diseases and biological pathways. Nucleic Acids Res. 2014, 42, D86–D91. [Google Scholar] [CrossRef]
- Liu, C.; Zhang, F.; Li, T.; Lu, M.; Wang, L.; Yue, W.; Zhang, D. MirSNP, a database of polymorphisms altering miRNA target sites, identifies miRNA-related SNPs in GWAS SNPs and eQTLs. BMC Genom. 2012, 13, 661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, V.; Bell, G.W.; Nam, J.-W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4, e05005. [Google Scholar] [CrossRef] [PubMed]
- Sabina, S.; Vecoli, C.; Borghini, A.; Guarino, R.; Andreassi, M.G. Analysis of miRNAs Targeting 3′UTR of H2AFX Gene: A General in Silico Approach. MicroRNA 2015, 4, 41–49. [Google Scholar] [CrossRef]
- Bhatnager, R.; Dang, A.S. Comprehensive in-silico prediction of damage associated SNPs in Human Prolidase gene. Sci. Rep. 2018, 8, 9430. [Google Scholar] [CrossRef] [Green Version]
- Ludwig, N.; Leidinger, P.; Becker, K.; Backes, C.; Fehlmann, T.; Pallasch, C.; Rheinheimer, S.; Meder, B.; Stähler, C.; Meese, E.; et al. Distribution of miRNA expression across human tissues. Nucleic Acids Res. 2016, 44, 3865–3877. [Google Scholar] [CrossRef]
- Martello, G.; Rosato, A.; Ferrari, F.; Manfrin, A.; Cordenonsi, M.; Dupont, S.; Enzo, E.; Guzzardo, V.; Rondina, M.; Spruce, T.; et al. A MicroRNA Targeting Dicer for Metastasis Control. Cell 2010, 141, 1195–1207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feinberg-Gorenshtein, G.; Guedj, A.; Shichrur, K.; Jeison, M.; Luria, D.; Kodman, Y.; Ash, S.; Feinmesser, M.; Edry, L.; Shomron, N.; et al. miR-192 Directly Binds and Regulates Dicer1 Expression in Neuroblastoma. PLoS ONE 2013, 8, e78713. [Google Scholar] [CrossRef] [Green Version]
- Forman, J.J.; Legesse-Miller, A.; Coller, H.A. A search for conserved sequences in coding regions reveals that the let-7 microRNA targets Dicer within its coding sequence. Proc. Natl. Acad. Sci. USA 2008, 105, 14879–14884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papadimitriou, S.; Gazzo, A.; Versbraegen, N.; Nachtegael, C.; Aerts, J.; Moreau, Y.; Van Dooren, S.; Nowe, A.; Smits, G.; Lenaerts, T. Predicting disease-causing variant combinations. Proc. Natl. Acad. Sci. USA 2019, 116, 11878. [Google Scholar] [CrossRef] [Green Version]
- Bejar, R.; Stevenson, K.; Abdel-Wahab, O.; Galili, N.; Nilsson, B.; Garcia-Manero, G.; Kantarjian, H.; Raza, A.; Levine, R.L.; Neuberg, D.; et al. Clinical Effect of Point Mutations in Myelodysplastic Syndromes. N. Engl. J. Med. 2011, 364, 2496–2506. [Google Scholar] [CrossRef] [Green Version]
- Papaemmanuil, E.; Gerstung, M.; Malcovati, L.; Tauro, S.; Gundem, G.; Van Loo, P.; Yoon, C.J.; Ellis, P.; Wedge, D.; Pellagatti, A.; et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 2013, 122, 3616–3627; quiz 3699. [Google Scholar] [CrossRef]
- Tsvetkov, N.Y.; Morozova, E.V.; Barkhatov, I.M.; Moiseev, I.S.; Barabanshchikova, M.V.; Tishkov, A.V.; Bug, D.S.; Petukhova, N.V.; Izmailova, E.A.; Bondarenko, S.N.; et al. Prognostic Value of Next-Generation Sequencing Data in Patients with Myelodysplastic Syndrome. Clin. Oncohematol. 2020, 13, 170–175. [Google Scholar] [CrossRef]
- Condrat, C.E.; Thompson, D.C.; Barbu, M.G.; Bugnar, O.L.; Boboc, A.; Cretoiu, D.; Suciu, N.; Cretoiu, S.M.; Voinea, S.C. miRNAs as Biomarkers in Disease: Latest Findings Regarding Their Role in Diagnosis and Prognosis. Cells 2020, 9, 276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nahand, J.S.; Taghizadeh-Boroujeni, S.; Karimzadeh, M.; Borran, S.; Pourhanifeh, M.H.; Moghoofei, M.; Bokharaei-Salim, F.; Karampoor, S.; Jafari, A.; Asemi, Z.; et al. microRNAs: New prognostic, diagnostic, and therapeutic biomarkers in cervical cancer. J. Cell. Physiol. 2019, 234, 17064–17099. [Google Scholar] [CrossRef] [PubMed]
- Mahdian-Shakib, A.; Dorostkar, R.; Tat, M.; Hashemzadeh, M.S.; Saidi, N. Differential role of microRNAs in prognosis, diagnosis, and therapy of ovarian cancer. Biomed. Pharmacother. 2016, 84, 592–600. [Google Scholar] [CrossRef] [PubMed]
- Asiaf, A.; Ahmad, S.T.; Arjumand, W.; Zargar, M.A. MicroRNAs in Breast Cancer: Diagnostic and Therapeutic Potential. Methods Mol. Biol. 2018, 1699, 23–43. [Google Scholar] [CrossRef] [PubMed]
- Parizadeh, S.M.; Jafarzadeh-Esfehani, R.; Ghandehari, M.; Parizadeh, S.M.R.; Hassanian, S.M.; Rezayi, M.; Ghayour-Mobarhan, M.; Ferns, G.A.; Avan, A. Circulating Exosomes as Potential Biomarkers in Cardiovascular Disease. Curr. Pharm. Des. 2018, 24, 4436–4444. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.-S.; Jin, J.-P.; Wang, J.-Q.; Zhang, Z.-G.; Freedman, J.H.; Zheng, Y.; Cai, L. miRNAS in cardiovascular diseases: Potential biomarkers, therapeutic targets and challenges. Acta Pharmacol. Sin. 2018, 39, 1073–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kingsley, S.M.K.; Bhat, B.V. Role of microRNAs in sepsis. Inflamm. Res. 2017, 66, 553–569. [Google Scholar] [CrossRef]
- Martínez-Hernández, R.; Serrano-Somavilla, A.; Ramos-Leví, A.; Sampedro-Nuñez, M.; Lens-Pardo, A.; Muñoz De Nova, J.L.; Triviño, J.C.; González, M.U.; Torné, L.; Casares-Arias, J.; et al. Integrated miRNA and mRNA expression profiling identifies novel targets and pathological mechanisms in autoimmune thyroid diseases. EBioMedicine 2019, 50, 329–342. [Google Scholar] [CrossRef] [Green Version]
- Moradifard, S.; Hoseinbeyki, M.; Ganji, S.M.; Minuchehr, Z. Analysis of microRNA and Gene Expression Profiles in Alzheimer’s Disease: A Meta-Analysis Approach. Sci. Rep. 2018, 8, 4767. [Google Scholar] [CrossRef]
- Yao, Y.; Jiang, C.; Wang, F.; Yan, H.; Long, D.; Zhao, J.; Wang, J.; Zhang, C.; Li, Y.; Tian, X.; et al. Integrative Analysis of miRNA and mRNA Expression Profiles Associated with Human Atrial Aging. Front. Physiol. 2019, 10, 1226. [Google Scholar] [CrossRef]
- Knox, B.; Wang, Y.; Rogers, L.J.; Xuan, J.; Yu, D.; Guan, H.; Chen, J.; Shi, T.; Ning, B.; Kadlubar, S.A.; et al. A functional SNP in the 3′-UTR of TAP2 gene interacts with microRNA hsa-miR-1270 to suppress the gene expression. Environ. Mol. Mutagen. 2018, 59, 134–143. [Google Scholar] [CrossRef] [Green Version]
- Skeeles, L.E.; Fleming, J.L.; Mahler, K.L.; Toland, A.E. The Impact of 3′UTR Variants on Differential Expression of Candidate Cancer Susceptibility Genes. PLoS ONE 2013, 8, e58609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pamuła-Piłat, J.; Tęcza, K.; Kalinowska-Herok, M.; Grzybowska, E. Genetic 3′UTR variations and clinical factors significantly contribute to survival prediction and clinical response in breast cancer patients. Sci. Rep. 2020, 10, 5736. [Google Scholar] [CrossRef]
- Zheng, X.; Chen, L.; Li, X.; Zhang, Y.; Xu, S.; Huang, X. Prediction of miRNA targets by learning from interaction sequences. PLoS ONE 2020, 15, e0232578. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Method | Variant Name | Change in the Number of Interacting miRNAs (ΔN) | Change in the Sum of Hybridization Energies (ΔW, kcal/mol) | Maximum Population Allele Frequency |
|---|---|---|---|---|
| IntaRNA | rs1479981622:C>T | 5 | −62.01 | 8 × 10−6 |
| rs536092006:T>C | 5 | −61.06 | 0.0003 | |
| rs1049043869:C>G | 6 | −55.91 | 6.4 × 10−5 | |
| MicroSNiPer | rs775376244:T>C | 2 | −24.60 | 0.0005 |
| rs550965893:T>G | 2 | −22.41 | 0.0002 | |
| rs771705336:C>G | 1 | −17.85 | 0.0003 | |
| rs766853644:T>C | 1 | −17.68 | 8 × 10−6 | |
| rs1362918185:G>A | 1 | −16.64 | 2.4 × 10−5 | |
| rs536221210:A>G | 1 | −16.57 | Not provided | |
| rs1221977896:A>C | 1 | −16.30 | 2.4 × 10−5 | |
| rs752035593:G>A | 1 | −15.81 | 3.2 × 10−5 | |
| rs1260437486:G>A | 1 | −15.56 | 8 × 10−6 | |
| rs1014491371:C>G | 1 | −15.29 | 8 × 10−6 | |
| rs761935330:G>A | 1 | −15.03 | 0.0003 | |
| rs1441949563:C>T | 1 | −14.07 | 3 × 10−5 | |
| rs1033831185:A>G | 1 | −11.70 | 3 × 10−5 | |
| rs1358226278:C>G | 1 | −11.38 | 8 × 10−6 | |
| rs1424327104:C>T | 1 | −11.11 | Not provided | |
| rs1288407577:T>G | 1 | −10.58 | 4 × 10−6 | |
| rs1187772510:A>G | 1 | −8.98 | 3.2 × 10−5 |
| Method | Variant Name | Change in the Number of Interacting miRNAs (ΔN) | Change in the Sum of Hybridization Energies (ΔW, kcal/mol) | Maximum Population Allele Frequency |
|---|---|---|---|---|
| IntaRNA | rs1296755923:T>G | 9 | −126.61 | 0.0003 |
| rs1207839989:A>G | 8 | −101.32 | 3 × 10−5 | |
| rs1041875974:G>T | 6 | −98.42 | Not provided | |
| rs981079616:T>G | 6 | −94.19 | 8 × 10−6 | |
| rs760246677:G>T | 6 | −91.69 | 8 × 10−6 | |
| rs989262025:A>G | 6 | −84.46 | 3.3 × 10−5 | |
| rs1281573015:A>T | 8 | −83.06 | 8 × 10−6 | |
| rs1248738927:G>C | 6 | −82.47 | Not provided | |
| rs1221590835:G>A | 7 | −79.94 | 3 × 10−5 | |
| rs1324216335:T>C | 7 | −74.46 | 8 × 10−6 | |
| rs1206166531:C>G | 5 | −71.61 | 0 | |
| rs1194042023:A>G | 5 | −68.88 | 3.2 × 10−5 | |
| rs889120755:C>G | 5 | −68.86 | Not provided | |
| rs571735282:A>G | 5 | −65.47 | 0.0012 | |
| COSN31961029:T>C | 5 | −65.08 | Not provided | |
| rs902240610:A>G | 5 | −64.83 | 8 × 10−5 | |
| rs552609115:T>C | 5 | −64.25 | Not provided | |
| rs930259829:T>G | 7 | −63.86 | 4.8 × 10−5 | |
| rs1206166531:C>T | 5 | −58.35 | 0.0002 | |
| rs535308545:A>C | 5 | −58.35 | 3.2 × 10−5 | |
| rs1381450764:G>A | 6 | −57.20 | Not provided | |
| rs923348310:C>T | 5 | −53.65 | 7.2 × 10−5 | |
| rs752666806:C>T | 5 | −45.40 | Not provided | |
| MicroSNiPer | rs565097712:G>A | 7 | −77.40 | 0.0004 |
| rs1396697573:A>G | 2 | −41.98 | Not provided | |
| rs1307365736:A>T | 2 | −30.12 | 8 × 10−6 | |
| rs895962647:A>T | 2 | −27.72 | 3.2 × 10−5 | |
| rs942463789:T>G | 1 | −24.94 | 0.0003 | |
| rs997751105:C>T | 2 | −22.40 | 1.6 × 10−5 | |
| rs926249853:T>G | 2 | −20.43 | 8 × 10−6 | |
| rs1478240035:G>T | 1 | −19.62 | 8 × 10−6 | |
| rs1405544334:G>T | 1 | −19.59 | 8 × 10−6 | |
| rs1273786688:T>G | 2 | −18.42 | 0.005 | |
| rs1045581145:A>T | 1 | −17.48 | 8 × 10−6 | |
| rs889120755:C>G | 1 | −16.79 | Not provided | |
| rs923753708:G>C | 1 | −16.70 | Not provided | |
| rs1461667204:T>A | 1 | −16.60 | 8 × 10−6 | |
| rs528282193:C>T | 1 | −16.60 | 0.001 | |
| rs1338875884:A>G | 1 | −16.32 | 3 × 10−5 | |
| rs1165916168:C>T | 1 | −16.04 | Not provided | |
| MU75396710:C>G | 1 | −15.66 | Not provided | |
| rs1394767633:A>G | 1 | −15.34 | 3.2 × 10−5 | |
| rs1288975386:T>A | 1 | −14.99 | Not provided | |
| rs752666806:C>T | 1 | −14.39 | Not provided | |
| rs1253019517:T>A | 1 | −14.31 | 1.6 × 10−5 | |
| rs1219762419:A>G | 1 | −13.75 | 6 × 10−5 | |
| rs1371551409:A>C | 1 | −13.69 | 8 × 10−6 | |
| rs1181773370:C>T | 1 | −12.71 | 3 × 10−5 | |
| rs1452738847:A>G | 1 | −12.03 | 8 × 10−6 | |
| rs1467042427:A>G | 1 | −11.51 | 8 × 10−6 | |
| COSN25075844:T>C | 1 | −11.48 | Not provided | |
| COSN25075844:T>C | 1 | −11.48 | Not provided | |
| MU85472748:T>C | 1 | −11.48 | Not provided | |
| MU85472748:T>C | 1 | −11.48 | Not provided | |
| rs972230345:A>G | 1 | −11.44 | 4 × 10−5 | |
| rs887676239:A>G | 1 | −11.12 | 5.6 × 10−5 | |
| rs959339279:A>G | 1 | −11.00 | 0.0001 | |
| rs971064154:A>G | 1 | −10.68 | 8 × 10−6 | |
| rs1205529398:G>A | 1 | −9.88 | 8 × 10−6 | |
| rs1306390419:A>G | 1 | −9.49 | 8 × 10−6 | |
| rs1292049568:G>T | 1 | −9.33 | 8 × 10−6 | |
| rs982013263:G>T | 1 | −9.17 | 6.4 × 10−5 | |
| rs914976738:C>T | 1 | −8.97 | Not provided | |
| rs1488494352:A>G | 1 | −8.60 | 8 × 10−6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bug, D.S.; Tishkov, A.V.; Moiseev, I.S.; Petukhova, N.V. Evaluating the Effect of 3′-UTR Variants in DICER1 and DROSHA on Their Tissue-Specific Expression by miRNA Target Prediction. Curr. Issues Mol. Biol. 2021, 43, 605-617. https://0-doi-org.brum.beds.ac.uk/10.3390/cimb43020044
Bug DS, Tishkov AV, Moiseev IS, Petukhova NV. Evaluating the Effect of 3′-UTR Variants in DICER1 and DROSHA on Their Tissue-Specific Expression by miRNA Target Prediction. Current Issues in Molecular Biology. 2021; 43(2):605-617. https://0-doi-org.brum.beds.ac.uk/10.3390/cimb43020044
Chicago/Turabian StyleBug, Dmitrii S., Artem V. Tishkov, Ivan S. Moiseev, and Natalia V. Petukhova. 2021. "Evaluating the Effect of 3′-UTR Variants in DICER1 and DROSHA on Their Tissue-Specific Expression by miRNA Target Prediction" Current Issues in Molecular Biology 43, no. 2: 605-617. https://0-doi-org.brum.beds.ac.uk/10.3390/cimb43020044