
Application of Combined Long Amplicon Sequencing (CoLAS) for Genetic Analysis of Neurofibromatosis Type 1: A Pilot Study



Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient and Sample
2.2. Genomic DNA and Total RNA Extraction and Full-Length cDNA Synthesis
2.3. Long-Range PCR and RT-PCR
2.4. Library Preparation and Sequencing
2.5. NGS DATA Analysis Pipeline
2.6. In Silico Analysis of Missense Variants
2.7. Heterozygosity Mapping of NF1 Region
2.8. Detection of Chromosomal-Level Large Deletion by Break Point-Specific Long-Range PCR
2.9. Accessing Features of NF1 Splicing Mutations
2.10. Sanger Sequencing
2.11. Statistical Analysis
2.12. DATA Deposition
3. Results
3.1. Clinical Features of the Patients with Neurofibromatosis Type 1
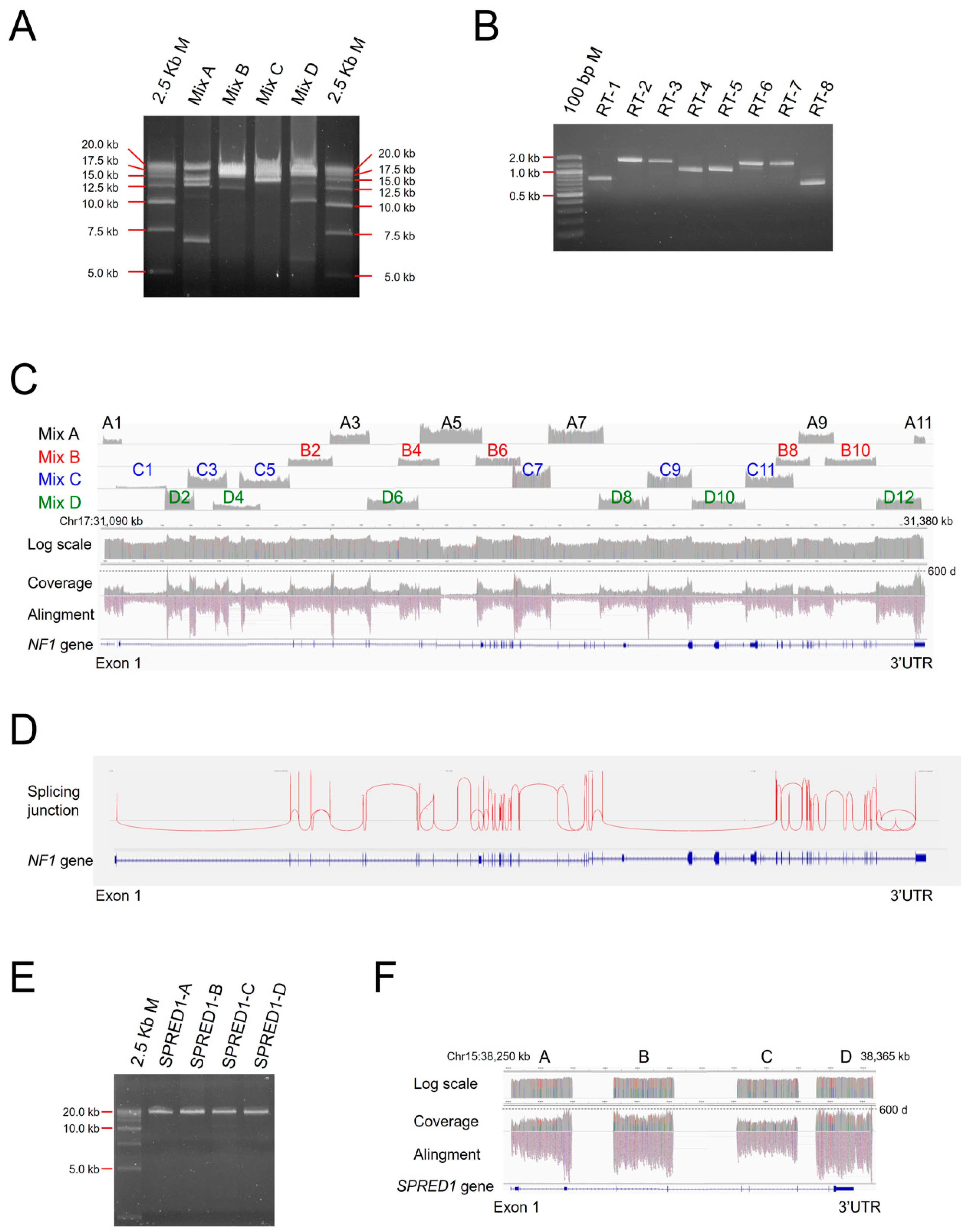
3.2. CoLAS for Genetic Analysis of NF1
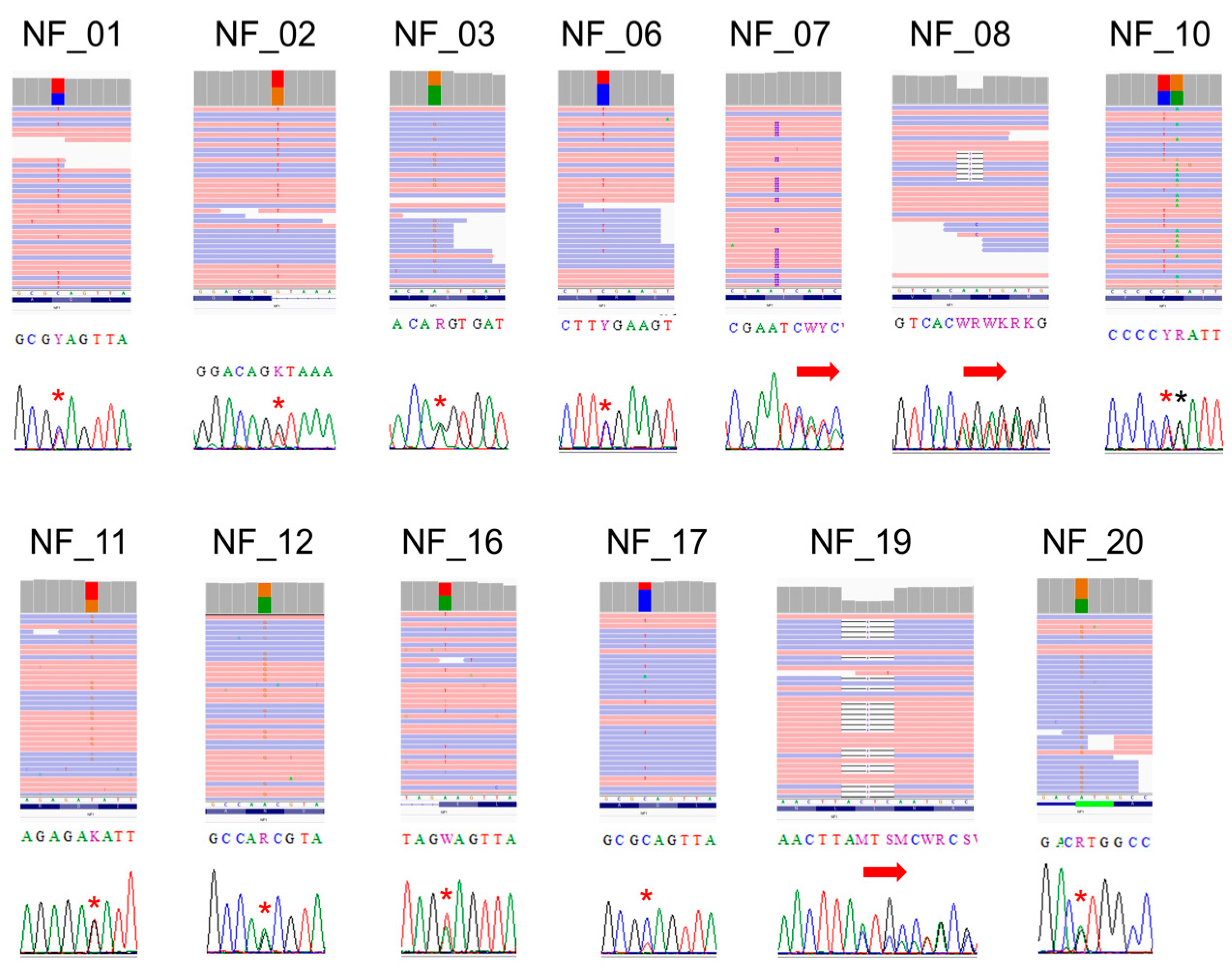
3.3. Point Mutation Detection of NF1
3.4. Heterozygosity Mapping of the NF1 Region
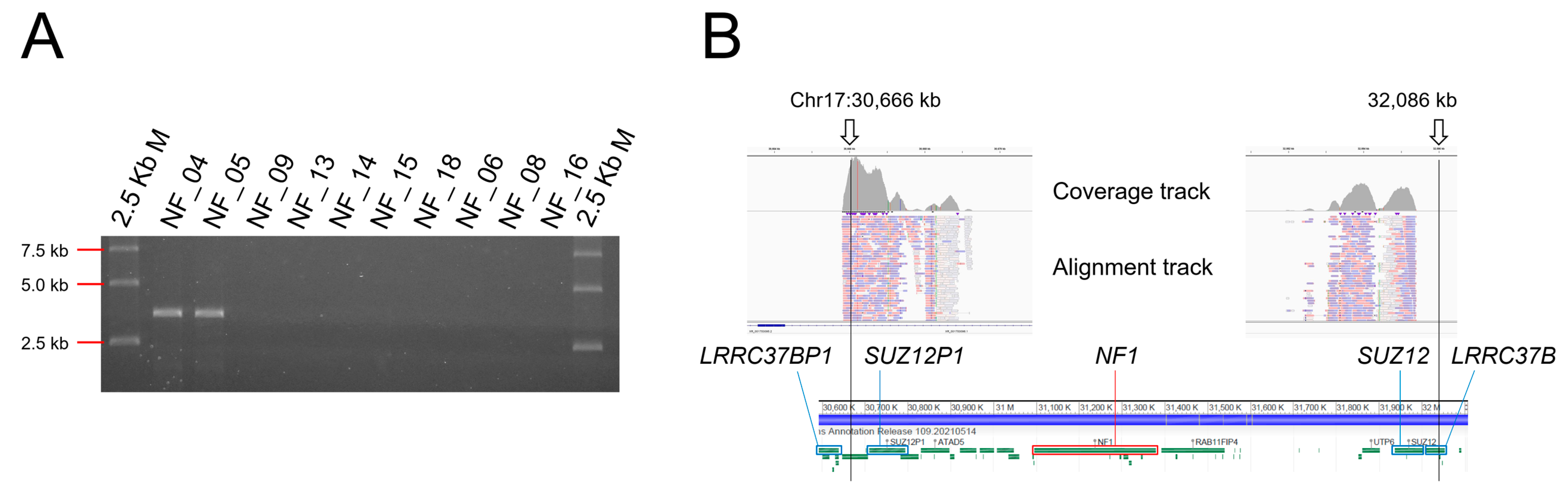
3.5. Detection of Chromosomal Level Large Deletion by Break Point-Specific Long-Range PCR
3.6. Accessing Features of NF1 Splicing Mutations
3.7. SPRED1 Mutation Analysis
3.8. No NF1 Mutation Detected Cases
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gutmann, D.H.; Aylsworth, A.; Carey, J.C.; Korf, B.; Marks, J.; Pyeritz, R.E.; Rubenstein, A.; Viskochil, D. The diagnostic evaluation and multidisciplinary management of neurofibromatosis 1 and neurofibromatosis 2. JAMA 1997, 278, 51–57. [Google Scholar] [CrossRef]
- Yohay, K.H. The genetic and molecular pathogenesis of NF1 and NF2. Semin. Pediatr. Neurol. 2006, 13, 21–26. [Google Scholar] [CrossRef]
- Hirbe, A.C.; Gutmann, D.H. Neurofibromatosis type 1: A multidisciplinary approach to care. Lancet Neurol. 2014, 13, 834–843. [Google Scholar] [CrossRef] [Green Version]
- Neurofibromatosis. Conference statement. National Institutes of Health Consensus Development Conference. Arch. Neurol. 1988, 45, 575–578. [Google Scholar]
- Wallace, M.R.; Marchuk, D.A.; Andersen, L.B.; Letcher, R.; Odeh, H.M.; Saulino, A.M.; Fountain, J.W.; Brereton, A.; Nicholson, J.; Mitchell, A.L. Type 1 neurofibromatosis gene: Identification of a large transcript disrupted in three NF1 patients. Science 1990, 249, 181–186. [Google Scholar] [CrossRef]
- Legius, E.; Messiaen, L.; Wolkenstein, P.; Pancza, P.; Avery, R.A.; Berman, Y.; Blakeley, J.; Babovic-Vuksanovic, D.; Cunha, K.S.; Ferner, R.; et al. Revised diagnostic criteria for neurofibromatosis type 1 and Legius syndrome: An international consensus recommendation. Genet. Med. 2021. [Google Scholar] [CrossRef] [PubMed]
- Denayer, E.; Legius, E. Legius Syndrome and its Relationship with Neurofibromatosis Type 1. Acta Derm. Venereol. 2020, 100, adv00093. [Google Scholar] [CrossRef]
- Wimmer, K.; Schamschula, E.; Wernstedt, A.; Traunfellner, P.; Amberger, A.; Zschocke, J.; Kroisel, P.; Chen, Y.; Callens, T.; Messiaen, L. AG-exclusion zone revisited: Lessons to learn from 91 intronic NF1 3’ splice site mutations outside the canonical AG-dinucleotides. Hum. Mutat. 2020, 41, 1145–1156. [Google Scholar] [CrossRef]
- Kang, E.; Kim, Y.M.; Seo, G.H.; Oh, A.; Yoon, H.M.; Ra, Y.S.; Kim, E.K.; Kim, H.; Heo, S.H.; Kim, G.H.; et al. Phenotype categorization of neurofibromatosis type I and correlation to NF1 mutation types. J. Hum. Genet. 2020, 65, 79–89. [Google Scholar] [CrossRef]
- Kehrer-Sawatzki, H.; Mautner, V.F.; Cooper, D.N. Emerging genotype-phenotype relationships in patients with large NF1 deletions. Hum. Genet. 2017, 136, 349–376. [Google Scholar] [CrossRef] [Green Version]
- García-Romero, M.T.; Parkin, P.; Lara-Corrales, I. Mosaic Neurofibromatosis Type 1: A Systematic Review. Pediatr. Dermatol. 2016, 33, 9–17. [Google Scholar] [CrossRef]
- Okumura, A.; Ozaki, M.; Niida, Y. Development of a practical NF1 genetic testing method through the pilot analysis of five Japanese families with neurofibromatosis type 1. Brain Dev. 2015, 37, 677–689. [Google Scholar] [CrossRef]
- Sabbagh, A.; Pasmant, E.; Imbard, A.; Luscan, A.; Soares, M.; Blanché, H.; Laurendeau, I.; Ferkal, S.; Vidaud, M.; Pinson, S.; et al. NF1 molecular characterization and neurofibromatosis type I genotype-phenotype correlation: The French experience. Hum. Mutat. 2013, 34, 1510–1518. [Google Scholar] [CrossRef]
- Pasmant, E.; Parfait, B.; Luscan, A.; Goussard, P.; Briand-Suleau, A.; Laurendeau, I.; Fouveaut, C.; Leroy, C.; Montadert, A.; Wolkenstein, P.; et al. Neurofibromatosis type 1 molecular diagnosis: What can NGS do for you when you have a large gene with loss of function mutations? Eur. J. Hum. Genet. 2015, 23, 596–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianchessi, D.; Ibba, M.C.; Saletti, V.; Blasa, S.; Langella, T.; Paterra, R.; Cagnoli, G.A.; Melloni, G.; Scuvera, G.; Natacci, F.; et al. Simultaneous Detection of. Genes 2020, 11, 671. [Google Scholar] [CrossRef]
- Supek, F.; Lehner, B.; Lindeboom, R.G.H. To NMD or Not To NMD: Nonsense-Mediated mRNA Decay in Cancer and Other Genetic Diseases. Trends Genet. 2020. [Google Scholar] [CrossRef]
- Togi, S.; Ura, H.; Niida, Y. Optimization and Validation of Multimodular, Long-Range PCR-Based Next-Generation Sequencing Assays for Comprehensive Detection of Mutation in Tuberous Sclerosis Complex. J. Mol. Diagn. 2021, 23, 424–446. [Google Scholar] [CrossRef]
- Lahiri, D.K.; Schnabel, B. DNA isolation by a rapid method from human blood samples: Effects of MgCl2, EDTA, storage time, and temperature on DNA yield and quality. Biochem. Genet. 1993, 31, 321–328. [Google Scholar] [CrossRef]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012. [Google Scholar] [CrossRef] [Green Version]
- Raedt, T.D.; Stephens, M.; Heyns, I.; Brems, H.; Thijs, D.; Messiaen, L.; Stephens, K.; Lazaro, C.; Wimmer, K.; Kehrer-Sawatzki, H.; et al. Conservation of hotspots for recombination in low-copy repeats associated with the NF1 microdeletion. Nat. Genet. 2006, 38, 1419–1423. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [Green Version]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.11–11.10.33. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherry, S.T.; Ward, M.H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [Green Version]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014, 42, D980–D985. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Thorvaldsdottir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, K.; Schulz, M.H.; Long, Q.; Apweiler, R.; Ning, Z. Pindel: A pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 2009, 25, 2865–2871. [Google Scholar] [CrossRef]
- Liu, X.; Wu, C.; Li, C.; Boerwinkle, E. dbNSFP v3.0: A One-Stop Database of Functional Predictions and Annotations for Human Nonsynonymous and Splice-Site SNVs. Hum. Mutat. 2016, 37, 235–241. [Google Scholar] [CrossRef] [Green Version]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef]
- Pasmant, E.; Sabbagh, A.; Spurlock, G.; Laurendeau, I.; Grillo, E.; Hamel, M.J.; Martin, L.; Barbarot, S.; Leheup, B.; Rodriguez, D.; et al. NF1 microdeletions in neurofibromatosis type 1: From genotype to phenotype. Hum. Mutat. 2010, 31, E1506–E1518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Correa, C.; Dorschner, M.; Brems, H.; Lázaro, C.; Clementi, M.; Upadhyaya, M.; Dooijes, D.; Moog, U.; Kehrer-Sawatzki, H.; Rutkowski, J.L.; et al. Recombination hotspot in NF1 microdeletion patients. Hum. Mol. Genet. 2001, 10, 1387–1392. [Google Scholar] [CrossRef] [Green Version]
- Van der Heiden, I.P.; van der Werf, M.; Lindemans, J.; van Schaik, R.H. Sequencing: Not always the “gold standard. Clin. Chem. 2004, 50, 248–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, J.M.; Sohn, Y.B.; Jeong, S.Y.; Kim, H.J.; Messiaen, L.M. Mutation spectrum of NF1 and clinical characteristics in 78 Korean patients with neurofibromatosis type 1. Pediatrc Neurol. 2013, 48, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Sharif, S.; Upadhyaya, M.; Ferner, R.; Majounie, E.; Shenton, A.; Baser, M.; Thakker, N.; Evans, D.G. A molecular analysis of individuals with neurofibromatosis type 1 (NF1) and optic pathway gliomas (OPGs), and an assessment of genotype-phenotype correlations. J. Med. Genet. 2011, 48, 256–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Set | Primer Name | Primer Seq | Primer Position | Product Size (bp) | Final Primer Conc. µM | Multiplex Group |
|---|---|---|---|---|---|---|
| Multiplex long PCR primers | NC_000017.11 (chr17, GRCh38.p13) | |||||
| NF1-A1 | NF1_1FN | 5′-TTTCATTGTCTTTCTCCAAAGCACAGG-3′ | g.31089750_31089776 | 6776 | 0.038 | A |
| NF1_1RN | 5′-CCTTAAACCATCCCCAACTACCTACAGC-3′ | g.31096498_31096525 | ||||
| NF1-C1 | NF1_In1_1F | 5′-TGGCTATGAGTTTTGCAGAGGAAAGC-3′ | g.31094323_31094348 | 17,828 | 0.5 | C |
| NF1_In1_1R | 5′-TGAGACAAAAACAGCTCAAGGGTTCC-3′ | g.31112125_31112150 | ||||
| NF1-D2 | NF1_In1N_1-2F | 5′-AAACGCTTAAAAGCAGCCAGAAGAGG-3′ | g.31111486_31111511 | 10,488 | 0.07 | D |
| NF1_In1N_1-2R | 5′-ACTGTGCTAAGCCTGGGGATATTAATGG-3′ | g.31121946_31121973 | ||||
| NF1-C3 | NF1_In1N_2S | 5′-TCTGGATATTAGCCCTTGGTCAGATGG-3′ | g.31119500_31119526 | 13,970 | 0.1 | C |
| NF1_In1N_2AS | 5′-GGGTCTTGAAGTGGTATCTGCACACC-3′ | g.31133444_31133469 | ||||
| NF1-D4 | NF1_In1_3F | 5′-GCATGGTTGCTTTGTAGTGTCACTGG-3′ | g.31128397_31128422 | 16,667 | 0.3 | D |
| NF1_In1_3R | 5′-GAACGAAGAGGAAAATCTAGGGGATGC-3′ | g.31145037_31145063 | ||||
| NF1-C5 | NF1_In1_4F | 5′-TGTGCAGCTATCATCCTGTGTTCTCC-3′ | g.31137591_31137616 | 17,819 | 0.2 | C |
| NF1_In1_4R | 5′-AGCACCCTCAGCTACACAATTGAAGG-3′ | g.31155384_31155409 | ||||
| NF1-B2 | NF1_2-5F | 5′-GCCCCCTCCTTTACACTCTAAAAATGC-3′ | g.31154673_31154699 | 15,742 | 0.042 | B |
| NF1_2-5R | 5′-ATCACAATCTCCTCGTTCCATTCTGC-3′ | g.31170389_31170414 | ||||
| NF1-A3 | NF1_5-8F | 5′-CCTGCCCTCACCTTGTATATCTCTTGC-3′ | g.31169247_31169273 | 14,116 | 0.038 | A |
| NF1_5-8R | 5′-CATGAAGGAAACCATCTCATGTTCAGC-3′ | g.31183336_31183362 | ||||
| NF1-D6 | NF1_In8_FN | 5′-GCCCTTGGGTTTTTACATAGTGTCAGC-3′ | g.31182416_31182442 | 18,077 | 0.12 | D |
| NF1_In8_RN | 5′-GCACTTTCTGTCAGCTGCCTACTTCC-3′ | g.31200467_31200492 | ||||
| NF1-B4 | NF1_9-10F | 5′-TGTGATGCACATTGAAGTTTGAGAACC-3′ | g.31193253_31193279 | 14,719 | 0.04 | B |
| NF1_9-10R | 5′-GAAAATTATGGGCAGCTGGTAAAGTGG-3′ | g.31207945_31207971 | ||||
| NF1-A5 | NF1_10-15F | 5′-GTATGGGTGCTTTGTGCTTCTTCTGG-3′ | g.31200898_31200923 | 21,982 | 0.1 | A |
| NF1_10-15R | 5′-ACTCCAGAGCTGCACTGTCTAACATGG-3′ | g.31222853_31222879 | ||||
| NF1-B6 | NF1_15-29F | 5′-CTCAAAAGGAAAAAGCTGCACACATAGG-3′ | g.31220468_31220495 | 15,765 | 0.05 | B |
| NF1_15-29R | 5′-TGTCCCTGGATCTAAGGCAAATAAAAGG-3′ | g.31236205_31236232 | ||||
| NF1-C7 | NF1_In29_F | 5′-GGTGATTTTTCAGCTGTAGGGAAGTGG-3′ | g.31233344_31233370 | 13,515 | 0.08 | C |
| NF1_In29_R | 5′-AAGAGATTCCGCAGGCAGACTTACTAGG-3′ | g.31246831_31246858 | ||||
| NF1-A7 | NF1_30-36F | 5′-ACTCAGCACTTCCAGAGATTCCAAGG-3′ | g.31245995_31246020 | 19,438 | 0.07 | A |
| NF1_30-36R | 5′-AAGTCAACTGGGAAAAACCAAACTTGC-3′ | g.31265406_31265432 | ||||
| NF1-D8 | NF1_In35_1F | 5′-TTTGACATCACTGAGGACATCCTAGCC-3′ | g.31263534_31263560 | 17,737 | 0.1 | D |
| NF1_In35_1R | 5′-TGAGAATAGCCCAGAAGTTCACACAGG-3′ | g.31281244_31281270 | ||||
| NF1-C9 | NF1_In35_2F | 5′-AGGCTGTGGGAAGGATATTGTAAGTGG-3′ | g.31280589_31280615 | 15,711 | 0.11 | C |
| NF1_In35_2R | 5′-CCTGTTCATCCTTCTGTTTCTCACACC-3′ | g.31296273_31296299 | ||||
| NF1-D10 | NF1_In35_3F | 5′-TCCAGGGTCCTCAGATTGGTATATTGG-3′ | g.31296081_31296107 | 18,983 | 0.1 | D |
| NF1_In35_3R | 5′-TACTCAGCACCACTGATGAGAGAAATGC-3′ | g.31315036_31315063 | ||||
| NF1-C11 | NF1_In35_4F | 5′-ACAGCGTACGAGAGTTCCATTTTCTCC-3′ | g.31314920_31314946 | 16,782 | 0.11 | C |
| NF1_In35_4R | 5′-CTTATGGTGTACGGTGAAGACGAAGACC-3′ | g.31331674_31331701 | ||||
| NF1-B8 | NF1_37-48FN | 5′-AGAGCTCCAGATGGGTCATTCCTACC-3′ | g.31325474_31325499 | 12,099 | 0.02 | B |
| NF1_37-40R | 5′-TCCAACAAAGCTTCTGTGACTGTTTCC-3′ | g.31337546_31337572 | ||||
| NF1-A9 | NF1_40-48FN | 5′-AGCATCCAGAGGGATATTCTCAGTACCC-3′ | g.31333463_31333490 | 12,591 | 0.013 | A |
| NF1_40-48RN | 5′-TTGCCACGTCTTTGTACTGTCTTAGCC-3′ | g.31346027_31346053 | ||||
| NF1-B10 | NF1_48-57F | 5′-ATTCTAGTGCCCTTTGGATGCTAGGC-3′ | g.31342682_31342707 | 18,097 | 0.09 | B |
| NF1_48-57R | 5′-CTGATCACAAGGGAATTCCTAATGTTGG-3′ | g.31360751_31360778 | ||||
| NF1-D12 | NF1_In56_F | 5′-AGAGCCAGCTTAGTATCACTGCCAACC-3′ | g.31360626_31360652 | 15,976 | 0.11 | D |
| NF1_In56_R | 5′-TCTCAAGGGACACATAGTGGTGAGAGG-3′ | g.31376575_31376601 | ||||
| NF1-A11 | NF1_58-UTR-F | 5′-TCCTAGAATGTGTCCCCGTTGTTAAGC-3′ | g.31373914_31373940 | 4102 | 0.015 | A |
| NF1_58-UTR-R | 5′-TTCTATAGCGGGAAAGCTGAAAAGTTGG-3′ | g.31377988_31378015 | ||||
| Large genomic deletion (type-1 microdeletion)-specific primers # | NC_000017.11 (chr17, GRCh38.p13) | |||||
| NF1-PRS2 | NF1-PRS2-F | 5′-TCAACCTCCCAGGCTCCCGAA-3′ | g.30665807_30665827 | 0.1 | ||
| NF1-PRS2-R | 5′-AGCCCCGAGGGAATGAAAAGC-3′ | g.32085390_32085410 | ||||
| RT-PCR primers | NM_000267.3 (transcript variant 2) | |||||
| NF1-RT1 | NF1_RT_1F | 5′-GACCCTCTCCTTGCCTCTTC-3′ | c.-109_-90 | 785 | 0.4 | |
| NF1_RT_1R | 5′-TTTCTACCCAGTTCCAAAATGC-3′ | c.655_676 | ||||
| NF1-RT2 | NF1_RT_2F | 5′-AAGAAGGTTGCGCAGTTAGC-3′ | c.613_632 | 1461 | 0.4 | |
| NF1_RT_3R | 5′-CAGGGCCACTTCTAGTTTGG-3′ | c.2004_2073 | ||||
| NF1-RT3 | NF1_RT_4F | 5′-CTCTCTCCGGAAGGGAAAAG-3′ | c.1968_1987 | 1410 | 0.4 | |
| NF1_RT_5R | 5′-TGCGCACTTTCATCTTCAAC-3′ | c.3358_3377 | ||||
| NF1-RT4 | NF1_RT_6F | 5′-TGATGGAAGCCAAATCACAG-3′ | c.3284_3303 | 1078 | 0.4 | |
| NF1_RT_31skipR | 5′-GCTGCATCAAAGTTGCTTTTC-3′ | c.4341_4361 | 1141 (variant 1) | |||
| NF1-RT5 | NF1_RT_7S | 5′-AATCCTGCCATTGTCTCACC-3′ | c.4180_4199 | 1141 | 0.4 | |
| NF1_RT_8AS | 5′-CTACTAGGCAGATTTCTTCAATTTCC-3′ | c.5295_5320 | ||||
| NF1-RT6 | NF1_RT_9F | 5′-CCAGAGCACAAACCTGTGG-3′ | c.5215_5237 | 1440 | 0.4 | |
| NF1_RT_10AS | 5′-CACCTGTTGCACTGGTTTTG-3′ | c.6634_6654 | ||||
| NF1-RT7 | NF1_RT_11F | 5′-TGCTTTGACATCCTTGGAAAC-3′ | c.6528_6548 | 1439 | 0.4 | |
| NF1_RT_12R | 5′-AATTTGGATCTTGGCACAATG-3′ | c.7946_7966 | ||||
| NF1-RT8 | NF1_RT_13F | 5′-TAGCAGAGGCCAGTGTTGTG-3′ | c.7865_7884 | 776 | 0.4 | |
| NF1_RT_13R | 5′-GCAGCATTAAATTTAGGCAAGG-3′ | c.*162_183 | ||||
| SPRED1 long PCR primers | NC_000015.10 (chr15, GRCh38.p13) | |||||
| SPRED1_A | SPRED1_A-F | 5′-ATTCCTGGAGAGGGATGGTAGAAGAGG-3′ | g.38250821_38250847 | 19,058 | 0.15 | |
| SPRED1_A-R | 5′-CCATCCAATACAGCAGTCAACACTGG-3′ | g.38269853_38269878 | ||||
| SPRED1_B | SPRED1_B-F | 5′-TGTCTGATGTAAAAGCCACTGATGTCG-3′ | g.38282586_38282612 | 18,830 | 0.15 | |
| SPRED1_B-R | 5′-ACGTGAAACATCAGGTGTTCTGATTCC-3′ | g.38301389_38301415 | ||||
| SPRED1_C | SPRED1_C-F | 5′-TCCTGGAATGGATCCTATACTGGAAGG-3′ | g.38320922_38320948 | 19,080 | 0.15 | |
| SPRED1_C-R | 5′-TTACTGTGTCTGGTAAAGGGCAGATGG-3′ | g.38339975_38340001 | ||||
| SPRED1_D | SPRED1_D-F | 5′-ACCTTGCCAAAAGGACTAGACAACTGC-3′ | g.38345452_38345478 | 17,718 | 0.15 | |
| SPRED1_D-R | 5′-TAAAATAGATAGGGTTGGACGCGATGG-3′ | g.38363143_38363169 | ||||
| #Patient | Age | Sex | Café au Lait Macules | Cutaneous/Subcutaneous Neurofibromas | Plexiform Neurofibroma | Axillary or Groin Freckling | Optic Pathway Glioma | Lisch Nodules | Bony Dysplasia | First Degree Relative with NF1 | Other Findings | Clinical Diagnosis # |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NF_01 * | 30 | F | Y | Y | N | Y | N | Y | Y | Y | Epi, MR | Y |
| NF_02 * | 16 | F | Y | N | N | Y | N | Y | Y | N | UBO | Y |
| NF_03 * | 12 | M | Y | N | N | N | N | N | N | Y | Epi, MR | Y |
| NF_04 * | 3 | M | Y | N | N | N | N | N | N | Y | MR | Y |
| NF_05 * | 21 | F | Y | Y | Y | Y | N | Y | N | N | MR | Y |
| NF_06 | 2 | M | Y | N | N | N | N | N | N | N | Epi | N |
| NF_07 | 13 | M | Y | N | N | N | N | Y | N | Y | DD | Y |
| NF_08 | 8 | M | Y | N | N | Y | N | N | N | N | MR, Mo | Y |
| NF_09 | 1 | F | Y | N | N | N | N | N | N | N | Epi | N |
| NF_10 | 9 | F | Y | Y | N | N | Y | N | N | N | Y | |
| NF_11 | 14 | M | Y | N | N | N | N | N | N | Y | Y | |
| NF_12 | 8 | M | Y | N | N | Y | N | N | N | N | Y | |
| NF_13 | 1 | M | Y | N | N | N | N | N | N | N | N | |
| NF_14 | 9 | F | Y | N | N | Y | N | N | N | N | Y | |
| NF_15 | 12 | M | Y | N | N | Y | N | N | N | N | Y | |
| NF_16 | 5 | M | Y | N | N | N | N | N | N | N | MR, UBO | N |
| NF_17 | 1 | M | Y | N | N | Y | N | N | Y | N | Y | |
| NF_18 | 35 | F | N | Y | Y | N | N | N | N | N | Y | |
| NF_19 | 4 | M | Y | Y | N | Y | N | Y | N | N | MR, Epi, Mo | Y |
| NF_20 | 57 | F | Y | Y | Y | Y | N | N | Y | N | Y |
| #Patient | #Chrom | Pos (hg38) | dbSNP_ID | Ref | Alt | HGVS_Format (NM_000267.3) | Minor Allele Frequency | ClinVar |
|---|---|---|---|---|---|---|---|---|
| NF_01 | chr17 | 31181460 | rs786203448 | C | T | c.625C>T p.Gln209Ter | NA | Pathogenic (known mutation) |
| NF_02 | chr17 | 31229466 | rs1131691122 | G | T | c.2850+1G>T | NA | Pathogenic (known mutation) |
| NF_03 | chr17 | 31260403 | rs199474743 | A | G | c.4402A>G r.4368_4402del p.Phe1457Ter | NA | Pathogenic (known mutation) |
| NF_06 | chr17 | 31249093 | rs137854560 | C | T | c.4084C>T p.Arg1362Ter | T = 0.000008 (1/121,394, ExAC) | Pathogenic (known mutation) |
| NF_07 | chr17 | 31201474 | NA | A | ATT | c.1250_1251insTT p.Ile418SerfsTer56 | NA | Pathogenic (SCV001571436) |
| NF_08 | chr17 | 31233143 | NA | CAA | C | c.3639_3640del p.Met1214AspfsTer3 | NA | Pathogenic (SCV001571437) |
| NF_10 | chr17 | 31226466 | rs17881753 | C | T | c.2033C>T p.Pro678Leu | T = 0.000298 (36/120,726, ExAC) | Benign (1); Likely benign (2); Uncertain significance (1) |
| NF_11 | chr17 | 31337833 | rs786203831 | T | G | c.6594T>G p.Asp2198Glu | G = 0.00005 (1/21,382, ALFA Project) | Uncertain significance |
| NF_12 | chr17 | 31232852 | rs199474764 | A | G | c.3467A>G p.Asn1156Ser | G = 0.000008 (1/121,268, ExAC) | Pathogenic (1); Uncertain significance (1) |
| NF_16 | chr17 | 31200422 | NA | A | T | c.889A>T p.Lys297Ter | NA | Pathogenic (SCV001571438) |
| NF_17 | chr17 | 31181460 | rs786203448 | C | T | c.625C>T p.Gln209Ter | NA | Pathogenic (known mutation) |
| NF_19 | chr17 | 31232840 | rs1321848637 | TACTC | T | c.3457_3460del p.Leu1153MetfsTer4 | delCTCA = 0.000008 (1/125,568, TOPMED) | Pathogenic (known mutation) |
| NF_20 | chr17 | 31095310 | rs1060500252 | A | G | c.1A>G p.Met1Val | NA | Pathogenic (known mutation) |
| PolyPhen-2 | ||||||||
|---|---|---|---|---|---|---|---|---|
| #Patient | dbSNP_ID | SIFT | HumDiv | HumVar | Mutation Taster | Mutation Assessor | Likelihood Ratio Test (LRT) | CADD PHRED-Like Scaled C-Score (>20) |
| NF_10 | rs17881753 | Damaging (0.016) | Benign (0.124) | Possibly damaging (0.816) | Disease causing (0.999915) | Low (1.2) | Deleterious (0.000005) | 21.2 |
| NF_11 | rs786203831 | Tolerated (0.146) | Probably damaging (0.98) | Probably damaging (0.99) | Disease causing (0.999638) | Low (1.59) | Deleterious (0.000000) | 22 |
| NF_12 | rs199474764 | Damaging (0.000) | Probably damaging (0.997) | Probably damaging (1.0) | Disease causing (0.999999) | Medium (2.98) | Deleterious (0.000000) | 25.6 |
| Primer/#Patient | A1 | C1 | D2 | C3 | D4 | C5 | B2 | A3 | D6 | B4 | A5 | B6 | C7 | A7 | D8 | C9 | D10 | C11 | B8 | A9 | B10 | D12 | A11 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NF_01 | 3/1 | 3/0 | 6/0 | 12/0 | 15/1 | 12/0 | 14/0 | 17/1 | 20/4 | 13/3 | 23/0 | 8/0 | 13/3 | 10/0 | 20/3 | 10/3 | 6/3 | 2/0 | 1/0 | 1/0 | 2/0 | 12/2 | 2/1 |
| NF_02 | 0/1 | 2/1 | 1/0 | 0/0 | 0/0 | 2/0 | 3/0 | 1/0 | 11/2 | 10/0 | 1/1 | 20/0 | 8/0 | 3/0 | 15/2 | 7/3 | 7/0 | 25/0 | 7/2 | 7/0 | 15/2 | 19/2 | 2/1 |
| NF_03 | 4/2 | 17/2 | 9/0 | 13/0 | 20/1 | 19/1 | 15/0 | 15/1 | 19/5 | 12/2 | 24/2 | 14/0 | 14/3 | 11/0 | 20/3 | 9/3 | 17/3 | 21/1 | 8/1 | 6/1 | 14/2 | 19/1 | 3/0 |
| NF_04 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 |
| NF_05 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 |
| NF_06 | 0/1 | 1/1 | 2/0 | 0/0 | 1/0 | 3/1 | 0/0 | 0/0 | 1/0 | 0/0 | 0/1 | 1/0 | 0/0 | 1/0 | 1/0 | 0/0 | 1/2 | 0/0 | 2/0 | 0/1 | 0/0 | 0/0 | 0/0 |
| NF_07 | 4/0 | 11/1 | 6/0 | 12/0 | 18/1 | 17/2 | 13/0 | 15/1 | 21/4 | 12/2 | 24/1 | 14/0 | 14/3 | 11/0 | 19/3 | 10/3 | 17/3 | 20/0 | 9/2 | 7/1 | 14/2 | 20/1 | 3/0 |
| NF_08 | 0/0 | 2/0 | 0/0 | 1/0 | 0/0 | 0/0 | 0/0 | 0/1 | 0/0 | 0/0 | 2/0 | 0/0 | 1/0 | 1/0 | 0/0 | 0/1 | 1/0 | 0/0 | 1/0 | 0/0 | 1/1 | 0/0 | 0/0 |
| NF_09 | 3/0 | 16/2 | 15/0 | 12/0 | 22/1 | 20/2 | 15/0 | 16/1 | 20/5 | 13/2 | 24/1 | 14/0 | 14/3 | 10/1 | 22/1 | 9/3 | 19/2 | 20/0 | 7/2 | 6/0 | 13/2 | 19/1 | 3/0 |
| NF_10 | 3/0 | 5/2 | 5/0 | 12/0 | 18/1 | 15/1 | 14/0 | 16/1 | 19/5 | 13/2 | 30/1 | 10/0 | 14/3 | 10/0 | 18/2 | 9/3 | 14/3 | 21/1 | 7/1 | 6/0 | 13/3 | 18/1 | 2/0 |
| NF_11 | 3/0 | 15/2 | 7/0 | 11/0 | 19/1 | 17/2 | 13/0 | 13/1 | 11/2 | 7/2 | 14/1 | 8/0 | 9/3 | 7/0 | 11/0 | 5/1 | 11/3 | 9/0 | 1/0 | 1/0 | 7/0 | 1/1 | 1/1 |
| NF_12 | 0/0 | 0/1 | 2/0 | 1/0 | 0/0 | 4/1 | 0/0 | 0/0 | 0/0 | 1/0 | 1/0 | 1/0 | 1/0 | 0/0 | 1/0 | 2/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 |
| NF_13 | 0/0 | 2/1 | 1/0 | 1/1 | 0/0 | 3/1 | 2/0 | 0/0 | 0/0 | 0/0 | 1/0 | 0/0 | 0/0 | 0/0 | 1/0 | 1/0 | 1/0 | 1/0 | 1/0 | 0/0 | 3/0 | 0/0 | 1/0 |
| NF_14 | 0/0 | 1/0 | 0/0 | 0/0 | 0/0 | 2/0 | 1/0 | 1/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/1 | 1/0 | 1/0 | 0/0 | 3/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 |
| NF_15 | 5/1 | 13/6 | 11/1 | 11/1 | 5/3 | 27/4 | 14/2 | 15/4 | 20/6 | 12/1 | 22/2 | 15/2 | 14/6 | 10/2 | 22/4 | 10/3 | 7/4 | 2/5 | 2/0 | 2/1 | 2/0 | 11/1 | 2/0 |
| NF_16 | 4/0 | 15/2 | 11/0 | 12/0 | 21/1 | 17/1 | 14/0 | 16/1 | 20/5 | 14/1 | 28/1 | 14/0 | 14/3 | 10/0 | 20/3 | 11/3 | 18/3 | 20/1 | 7/1 | 6/1 | 14/2 | 20/1 | 3/0 |
| NF_17 | 2/1 | 14/2 | 8/0 | 9/0 | 19/2 | 17/2 | 14/0 | 14/1 | 12/2 | 6/1 | 13/1 | 6/0 | 8/3 | 7/0 | 11/0 | 7/1 | 10/3 | 10/0 | 1/0 | 1/0 | 4/0 | 1/1 | 1/1 |
| NF_18 | 2/0 | 8/2 | 11/0 | 11/1 | 18/1 | 17/2 | 12/0 | 13/1 | 11/4 | 6/0 | 6/0 | 3/0 | 7/3 | 7/0 | 11/0 | 5/1 | 7/3 | 9/0 | 1/0 | 1/0 | 3/0 | 1/0 | 1/1 |
| NF_19 | 2/0 | 6/0 | 6/0 | 12/0 | 17/1 | 14/0 | 7/0 | 16/1 | 15/4 | 5/0 | 2/0 | 7/0 | 13/3 | 1/0 | 0/0 | 11/3 | 0/0 | 12/0 | 7/1 | 6/0 | 2/0 | 18/2 | 4/1 |
| NF_20 | 4/1 | 13/2 | 9/0 | 11/0 | 19/1 | 18/2 | 14/0 | 15/1 | 19/0 | 25/6 | 1/0 | 14/0 | 14/3 | 12/0 | 20/2 | 10/4 | 15/3 | 21/0 | 9/3 | 8/0 | 13/1 | 19/1 | 3/0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Togi, S.; Ura, H.; Niida, Y. Application of Combined Long Amplicon Sequencing (CoLAS) for Genetic Analysis of Neurofibromatosis Type 1: A Pilot Study. Curr. Issues Mol. Biol. 2021, 43, 782-801. https://0-doi-org.brum.beds.ac.uk/10.3390/cimb43020057
Togi S, Ura H, Niida Y. Application of Combined Long Amplicon Sequencing (CoLAS) for Genetic Analysis of Neurofibromatosis Type 1: A Pilot Study. Current Issues in Molecular Biology. 2021; 43(2):782-801. https://0-doi-org.brum.beds.ac.uk/10.3390/cimb43020057
Chicago/Turabian StyleTogi, Sumihito, Hiroki Ura, and Yo Niida. 2021. "Application of Combined Long Amplicon Sequencing (CoLAS) for Genetic Analysis of Neurofibromatosis Type 1: A Pilot Study" Current Issues in Molecular Biology 43, no. 2: 782-801. https://0-doi-org.brum.beds.ac.uk/10.3390/cimb43020057