
A High-Cholesterol Diet Increases Toll-like Receptors and Other Harmful Factors in the Rabbit Myocardium: The Beneficial Effect of Statins

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Experimental Protocol
2.2. Tissue Preparation
2.3. mRNA Analysis
2.4. Statistical Analysis
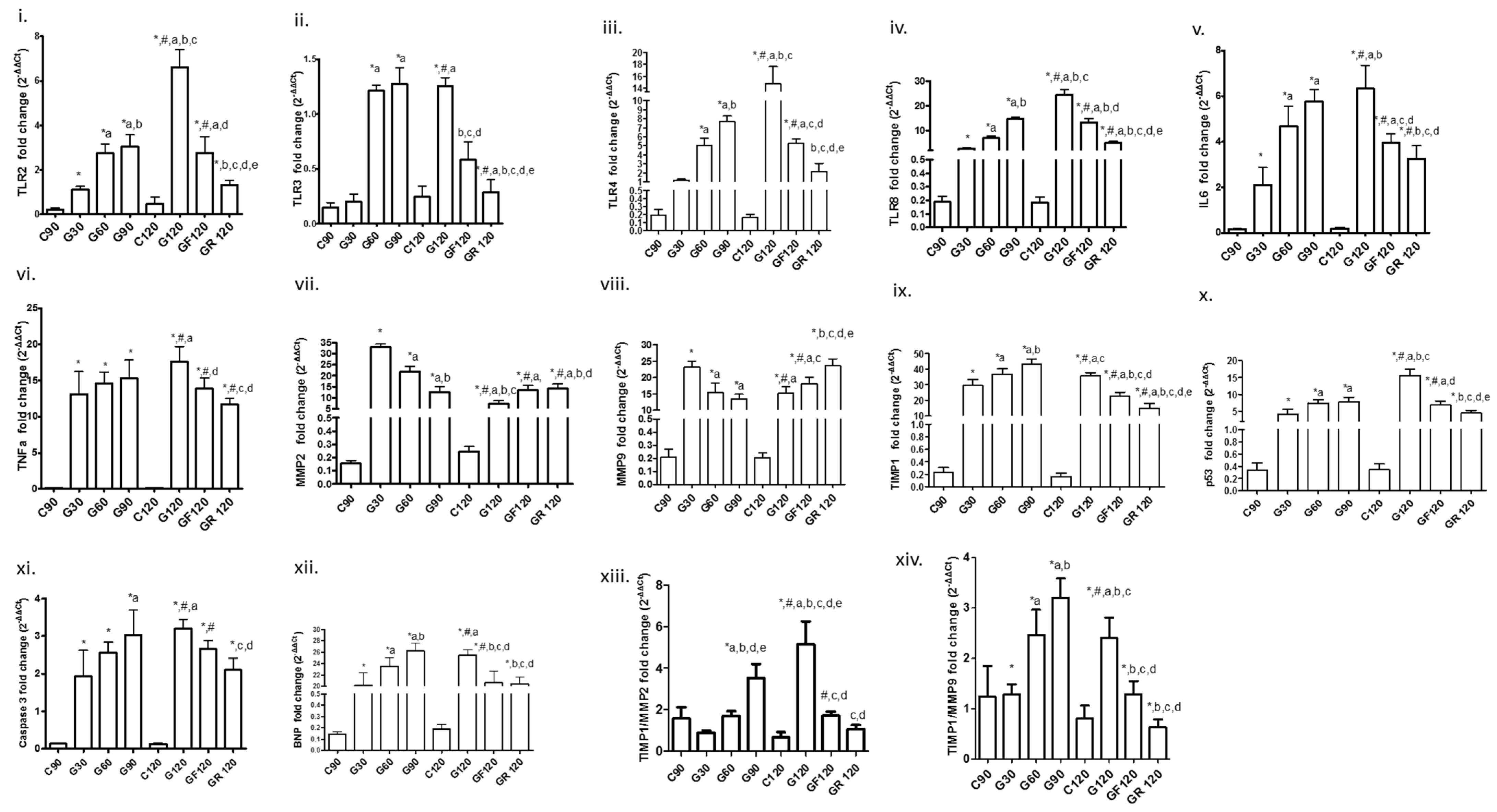
3. Results
3.1. Toll-Like Receptors
3.2. Inflammatory Markers
3.3. ΜΜP2, MMP9, Tissue Inhibitor of Metalloproteinase-1 (TIMP1) and Their Ratio
3.4. Apoptotic Factors
3.5. Biomarker of Myocardial Dysfunction
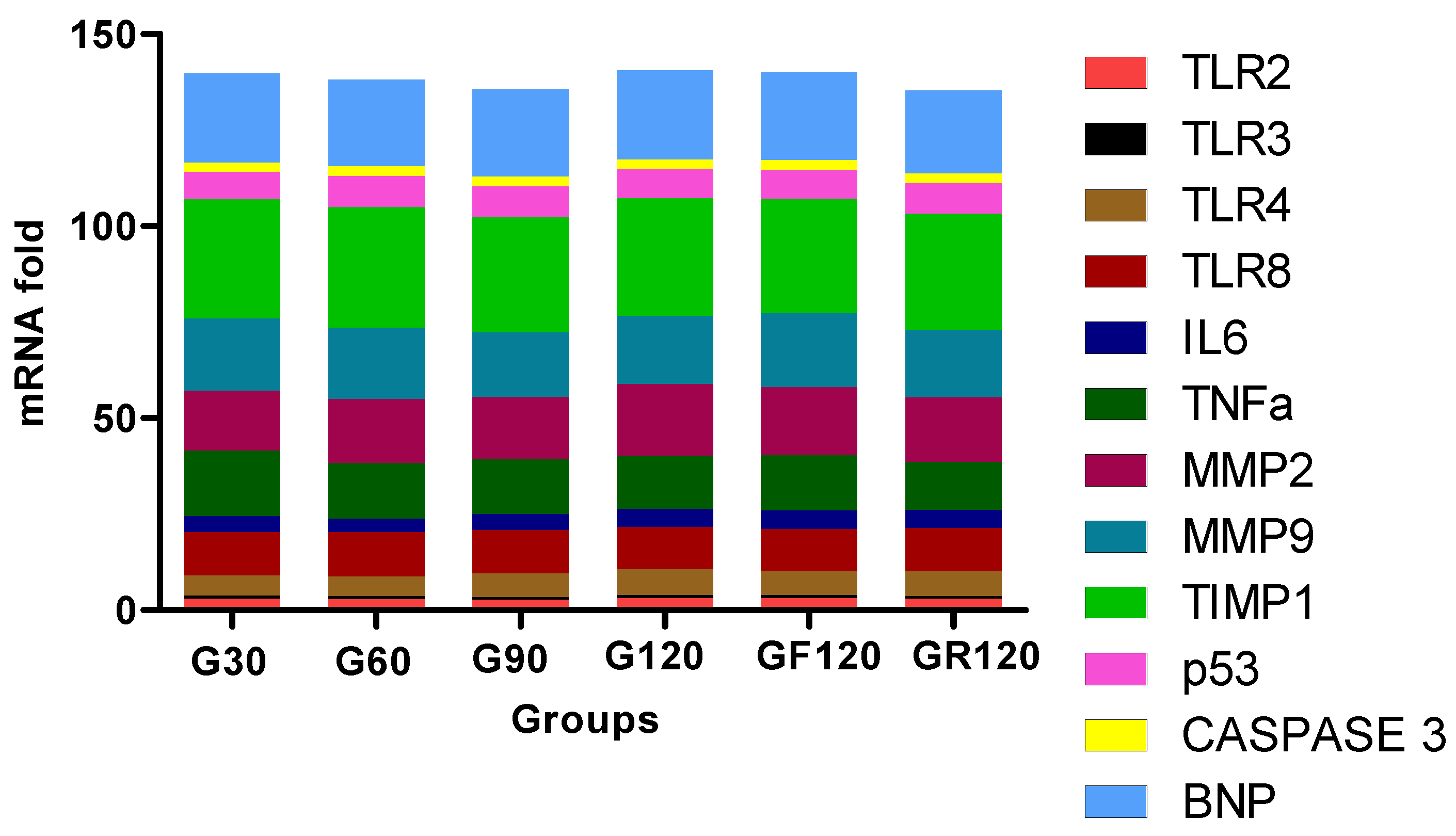
3.6. Trend of Biomarkers
4. Discussion
4.1. TLR Overexpression
4.2. Other Harmful Factors
4.3. Study Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Fayad, Z.A.; Swirski, F.K.; Calcagno, C.; Robbins, C.S.; Mulder, W.; Kovacic, J.C. Monocyte and macrophage dynamics in the cardiovascular system: JACC Macrophage in CVD Series (Part 3). J. Am. Coll. Cardiol. 2018, 72, 2198–2212. [Google Scholar] [CrossRef] [PubMed]
- Goulopoulou, S.; McCarthy, C.G.; Webb, R.C. Toll-like Receptors in the Vascular System: Sensing the dangers within. Pharmacol. Rev. 2016, 68, 142–167. [Google Scholar] [CrossRef] [PubMed]
- Kaczorowski, D.J.; Nakao, A.; McCurry, K.R.; Billiar, T.R. Toll-like receptors and myocardial ischemia/reperfusion, inflammation, and injury. Curr. Cardiol. Rev. 2009, 5, 196–202. [Google Scholar] [CrossRef] [Green Version]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [Green Version]
- Kapelouzou, A.; Giaglis, S.; Peroulis, M.; Katsimpoulas, M.; Moustardas, P.; Aravanis, C.V.; Kostakis, A.; Karayannakos, P.E.; Cokkinos, D.V. Overexpression of Toll-Like Receptors 2, 3, 4, and 8 Is Correlated to the Vascular Atherosclerotic Process in the Hyperlipidemic Rabbit Model: The Effect of Statin Treatment. J. Vasc. Res. 2017, 54, 156–169. [Google Scholar] [CrossRef]
- Tziakas, D.; Chalikias, G.; Kapelouzou, A.; Tentes, I.; Schäfer, K.; Karayannakos, P.; Kostakis, A.; Boudoulas, H.; Konstantinides, S. Erythrocyte membrane cholesterol and lipid core growth in a rabbit model of atherosclerosis: Modulatory effects of rosuvastatin. Int. J. Cardiol. 2013, 170, 173–181. [Google Scholar] [CrossRef]
- Chomczynski, P. Sacchi N: Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 1987, 162, 156–159. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Hardarson, H.S.; Baker, J.S.; Yang, Z.; Purevjav, E.; Huang, C.H.; Alexopoulou, L.; Li, N.; Flavell, R.A.; Bowles, N.E.; Vallejo, J.G. Toll-like receptor 3 is an essential component of the innate stress response in virus-induced cardiac injury. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H251–H258. [Google Scholar] [CrossRef] [Green Version]
- Cole, J.E.; Navin, T.J.; Cross, A.J.; Goddard, M.E.; Alexopoulou, L.; Mitra, A.T.; Davies, A.H.; Flavell, R.A.; Feldmann, M.; Monaco, C. Unexpected protective role for Toll-like receptor 3 in the arterial wall. Proc. Natl. Acad. Sci. USA 2011, 108, 2372–2377. [Google Scholar] [CrossRef] [Green Version]
- Fattahi, F.; Russell, M.W.; Malan, E.A.; Parlett, M.; Abe, E.; Zetoune, F.S.; Ward, P.A. Harmful Roles of TLR3 and TLR9 in Cardiac Dysfunction Developing during Polymicrobial Sepsis. BioMed Res. Int. 2018, 2018, 4302726. [Google Scholar] [CrossRef]
- Gao, T.; Zhang, S.P.; Wang, J.F.; Liu, L.; Wang, Y.; Cao, Z.Y.; Hu, Q.K.; Yuan, W.J.; Lin, L. TLR3 contributes to persistent autophagy and heart failure in mice after myocardial infarction. J. Cell Mol. Med. 2018, 22, 395–408. [Google Scholar] [CrossRef] [Green Version]
- Jurk, M.; Heil, F.; Vollmer, J.; Schetter, C.; Krieg, A.M.; Wagner, H.; Lipford, G.; Bauer, S. Human TLR7 or TLR8 independently confer responsiveness to the antiviral compound R-848. Nat. Immunol. 2002, 3, 499. [Google Scholar] [CrossRef] [PubMed]
- Salagianni, M.; Galani, I.E.; Lundberg, A.M.; Davos, C.H.; Varela, A.; Gavriil, A.; Lyytikäinen, L.P.; Lehtimäki, T.; Sigala, F.; Folkersen, L.; et al. Andreakos EToll-like receptor 7 protects from atherosclerosis by constraining “inflammatory” macrophage activation. Circulation 2012, 126, 952–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Triantafilou, K.; Orthopoulos, G.; Vakakis, E.; Ahmed, M.A.; Golenbock, D.T.; Lepper, P.M.; Triantafilou, M. Human cardiac inflammatory responses triggered by Coxsackie B viruses are mainly Toll-like receptor (TLR) 8-dependent. Cell Microbiol. 2005, 7, 1117–1126. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, T.V.; Okun, E.; Tang, S.C.; Thundyil, J.; Taylor, S.M.; Woodruff, T.M. Toll-like receptors in ischemia-reperfusion injury. Shock 2009, 32, 4–16. [Google Scholar] [CrossRef]
- Spirig, R.; Tsui, J.; Shaw, S. The Emerging Role of TLR and Innate Immunity in Cardiovascular Disease. Cardiol. Res. Pract. 2012, 2012, 181394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruysschaert, J.-M.; Lanez, C. Role of chol and TLR. Microdomains in TLR-mediated signaling. Biochim. Biophys. Acta 2015, 1848, 1860–1867. [Google Scholar] [CrossRef] [PubMed]
- Methe, H.; Kim, J.-O.; Koffler, S.; Nabauer, M.; Weis, M. Statins decrease Toll-like receptor 4 expression and downstream signaling in human CD14+ monocytes. Thromb. Vasc. Biol. 2005, 25, 1439–1445. [Google Scholar] [CrossRef] [Green Version]
- Hua, F.; Ha, T.; Ma, J.; Li, Y.; Kelley, J.; Gao, X.; Browder, I.W.; Kao, R.L.; Williams, D.L.; Li, C. Protection against myocardial ischemia/reperfusion injury in TLR4-deficient mice is mediated through a phosphoinositide 3-kinase-dependent mechanism. J. Immunol. 2007, 178, 7317–7324. [Google Scholar] [CrossRef] [Green Version]
- Shimamoto, A.; Chong, A.J.; Yada, M.; Shomura, S.; Takayama, H.; Fleisig, A.J.; Agnew, M.L.; Hampton, C.R.; Rothnie, C.L.; Spring, D.J.; et al. Inhibition of Toll-like receptor 4 with eritoran attenuates myocardial ischemia-reperfusion injury. Circulation 2006, 114, I270–I274. [Google Scholar] [CrossRef] [Green Version]
- Shishido, T.; Nozaki, N.; Yamaguchi, S.; Shibata, Y.; Nitobe, J.; Miyamoto, T.; Takahashi, H.; Arimoto, T.; Maeda, K.; Yamakawa, M.; et al. Toll-like receptor-2 modulates ventricular remodeling after myocardial infarction. Circulation 2003, 108, 2905–2910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timmers, L.; Sluijter, J.P.; van Keulen, J.K.; Hoefer, I.E.; Nederhoff, M.G.; Goumans, M.J.; Doevendans, P.A.; van Echteld, C.J.; Joles, J.A.; Quax, P.H.; et al. Toll-like receptor 4 mediates maladaptive left ventricular remodeling and impairs cardiac function after myocardial infarction. Circ. Res. 2008, 102, 257–264. [Google Scholar] [CrossRef] [Green Version]
- Frantz, S.; Kobzik, L.; Kim, Y.D.; Fukazawa, R.; Medzhitov, R.; Lee, R.T.; Kelly, R.A. Toll4 (TLR4) expression in cardiac myocytes in normal and failing myocardium. J. Clin. Investig. 1999, 104, 271–280. [Google Scholar] [CrossRef] [Green Version]
- Földes, G.; von Haehling, S.; Okonko, D.O.; Jankowska, E.A.; Poole-Wilson, P.A.; Anker, S.D. Fluvastatin reduces increased blood monocyte Toll-like receptor 4 expression in whole blood from patients with chronic heart failure. Int. J. Cardiol. 2008, 124, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Aliprantis, A.O.; Yang, R.B.; Weiss, D.S.; Godowski, P.; Zychlinsky, A. The apoptotic signaling pathway activated by Toll-like receptor-2. EMBO J. 2000, 19, 3325–3336. [Google Scholar] [CrossRef] [Green Version]
- Ruckdeschel, K.; Pfaffinger, G.; Haase, R.; Sing, A.; Weighardt, H.; Häcker, G.; Holzmann, B.; Heesemann, J. Signaling of apoptosis through TLRs critically involves toll/IL-1 receptor domain-containing adapter inducing IFN-beta, but not MyD88, in bacteria-infected murine macrophages. J. Immunol. 2004, 173, 3320–3328. [Google Scholar] [CrossRef] [Green Version]
- Ternacle, J.; Wan, F.; Sawaki, D.; Surenaud, M.; Pini, M.; Mercedes, R.; Ernande, L.; Audureau, E.; Dubois-Rande, J.L.; Adnot, S.; et al. Short-term high-fat diet compromises myocardial function: A radial strain rate imaging study. Eur. Heart J. Cardiovasc. Imaging 2017, 18, 1283–1291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartekova, M.; Radosinska, J.; Jelemensky, M.; Dhalla, N.S. Role of cytokines and inflammation in heart function during health and disease. Heart Fail. Rev. 2018, 23, 733–758. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, S.; Hartung, D.; Ohshima, S.; Edwards, D.S.; Zhou, J.; Yalamanchili, P.; Azure, M.; Fujimoto, A.; Isobe, S.; Matsumoto, Y.; et al. Molecular imaging of matrix metalloproteinase in atherosclerotic lesions: Resolution with dietary modification and statin therapy. J. Am. Coll. Cardiol. 2008, 52, 1847–1857. [Google Scholar] [CrossRef] [Green Version]
- Galis, Z.S.; Sukhova, G.K.; Lark, M.W.; Libby, P. Increased expression of matrix metalloproteinases and matrix degrading activity in vulnerable regions of human atherosclerotic plaques. J. Clin. Investig. 1994, 94, 2493–2503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeda, U.; Shimada, K. Matrix metalloproteinases and coronary artery diseases. Clin. Cardiol. 2003, 26, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Orbe, J.; Fernandez, L.; Rodríguez, J.A.; Rábago, G.; Belzunce, M.; Monasterio, A.; Roncal, C.; Páramo, J.A. Different expression of MMPs/TIMP-1 in human atherosclerotic lesions. Relation to plaque features and vascular bed. Atherosclerosis 2003, 170, 269–276. [Google Scholar] [CrossRef] [Green Version]
- Galis, Z.S.; Khatri, J.J. Matrix metalloproteinases in vascular remodeling and atherogenesis: The good, the bad, and the ugly. Circ. Res. 2002, 90, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Antonov, I.B.; Kozlov, K.L.; Pal’tseva, E.M.; Polyakova, O.V.; Lin’kova, N.S. Matrix Metalloproteinases MMP-1 and MMP-9 and Their Inhibitor TIMP-1 as Markers of Dilated Cardiomyopathy in Patients of Different Age. Bull. Exp. Biol. Med. 2018, 164, 550–553. [Google Scholar] [CrossRef]
- DeLeon-Pennell, K.Y.; Meschiari, C.A.; Jung, M.; Lindsey, M.L. Matrix Metalloproteinases in Myocardial Infarction and Heart Failure. Prog. Mol. Biol. Transl. Sci. 2017, 147, 75–100. [Google Scholar]
- Becirovic-Agic, M.; Chalise, U.; Daseke, M.J.; Konfrst, S.; Salomon, J.D.; Mishra, P.K.; Lindsey, M.L. Infarct in the Heart: What’s MMP-9 Got to Do with It? Biomolecules 2021, 11, 491. [Google Scholar] [CrossRef]
- Wang, W.; Schulze, C.J.; Suarez-Pinzon, W.L.; Dyck, J.R.; Sawicki, G.; Schulz, R. Intracellular action of matrix metalloproteinase-2 accounts for acute myocardial ischemia and reperfusion injury. Circulation 2002, 106, 1543–1549. [Google Scholar] [CrossRef] [Green Version]
- Heymans, S.; Schroen, B.; Vermeersch, P.; Milting, H.; Gao, F.; Kassner, A.; Gillijns, H.; Herijgers, P.; Flameng, W.; Carmeliet, P.; et al. Increased cardiac expression of tissue inhibitor of metalloproteinase-1 and tissue inhibitor of metalloproteinase-2 is related to cardiac fibrosis and dysfunction in the chronic pressure-overloaded human heart. Circulation 2005, 112, 1136–1144. [Google Scholar] [CrossRef] [Green Version]
- Takawale, A.; Zhang, P.; Patel, V.B.; Wang, X.; Oudit, G.; Kassiri, Z. Tissue Inhibitor of Matrix Metalloproteinase-1 Promotes Myocardial Fibrosis by Mediating CD63-Integrin β1 Interaction. Hypertension 2017, 69, 1092–1103. [Google Scholar] [CrossRef]
- Sundström, J.; Evans, J.C.; Benjamin, E.J.; Levy, D.; Larson, M.G.; Sawyer, D.B.; Siwik, D.A.; Colucci, W.S.; Sutherland, P.; Wilson, P.W.; et al. Relations of plasma matrix metalloproteinase-9 to clinical cardiovascular risk factors and echocardiographic left ventricular measures: The Framingham Heart Study. Circulation 2004, 109, 2850–2856. [Google Scholar] [CrossRef] [PubMed]
- Hansson, J.; Vasan, R.S.; Ärnlöv, J.; Ingelsson, E.; Lind, L.; Larsson, A.; Michaëlsson, K.; Sundström, J. Biomarkers of extracellular matrix metabolism (MMP-9 and TIMP-1) and risk of stroke, myocardial infarction, and cause-specific mortality: Cohort study. PLoS ONE 2011, 6, e16185. [Google Scholar] [CrossRef]
- Hansson, J.; Lind, L.; Hulthe, J.; Sundström, J. Relations of serum MMP-9 and TIMP-1 levels to left ventricular measures and cardiovascular risk factors: A population-based study. Eur. J. Cardiovasc. Prev. Rehabil. 2009, 16, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J. p53, the cellular gatekeeper for growth and division. Cell 1997, 88, 323–331. [Google Scholar] [CrossRef] [Green Version]
- Dixon, L.J.; Flask, C.A.; Papouchado, B.G.; Feldstein, A.E.; Nagy, L.E. Caspase-1 as a central regulator of high fat diet-induced non-alcoholic steatohepatitis. PLoS ONE 2013, 8, e56100. [Google Scholar] [CrossRef] [Green Version]
- Kehat, I.; Molkentin, J.D. Molecular pathways underlying cardiac remodeling during pathophysiologic stimulation. Circulation 2010, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarai, R.; Kaun, C.; Weiss, T.W.; Speidl, W.S.; Rychli, K.; Maurer, G.; Huber, K.; Wojta, J. Human cardiac fibroblasts express B-type natriuretic peptide: Fluvastatin ameliorates its up-regulation by interleukin-1alpha, tumour necrosis factor-alpha and transforming growth factor-beta. J. Cell Mol. Med. 2009, 13, 4415–4421. [Google Scholar] [CrossRef] [Green Version]
- Tsuruda, T.; Boerrigter, G.; Huntley, B.K.; Noser, J.A.; Cataliotti, A.; Costello-Boerrigter, L.C.; Chen, H.H.; Burnett, J.C.J. Brain natriuretic Peptide is produced in cardiac fibroblasts and induces matrix metalloproteinases. Circ. Res. 2002, 91, 1127–1134. [Google Scholar] [CrossRef] [Green Version]
- Carbone, S.; Mauro, A.G.; Mezzaroma, E.; Kraskauskas, D.; Marchetti, C.; Buzzetti, R.; Van Tassell, B.W.; Abbate, A.; Toldo, S. A high-sugar and high-fat diet impairs cardiac systolic and diastolic function in mice. Int. J. Cardiol. 2015, 198, 66–69. [Google Scholar] [CrossRef]
- Carbone, S.; Lee PJ, H.; Mauro, A.G.; Mezzaroma, E.; Buzzetti, R.; Van Tassell, B.; Abbate, A.; Toldo, S. Interleukin-18 mediates cardiac dysfunction induced by western diet independent of obesity and hyperglycemia in the mouse. Nutr. Diabetes 2017, 7, e258. [Google Scholar] [CrossRef] [Green Version]
- Drosatos, K.; Schulze, P.C. Cardiac lipotoxicity: Molecular pathways and therapeutic implications. Curr. Heart Fail. Rep. 2013, 10, 109–121. [Google Scholar] [CrossRef] [Green Version]
- Battault, S.; Renguet, E.; Van Steenbergen, A.; Horman, S.; Beauloye, C.; Bertrand, L. Myocardial glucotoxicity: Mechanisms and potential therapeutic targets. Arch. Cardiovasc. Dis. 2020, 113, 736–748. [Google Scholar] [CrossRef]
- Malmgren, S.; Spégel, S.; Danielsson, A.P.H.; Nagorny, C.L.; Andersson, L.E.; Dekker Nitert, M.; Ridderstråle, M.; Mulder, H.; Ling, C. Coordinate changes in histone modifications, mRNA levels, and metabolite profiles in clonal INS-1 832/13 β-cells accompany functional adaptations to lipotoxicity. J. Biol. Chem. 2013, 288, 11973–11987. [Google Scholar] [CrossRef] [Green Version]
- Cencioni, C.; Spallotta, F.; Greco, S.; Martelli, F.; Zeiher, A.M.; Gaetano, C. Epigenetic mechanisms of hyperglycemic memory. Int. J. Biochem. Cell Biol. 2014, 51, 155–158. [Google Scholar] [CrossRef] [PubMed]
- Keating, S.T.; El-Osta, A. Epigenetics and metabolism. Circ. Res. 2015, 116, 715–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, T.-M.; Chai, T.-F.; Tsai, C.-H. Association of pravastatin and left ventricular mass in hypercholesterolemic patients: Role of 8-150 prostaglandin fzalpha formation. Clin. Trial J. Cardiovasc. Pharmacol. 2002, 40, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Deo, S.V.; Rababa’h, A.; Altarabsheh, S.E.; Lim, J.Y.; Cho, Y.H.; Park, S.J. Statin therapy improves long-term survival in non-ischaemic cardiomyopathy: A pooled analysis of 4500 patients. Heart Lung Circ. 2014, 23, 985–987. [Google Scholar] [CrossRef]
- Gastelurrutia, P.; Lupon, J.; de Antonio, M.; Urrutia, A.; Díez, C.; Coll, R.; Altimir, S.; Bayes-Genis, A. Statins in heart failure: The paradox between large randomized clinical trials and real life. Mayo Clin. Proc. 2012, 87, 555–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krum, H.; McMurray, J.J. Statins and chronic heart failure: Do we need a large-scale outcome trial? J. Am. Coll. Cardiol. 2002, 39, 1567–1573. [Google Scholar] [CrossRef] [Green Version]
- Ward, N.C.; Watts, G.F.; Eckel, R.H. Statin Toxicity. Circ. Res. 2019, 124, 328–350. [Google Scholar] [CrossRef]
- Yu, L.; Feng, Z. The Role of Toll-Like Receptor Signaling in the Progression of Heart Failure. Mediat. Inflamm. 2018, 2018, 9874109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimura, M.; Naito, S. Tissue-specific mRNA expression profiles of human toll-like receptors and related genes. Biol. Pharm. Bull. 2005, 28, 886–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zarember, K.A.; Godowski, P.J. Tissue expression of human Toll-like receptors and differential regulation of Toll-like receptor mRNAs in leukocytes in response to microbes, their products, and cytokines. J. Immunol. 2002, 168, 554–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akbal, E.; Koçak, E.; Köklü, S.; Ergül, B.; Akyürek, Ö.; Yılmaz, F.M. Serum Toll-Like Receptor-2, Toll-Like Receptor-4 Levels in Patients with HBeAg-Negative Chronic Viral Hepatitis B. Viral Immunol. 2017, 30, 278–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Gene | Forward | Reserve |
|---|---|---|
| TLR2 | 5′-CTCCTGCTGACGCTGCTC-3′ | 5′-TTCCTCGGCTTCCTCTTGG-3′ |
| TLR3 | 5′-ATCTCCTCTCTTTGGGGACTGTTG-3′ | 5′-TGTTGGTGGGCAGGTCATCAGG-3′ |
| TLR4 | 5′-CTCACATCCGAGTTGCCTTCCG-3′ | 5′-AAATGCTCCCTGGTACACCTGTTC-3′ |
| TLR8 | 5′-ATCTTGTTCTTCTTCTCGTTCTC-3′ | 5′-CCTGTAACCTCTGACCTTGG-3′ |
| IL6 | 5′-CTACCGCTTTCCCCACTTCAG-3′ | 5′-TCCTCAGCTCCTTGATGGTCTC-3′ |
| TNFa | 5′-AGCCCACGTAGTAGCAAACCC-3′ | 5′-TTGATGGCAGAGAGGAGGTTGA-3′ |
| MMP2 | 5′-GAAGGTCAAGTGGTCCGTGT-3′ | 5′-CCGTACTTGCCATCCTTCTC-3′ |
| MMP9 | 5-TGCCA GAGTACCTGTTCCGCTATG-3 | 5-TGCCACTTGAGGTCACCCTCGAA-3 |
| TIMP1 | 5′-TTCTCATCGCTGGACAACTG-3′ | 5′-AGCGTAGGTCTTGGTGAAGC-3′ |
| p53 | 5′-ATGCCTACCTCACGGGGTCT-3′ | 5′-AGGGTAGGGAACCAGCACCAT-3′ |
| BNP | 5′-TGC TCT TCT TGC ACC TGT-3′ | 5′-GCA GCT GCT GTA TCT CAG AAA-3′ |
| CASP3 | 5′-GGTAGCGACAGAGTTCGAGT-3′ | 5′-TGAGAGGGAAGCAGAGTAACAG-3′ |
| b-ACTIN | 5′-CCATGTACGTGGCCATCCAG-3′ | 5′-TCTTCATGAGGTAGTCGGTCAGGTC-3′ |
| Groups | C90 | G30 | G60 | G90 | C120 | G120 | GF120 | GR120 |
|---|---|---|---|---|---|---|---|---|
| TLR2 | 0.211 ± 0.078 | 1.120 ± 0.135 *, a | 2.743 ± 0.422 *, a | 3.038 ± 0.544 *, a, b | 0.478 ± 0.303 | 6.605 ± 0.800 *, #, a, b, c | 2.768 ± 0.719 *, #, a, d | 1.328 ± 0.195 *, b, c, d, e |
| TLR3 | 0.15 ± 0.04 | 0.2 ± 0.067 | 1.212 ± 0.05 *, a | 1.273 ± 0.147 *, a | 0.246 ± 0.097 | 1.252 ± 0.078 *, #, a | 0.583 ± 0.161 b, c, d | 0.285 ± 0.116 *, a, b, c, d, e |
| TLR4 | 0.19 ± 0.07 | 1.213 ± 0.092 | 5.017 ± 0.845 *, a | 7.705 ± 0.613 *, a, b | 0.165 ± 0.036 | 14.67 ± 2.925 *, #, a, b, c | 5.277 ± 0.481 *, #, c, d | 2.18 ± 0.886 b, c, d, e |
| TLR8 | 0.188 ± 0.041 | 2.643 ± 0.469 * | 6.98 ± 0.769 *, a | 14.68 ± 0.876 *, a, b | 0.185 ± 0.04 | 24.47 ± 2.169 *, #, a, b, c | 13.38 ± 1.589 *, #, b, d | 4.962 ± 0.674 *, #, a, b, c, d, e |
| IL6 | 0.16 ± 0.034 | 2.112 ± 0.759 * | 4.687 ± 0.866 *, a | 5.768 ± 0.522 *, a | 0.198 ± 0.047 | 6.324 ± 1.027 *, #, a, b | 3.957 ± 0.389 *, #, c, d | 3.25 ± 0.579 *, #, b, c, d |
| TNFa | 0.141 ± 0.034 | 13.15 ± 3.073 * | 14.64 ± 1.476 * | 15.32 ± 2.553 * | 0.13 ± 0.028 | 17.62 ± 2.113 *, #, a | 13.89 ± 1.423 *, #, d | 11.67 ± 0.834 *, #, c, d |
| MMP2 | 0.153 ± 0.023 | 32.88 ± 1.516 * | 21.8 ± 2.492 *, a | 12.57 ± 2.514 *, a, b | 0.243 ± 0.038 | 7.195 ± 1.428 *, #, a, b, c | 13.43 ± 2.279 *, #, a | 14.06 ± 2.187 *, #, a, b, d |
| MMP9 | 0.206 ± 0.063 | 23.16 ± 1.72 * | 15.37 ± 2.949 *, a | 13.53 ± 1.408 *, a | 0.205 ± 0.037 | 15.18 ± 2.002 *, #, a | 18.03 ± 2.124 *, #, a, c | 23.59 ± 2.1 *, b, c, d, e |
| TIMP1 | 0.235 ± 0.073 | 29.53 ± 3.81 * | 36.84 ± 3.336 *, a | 43.06 ± 3.411 *, a, b | 0.163 ± 0.059 | 35.77 ± 2.033 *, #, a, c | 22.83 ± 2.341 *, #, a, b, c, d | 14.83 ± 3.143 *, #, a, b, c, d, e |
| p53 | 0.333 ± 0.121 | 4.163 ± 1.498 * | 7.413 ± 0.962 *, a | 7.803 ± 1.304 *, a | 0.348 ± 0.09 | 15.53 ± 1.897 *, #, a, b, c | 6.785 ± 1.266 *, #, a, d | 4.42 ± 0.841 *, b, c, d, e |
| CASP3 | 0.13 ± 0.014 | 1.937 ± 0.682 * | 2.552 ± 0.285 * | 3.033 ± 0.665 *, a | 0.128 ± 0.024 | 3.195 ± 0.258 *, #, a | 2.65 ± 0.237 *, #, a | 2.103 ± 0.316 *, c, d |
| BNP | 0.145 ± 0.018 | 20.12 ± 2.34 * | 23.52 ± 1.424 *, a | 26.21 ± 1.328 *, a, b | 0.19 ± 0.041 | 25.44 ± 0.998 *, #, a | 20.63 ± 2.049 *, #, b, c, d | 20.4 ± 1.259 *, b, c, d |
| TIMP1/MMP2 | 1.53 ± 0.53 | 0.89 ± 0.09 | 1.7 ± 0.22 | 3.53 ± 0.67 *, a, b, d, e | 0.67 ± 0.24 | 5.14 ± 1.01 *, #, a, b, c, d, e | 1.72 ± 0.19 *, #, c, d | 1.06 ± 0.19 c, d |
| TIMP1/MMP9 | 1.24 ± 0.6 | 1.28 ± 0.2 * | 2.46 ± 0.49 *, a | 3.2 ± 0.37 *, a, b | 0.8 ± 0.25 | 2.4 ± 0.4 *, #, a, b, c | 1.29 ± 0.25 *, b, c, d | 0.63 ± 0.15 *, b, c, d |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kapelouzou, A.; Katsimpoulas, M.; Kontogiannis, C.; Lidoriki, I.; Georgiopoulos, G.; Kourek, C.; Papageorgiou, C.; Mylonas, K.S.; Dritsas, S.; Charalabopoulos, A.; et al. A High-Cholesterol Diet Increases Toll-like Receptors and Other Harmful Factors in the Rabbit Myocardium: The Beneficial Effect of Statins. Curr. Issues Mol. Biol. 2021, 43, 818-830. https://0-doi-org.brum.beds.ac.uk/10.3390/cimb43020059
Kapelouzou A, Katsimpoulas M, Kontogiannis C, Lidoriki I, Georgiopoulos G, Kourek C, Papageorgiou C, Mylonas KS, Dritsas S, Charalabopoulos A, et al. A High-Cholesterol Diet Increases Toll-like Receptors and Other Harmful Factors in the Rabbit Myocardium: The Beneficial Effect of Statins. Current Issues in Molecular Biology. 2021; 43(2):818-830. https://0-doi-org.brum.beds.ac.uk/10.3390/cimb43020059
Chicago/Turabian StyleKapelouzou, Alkistis, Michalis Katsimpoulas, Christos Kontogiannis, Irene Lidoriki, Georgios Georgiopoulos, Christos Kourek, Christos Papageorgiou, Konstantinos S. Mylonas, Spyridon Dritsas, Alexandros Charalabopoulos, and et al. 2021. "A High-Cholesterol Diet Increases Toll-like Receptors and Other Harmful Factors in the Rabbit Myocardium: The Beneficial Effect of Statins" Current Issues in Molecular Biology 43, no. 2: 818-830. https://0-doi-org.brum.beds.ac.uk/10.3390/cimb43020059