
Farnesol Ameliorates Demyelinating Phenotype in a Cellular and Animal Model of Charcot-Marie-Tooth Disease Type 1A

 , and
, and 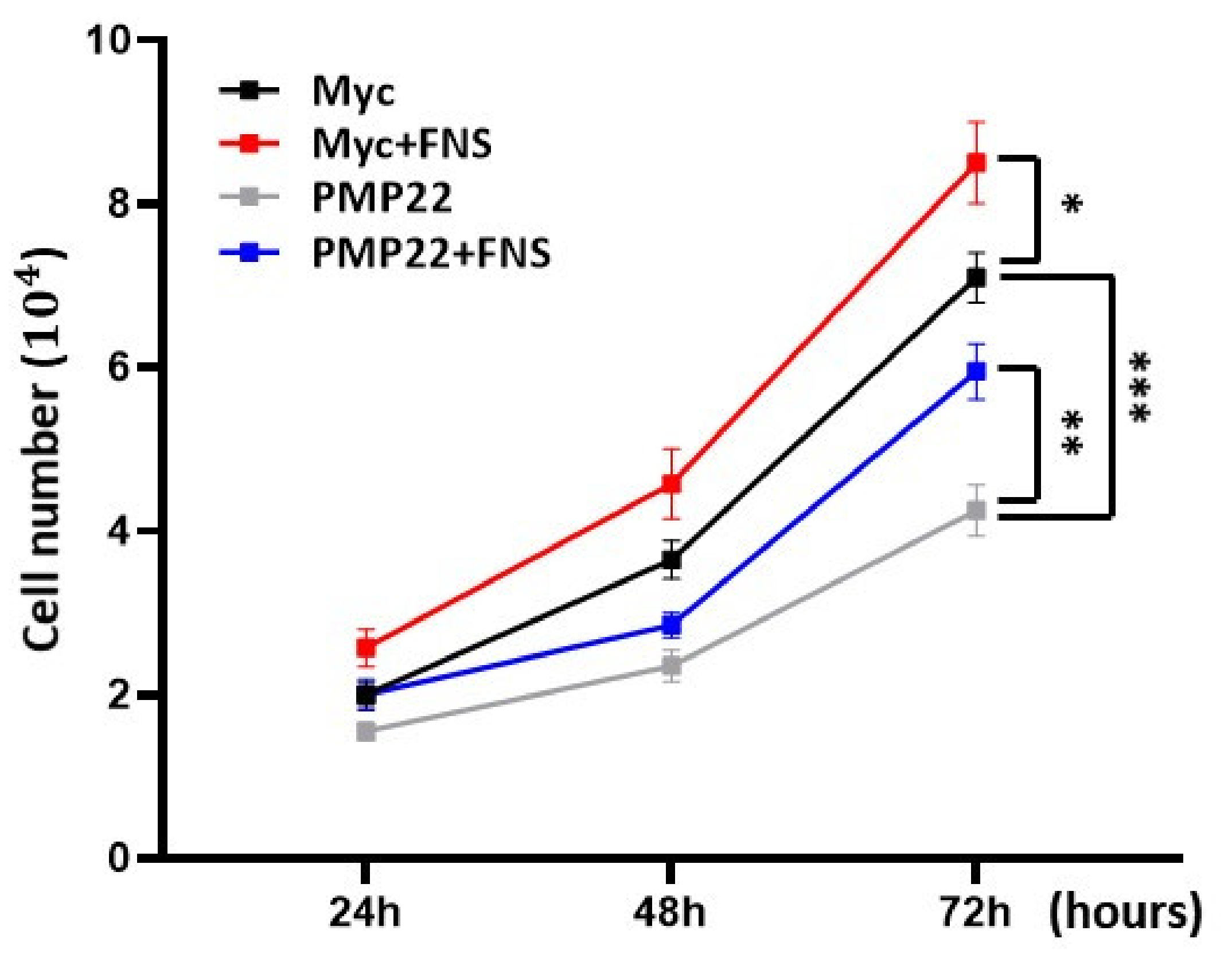{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Quantitation of mRNA
2.3. Mouse Drug Administration and Phenotype Evaluation
2.4. Electron Microscopy and Determination of Axon Diameter
2.5. Immunohistochemistry
2.6. Statistical Analysis
3. Results
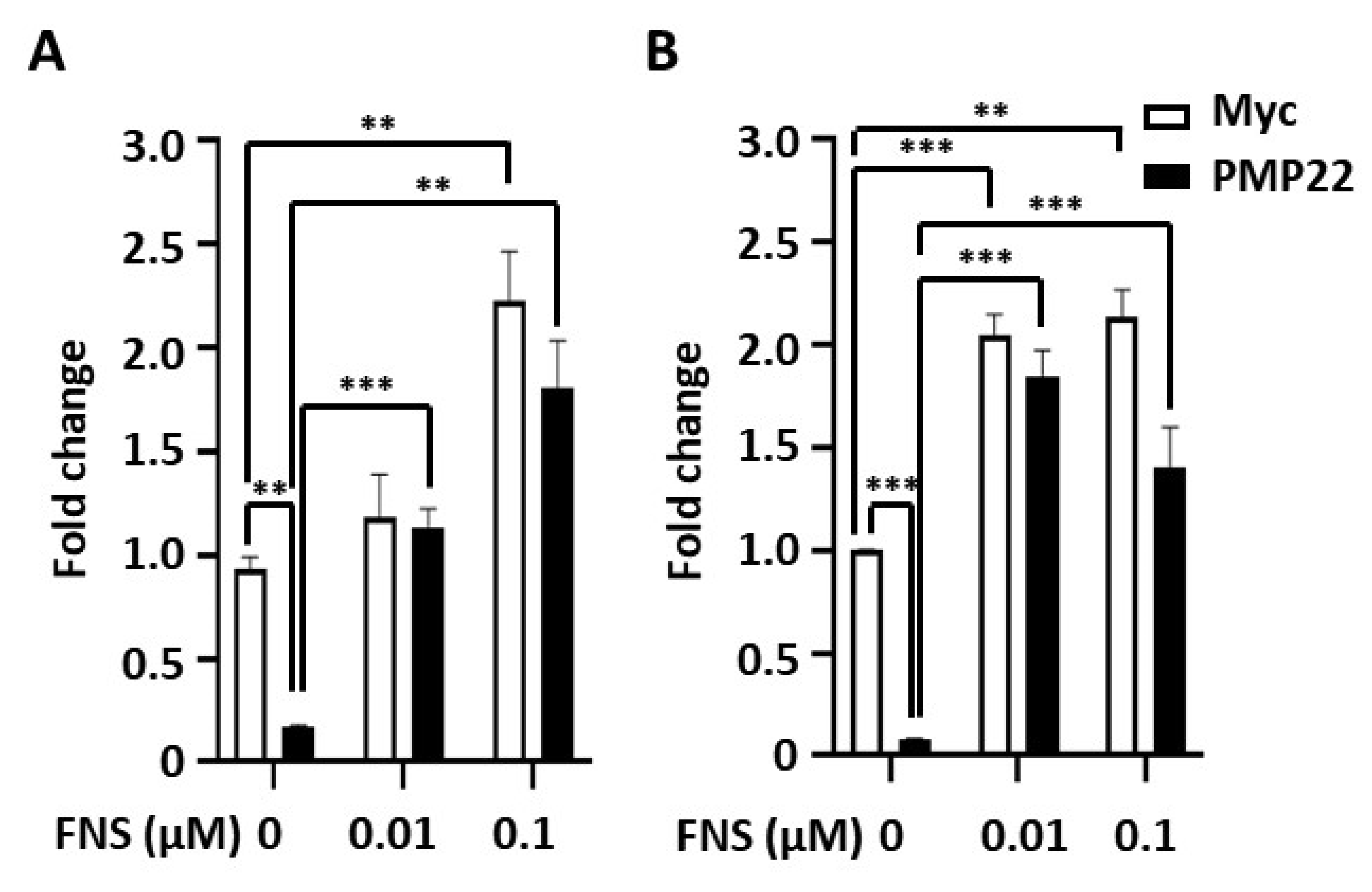
3.1. Farnesol Increases Myelination In Vitro
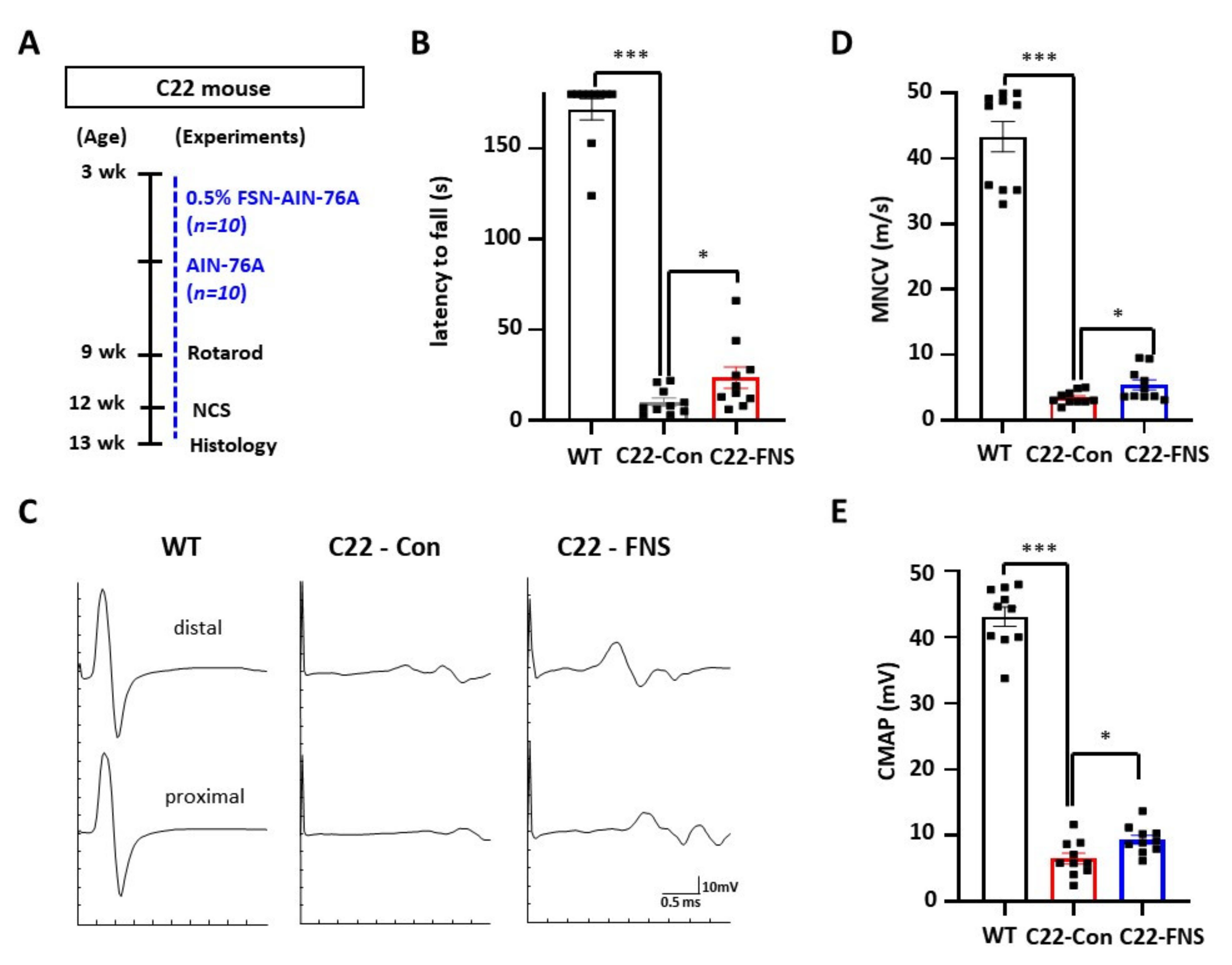
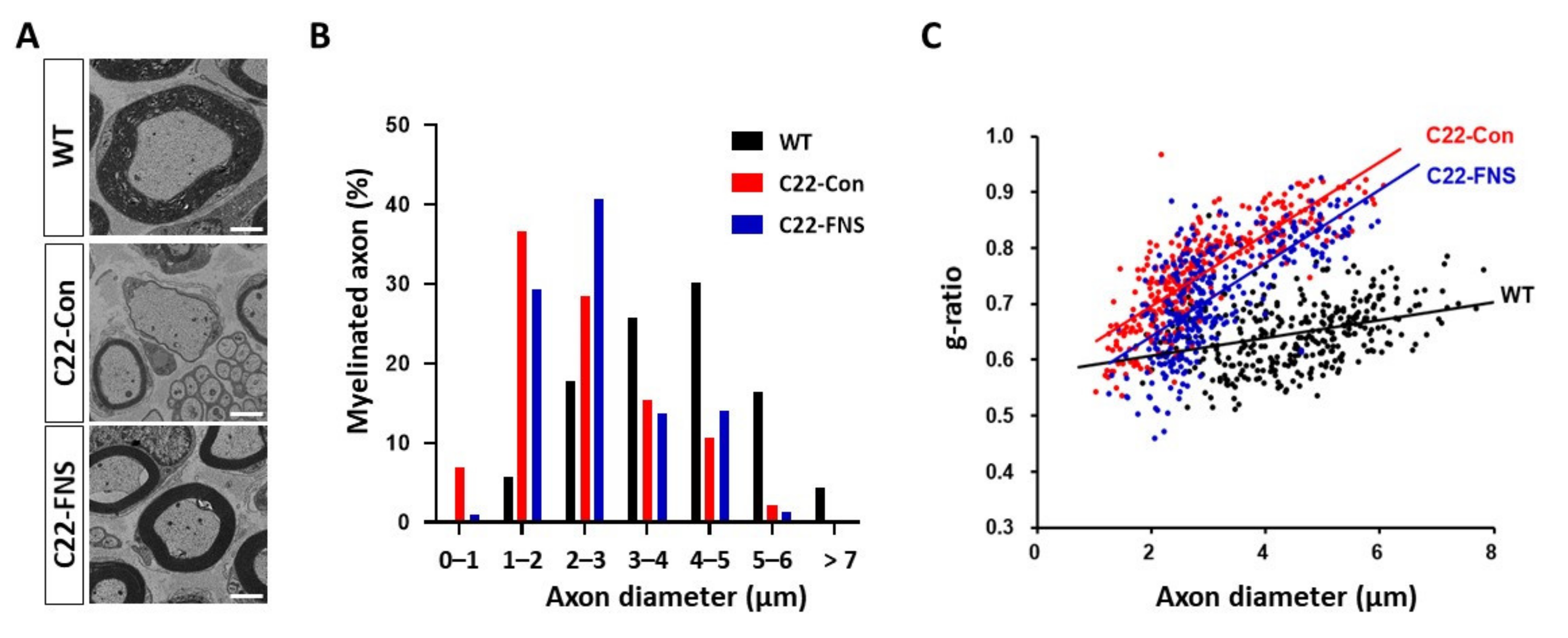
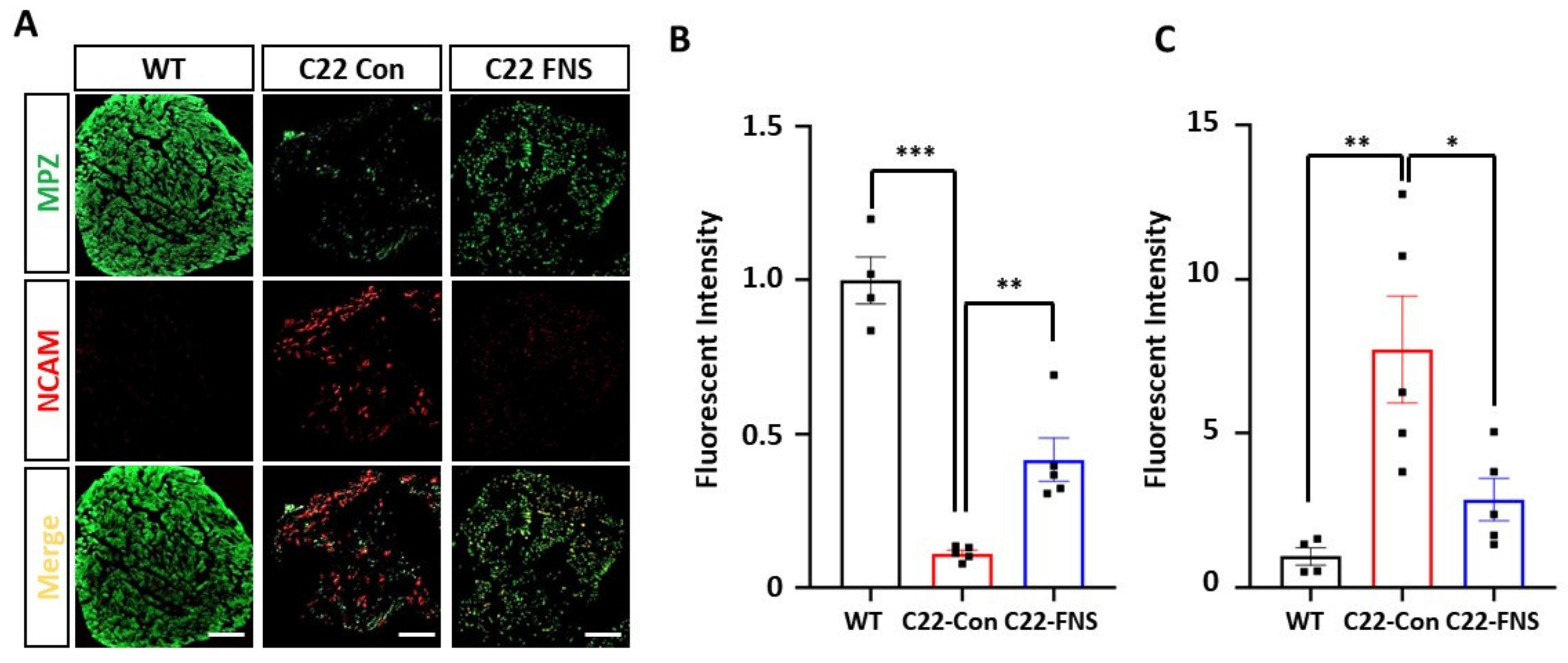
3.2. Farnesol Ameliorates Demyelinating Phenotype of CMT1A Mouse Model
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Skre, H. Genetic and clinical aspects of Charcot-Marie-Tooth’s disease. Clin. Genet. 1974, 6, 98–118. [Google Scholar] [CrossRef] [PubMed]
- Laurá, M.; Pipis, M.; Rossor, A.M.; Reilly, M.M. Charcot-Marie-Tooth disease and related disorders: An evolving landscape. Curr. Opin. Neurol. 2019, 32, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Harding, A.E.; Thomas, P.K. Genetic aspects of hereditary motor and sensory neuropathy (types I and II). J. Med. Genet. 1980, 17, 329–336. [Google Scholar] [CrossRef] [Green Version]
- Pareyson, D.; Marchesi, C.; Salsano, E. Dominant Charcot-Marie-Tooth syndrome and cognate disorders. Handb. Clin. Neurol. 2013, 115, 817–845. [Google Scholar] [PubMed]
- Miniou, P.; Fontes, M. Therapeutic Development in Charcot Marie Tooth Type 1 Disease. Int. J. Mol. Sci. 2021, 22, 6755. [Google Scholar] [CrossRef] [PubMed]
- Eggermann, K.; Gess, B.; Hausler, M.; Weis, J.; Hahn, A.; Kurth, I. Hereditary Neuropathies. Dtsch. Arztebl. Int. 2018, 115, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Pipis, M.; Rossor, A.M.; Laura, M.; Reilly, M.M. Next-generation sequencing in Charcot-Marie-Tooth disease: Opportunities and challenges. Nat. Rev. Neurol. 2019, 15, 644–656. [Google Scholar] [CrossRef] [PubMed]
- McCray, B.A.; Scherer, S.S. Axonal Charcot-Marie-Tooth Disease: From Common Pathogenic Mechanisms to Emerging Treatment Opportunities. Neurotherapeutics 2021, 1–17. [Google Scholar] [CrossRef]
- Verhoeven, K.; De Jonghe, P.; Coen, K.; Verpoorten, N.; Auer-Grumbach, M.; Kwon, J.M.; FitzPatrick, D.; Schmedding, E.; De Vriendt, E.; Jacobs, A.; et al. Mutations in the small GTP-ase late endosomal protein RAB7 cause Charcot-Marie-Tooth type 2B neuropathy. Am. J. Hum. Genet. 2003, 72, 722–727. [Google Scholar] [CrossRef] [Green Version]
- Puls, I.; Jonnakuty, C.; LaMonte, B.H.; Holzbaur, E.L.; Tokito, M.; Mann, E.; Floeter, M.K.; Bidus, K.; Drayna, D.; Oh, S.J.; et al. Mutant dynactin in motor neuron disease. Nat. Genet. 2003, 33, 455–456. [Google Scholar] [CrossRef] [Green Version]
- Crimella, C.; Baschirotto, C.; Arnoldi, A.; Tonelli, A.; Tenderini, E.; Airoldi, G.; Martinuzzi, A.; Trabacca, A.; Losito, L.; Scarlato, M.; et al. Mutations in the motor and stalk domains of KIF5A in spastic paraplegia type 10 and in axonal Charcot-Marie-Tooth type 2. Clin. Genet. 2012, 82, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Evgrafov, O.V.; Mersiyanova, I.; Irobi, J.; Van Den Bosch, L.; Dierick, I.; Leung, C.L.; Schagina, O.; Verpoorten, N.; Van Impe, K.; Fedotov, V.; et al. Mutant small heat-shock protein 27 causes axonal Charcot-Marie-Tooth disease and distal hereditary motor neuropathy. Nat. Genet. 2004, 36, 602–606. [Google Scholar] [CrossRef] [Green Version]
- Fontaine, J.M.; Sun, X.K.; Hoppe, A.D.; Simon, S.; Vicart, P.; Welsh, M.J.; Benndorf, R. Abnormal small heat shock protein interactions involving neuropathy-associated HSP22 (HSPB8) mutants. FASEB J. 2006, 20, 2168. [Google Scholar] [CrossRef] [PubMed]
- Zimoń, M.; Baets, J.; Fabrizi, G.M.; Jaakkola, E.; Kabzińska, D.; Pilch, J.; Schindler, A.B.; Cornblath, D.R.; Fischbeck, K.H.; Auer-Grumbach, M.; et al. Dominant GDAP1 mutations cause predominantly mild CMT phenotypes. Neurology 2011, 77, 540–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu-Wai-Man, P.; Griffiths, P.G.; Gorman, G.S.; Lourenco, C.M.; Wright, A.F.; Auer-Grumbach, M.; Toscano, A.; Musumeci, O.; Valentino, M.L.; Caporali, L.; et al. Multi-system neurological disease is common in patients with OPA1 mutations. Brain 2010, 133, 771–786. [Google Scholar] [CrossRef]
- Zuchner, S.; De Jonghe, P.; Jordanova, A.; Claeys, K.G.; Guergueltcheva, V.; Cherninkova, S.; Hamilton, S.R.; Van Stavern, G.; Krajewski, K.M.; Stajich, J.; et al. Axonal neuropathy with optic atrophy is caused by mutations in mitofusin 2. Annu. Neurol. 2006, 59, 276–281. [Google Scholar] [CrossRef] [PubMed]
- Antonellis, A.; Ellsworth, R.E.; Sambuughin, N.; Puls, I.; Abel, A.; Lee-Lin, S.Q.; Jordanova, A.; Kremensky, I.; Christodoulou, K.; Middleton, L.T.; et al. Glycyl tRNA synthetase mutations in Charcot-Marie-Tooth disease type 2D and distal spinal muscular atrophy type V. Am. J. Hum. Genet. 2003, 72, 1293–1299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordanova, A.; Irobi, J.; Thomas, F.P.; Van Dijck, P.; Meerschaert, K.; Dewil, M.; Dierick, I.; Jacobs, A.; De Vriendt, E.; Guergueltcheva, V.; et al. Disrupted function and axonal distribution of mutant tyrosyl-tRNA synthetase in dominant intermediate Charcot-Marie-Tooth neuropathy. Nat. Genet. 2006, 38, 197–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boczonadi, V.; Jennings, M.J.; Horvath, R. The role of tRNA synthetases in neurological and neuromuscular disorders. FEBS Lett. 2018, 592, 703–717. [Google Scholar] [CrossRef] [Green Version]
- Lin, W.; Popko, B. Endoplasmic reticulum stress in disorders of myelinating cells. Nat. Neurosci. 2009, 12, 379–385. [Google Scholar] [CrossRef] [Green Version]
- Pennuto, M.; Tinelli, E.; Malaguti, M.; Del Carro, U.; D’Antonio, M.; Ron, D.; Quattrini, A.; Feltri, M.L.; Wrabetz, L. Ablation of the UPR-mediator CHOP restores motor function and reduces demyelination in Charcot-Marie-Tooth 1B mice. Neuron 2008, 57, 393–405. [Google Scholar] [CrossRef] [Green Version]
- Pisciotta, C.; Saveri, P.; Pareyson, D. Updated review of therapeutic strategies for Charcot-Marie-Tooth disease and related neuropathies. Expert. Rev. Neurother. 2021, 21, 701–713. [Google Scholar] [CrossRef]
- D’Ydewalle, C.; Krishnan, J.; Chiheb, D.M.; Van Damme, P.; Irobi, J.; Kozikowski, A.P.; Vanden Berghe, P.; Timmerman, V.; Robberecht, W.; Van Den Bosch, L. HDAC6 inhibitors reverse axonal loss in a mouse model of mutant HSPB1-induced Charcot-Marie-Tooth disease. Nat. Med. 2011, 17, 968–974. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Woo, S.Y.; Hong, Y.B.; Choi, H.; Kim, J.; Choi, H.; Mook-Jung, I.; Ha, N.; Kyung, J.; Koo, S.K.; et al. HDAC6 Inhibitors Rescued the Defective Axonal Mitochondrial Movement in Motor Neurons Derived from the Induced Pluripotent Stem Cells of Peripheral Neuropathy Patients with HSPB1 Mutation. Stem Cells Int. 2016, 2016, 9475981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, R.A.; McDermott, M.P.; Herrmann, D.N.; Hoke, A.; Clawson, L.L.; Siskind, C.; Feely, S.M.; Miller, L.J.; Barohn, R.J.; Smith, P.; et al. High-dosage ascorbic acid treatment in Charcot-Marie-Tooth disease type 1A: Results of a randomized, double-masked, controlled trial. JAMA Neurol. 2013, 70, 981–987. [Google Scholar] [CrossRef] [Green Version]
- Rocha, A.G.; Franco, A.; Krezel, A.M.; Rumsey, J.M.; Alberti, J.M.; Knight, W.C.; Biris, N.; Zacharioudakis, E.; Janetka, J.W.; Baloh, R.H.; et al. MFN2 agonists reverse mitochondrial defects in preclinical models of Charcot-Marie-Tooth disease type 2A. Science 2018, 360, 336–341. [Google Scholar] [CrossRef] [Green Version]
- Attarian, S.; Young, P.; Brannagan, T.H.; Adams, D.; Van Damme, P.; Thomas, F.P.; Casanovas, C.; Tard, C.; Walter, M.C.; Péréon, Y.; et al. A double-blind, placebo-controlled, randomized trial of PXT3003 for the treatment of Charcot-Marie-Tooth type 1A. Orphanet J. Rare Dis. 2021, 16, 433. [Google Scholar] [CrossRef]
- Morena, J.; Gupta, A.; Hoyle, J.C. Charcot-Marie-Tooth: From Molecules to Therapy. Int. J. Mol. Sci. 2019, 20, 3419. [Google Scholar] [CrossRef] [Green Version]
- Jung, Y.Y.; Hwang, S.T.; Sethi, G.; Fan, L.; Arfuso, F.; Ahn, K.S. Potential Anti-Inflammatory and Anti-Cancer Properties of Farnesol. Molecules 2018, 23, 2827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lateef, A.; Rehman, M.U.; Tahir, M.; Khan, R.; Khan, A.Q.; Qamar, W.; Sultana, S. Farnesol protects against intratracheally instilled cigarette smoke extract-induced histological alterations and oxidative stress in prostate of wistar rats. Toxicol. Int. 2013, 20, 35–42. [Google Scholar]
- Santhanasabapathy, R.; Sudhandiran, G. Farnesol attenuates lipopolysaccharide-induced neurodegeneration in Swiss albino mice by regulating intrinsic apoptotic cascade. Brain Res. 2015, 1620, 42–56. [Google Scholar] [CrossRef]
- Santhanasabapathy, R.; Vasudevan, S.; Anupriya, K.; Pabitha, R.; Sudhandiran, G. Farnesol quells oxidative stress, reactive gliosis and inflammation during acrylamide-induced neurotoxicity: Behavioral and biochemical evidence. Neuroscience 2015, 308, 212–227. [Google Scholar] [CrossRef] [PubMed]
- Sell, L.B.; Ramelow, C.C.; Kohl, H.M.; Hoffman, K.; Bains, J.K.; Doyle, W.J.; Strawn, K.D.; Hevrin, T.; Kirby, T.O.; Gibson, K.M.; et al. Farnesol induces protection against murine CNS inflammatory demyelination and modifies gut microbiome. Clin. Immunol. 2021, 108766. [Google Scholar] [CrossRef]
- Choi, Y.-R.; Jung, S.-C.; Shin, J.; Yoo, S.Y.; Lee, J.-S.; Lee, J.; Hong, Y.B.; Choi, B.-O. Development of cell models for high-throughput screening system of Charcot-Marie-Tooth disease type 1. J. Genet. Med. 2015, 12, 25–30. [Google Scholar] [CrossRef]
- Huxley, C.; Passage, E.; Robertson, A.M.; Youl, B.; Huston, S.; Manson, A.; Saberan-Djoniedi, D.; Figarella-Branger, D.; Pellissier, J.F.; Thomas, P.K.; et al. Correlation between varying levels of PMP22 expression and the degree of demyelination and reduction in nerve conduction velocity in transgenic mice. Hum. Mol. Genet. 1998, 7, 449–458. [Google Scholar] [CrossRef] [Green Version]
- Ku, C.M.; Lin, J.Y. Farnesol, a sesquiterpene alcohol in herbal plants, exerts anti-inflammatory and antiallergic effects on ovalbumin-sensitized and -challenged asthmatic mice. Evid. Based Complement. Alternat. Med. 2015, 2015, 387357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, A.; Lee, Y.; Kam, T.I.; Kang, S.U.; Neifert, S.; Karuppagounder, S.S.; Khang, R.; Kang, H.; Park, H.; Chou, S.C.; et al. PARIS farnesylation prevents neurodegeneration in models of Parkinson’s disease. Sci. Transl. Med. 2021, 13, eaax8891. [Google Scholar] [CrossRef]
- Lee, J.S.; Kwak, G.; Kim, H.J.; Park, H.T.; Choi, B.O.; Hong, Y.B. miR-381 Attenuates Peripheral Neuropathic Phenotype Caused by Overexpression of PMP22. Exp. Neurobiol. 2019, 28, 279–288. [Google Scholar] [CrossRef]
- Hwang, S.H.; Chang, E.H.; Kwak, G.; Jeon, H.; Choi, B.O.; Hong, Y.B. Gait parameters as tools for analyzing phenotypic alterations of a mouse model of Charcot-Marie-Tooth disease. Anim. Cells Syst. 2021, 25, 11–18. [Google Scholar] [CrossRef]
- Kim, Y.H.; Kim, Y.H.; Shin, Y.K.; Jo, Y.R.; Park, D.K.; Song, M.Y.; Yoon, B.A.; Nam, S.H.; Kim, J.H.; Choi, B.O.; et al. p75 and neural cell adhesion molecule 1 can identify pathologic Schwann cells in peripheral neuropathies. Annu. Clin. Transl. Neurol. 2019, 6, 1292–1301. [Google Scholar] [CrossRef]
- Brancolini, C.; Marzinotto, S.; Edomi, P.; Agostoni, E.; Fiorentini, C.; Muller, H.W.; Schneider, C. Rho-dependent regulation of cell spreading by the tetraspan membrane protein Gas3/PMP22. Mol. Biol. Cell. 1999, 10, 2441–2459. [Google Scholar] [CrossRef] [Green Version]
- Nobbio, L.; Vigo, T.; Abbruzzese, M.; Levi, G.; Brancolini, C.; Mantero, S.; Grandis, M.; Benedetti, L.; Mancardi, G.; Schenone, A. Impairment of PMP22 transgenic Schwann cells differentiation in culture: Implications for Charcot-Marie-Tooth type 1A disease. Neurobiol. Dis. 2004, 16, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Sancho, S.; Young, P.; Suter, U. Regulation of Schwann cell proliferation and apoptosis in PMP22-deficient mice and mouse models of Charcot-Marie-Tooth disease type 1A. Brain 2001, 124, 2177–2187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoidl, G.; Blass-Kampmann, S.; D’Urso, D.; Schmalenbach, C.; Muller, H.W. Retroviral-mediated gene transfer of the peripheral myelin protein PMP22 in Schwann cells: Modulation of cell growth. EMBO J. 1995, 14, 1122–1128. [Google Scholar] [CrossRef] [PubMed]
- Komyathy, K.; Neal, S.; Feely, S.; Miller, L.J.; Lewis, R.A.; Trigge, G.; Siskind, C.E.; Shy, M.E.; Ramchandren, S. Anterior tibialis CMAP amplitude correlations with impairment in CMT1A. Muscle Nerve 2013, 47, 493–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jahangir, T.; Khan, T.H.; Prasad, L.; Sultana, S. Alleviation of free radical mediated oxidative and genotoxic effects of cadmium by farnesol in Swiss albino mice. Redox Rep. 2005, 10, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Lizarbe, S.; Civera-Tregon, A.; Cantarero, L.; Herrer, I.; Juarez, P.; Hoenicka, J.; Palau, F. Neuroinflammation in the pathogenesis of axonal Charcot-Marie-Tooth disease caused by lack of GDAP1. Exp. Neurol. 2019, 320, 113004. [Google Scholar] [CrossRef]
- Shy, M.E.; Arroyo, E.; Sladky, J.; Menichella, D.; Jiang, H.; Xu, W.; Kamholz, J.; Scherer, S.S. Heterozygous P0 knockout mice develop a peripheral neuropathy that resembles chronic inflammatory demyelinating polyneuropathy (CIDP). J. Neuropathol. Exp. Neurol. 1997, 56, 811–821. [Google Scholar] [CrossRef] [PubMed]
- Luft, U.C.; Bychkov, R.; Gollasch, M.; Gross, V.; Roullet, J.B.; McCarron, D.A.; Ried, C.; Hofmann, F.; Yagil, Y.; Yagil, C.; et al. Farnesol blocks the L-type Ca2+ channel by targeting the alpha 1C subunit. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 959–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roullet, J.B.; Luft, U.C.; Xue, H.; Chapman, J.; Bychkov, R.; Roullet, C.M.; Luft, F.C.; Haller, H.; McCarron, D.A. Farnesol inhibits L-type Ca2+ channels in vascular smooth muscle cells. J. Biol. Chem. 1997, 272, 32240–32246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roullet, J.B.; Spaetgens, R.L.; Burlingame, T.; Feng, Z.P.; Zamponi, G.W. Modulation of neuronal voltage-gated calcium channels by farnesol. J. Biol. Chem. 1999, 274, 25439–25446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nobbio, L.; Sturla, L.; Fiorese, F.; Usai, C.; Basile, G.; Moreschi, I.; Benvenuto, F.; Zocchi, E.; De Flora, A.; Schenone, A.; et al. P2X7-mediated increased intracellular calcium causes functional derangement in Schwann cells from rats with CMT1A neuropathy. J. Biol. Chem. 2009, 284, 23146–23158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanoye, C.G.; Sakakura, M.; Follis, R.M.; Trevisan, A.J.; Narayan, M.; Li, J.; Sanders, C.R.; Carter, B.D. Peripheral myelin protein 22 modulates store-operated calcium channel activity, providing insights into Charcot-Marie-Tooth disease etiology. J. Biol. Chem. 2019, 294, 12054–12065. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, N.-Y.; Kwak, G.; Doo, H.-M.; Kim, H.-J.; Jang, S.-Y.; Lee, Y.-I.; Choi, B.-O.; Hong, Y.-B. Farnesol Ameliorates Demyelinating Phenotype in a Cellular and Animal Model of Charcot-Marie-Tooth Disease Type 1A. Curr. Issues Mol. Biol. 2021, 43, 2011-2021. https://0-doi-org.brum.beds.ac.uk/10.3390/cimb43030138
Park N-Y, Kwak G, Doo H-M, Kim H-J, Jang S-Y, Lee Y-I, Choi B-O, Hong Y-B. Farnesol Ameliorates Demyelinating Phenotype in a Cellular and Animal Model of Charcot-Marie-Tooth Disease Type 1A. Current Issues in Molecular Biology. 2021; 43(3):2011-2021. https://0-doi-org.brum.beds.ac.uk/10.3390/cimb43030138
Chicago/Turabian StylePark, Na-Young, Geon Kwak, Hyun-Myung Doo, Hye-Jin Kim, So-Young Jang, Yun-Il Lee, Byung-Ok Choi, and Young-Bin Hong. 2021. "Farnesol Ameliorates Demyelinating Phenotype in a Cellular and Animal Model of Charcot-Marie-Tooth Disease Type 1A" Current Issues in Molecular Biology 43, no. 3: 2011-2021. https://0-doi-org.brum.beds.ac.uk/10.3390/cimb43030138