Human Lupus Plasma Pro-Atherogenic Effects on Cultured Macrophages Are Not Mitigated by Statin Therapy: A Mechanistic LAPS Substudy

 ,
, 
Abstract
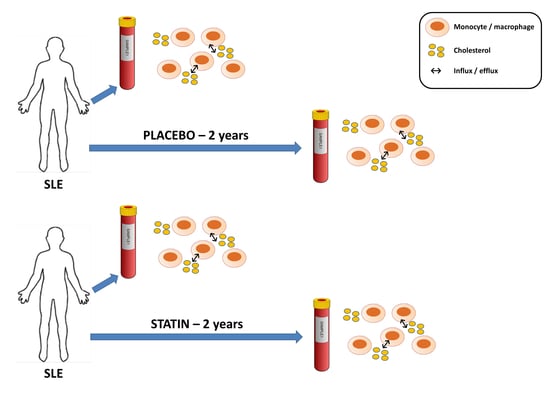
:
1. Introduction
2. Materials and Methods
2.1. Subject Inclusion and Exclusion Criteria
- Patients with a clinical diagnosis of SLE, confirmed by a faculty rheumatologist at Johns Hopkins.
- Age 18 years or older.
- Given informed consent.
- SLE patients with a known atherosclerotic event, such as angina, myocardial infarction, or stroke, with an abnormal lipid profile for which a statin would be standard of care.
- Pregnant patients (or patients planning to become pregnant in the next two years).
- Patients with known chronic liver disease, unexplained elevation of their liver enzymes greater than 2-times the upper limit of normal, or an elevated creatine phosphokinase greater than 1.5-times the upper limit of the normal value for the patient’s racial group.
- Patients with triglycerides >500 mg/dl or LDL >190 in the absence of two risk factors (and who were unwilling to participate in a formal nutritional/lifestyle modification program that was recommended for them).
2.2. Statin or Placebo Treatment
2.3. Cell Culture and Experimental Conditions
2.4. RNA Isolation and Gene Expression Analysis by QRT-PCR
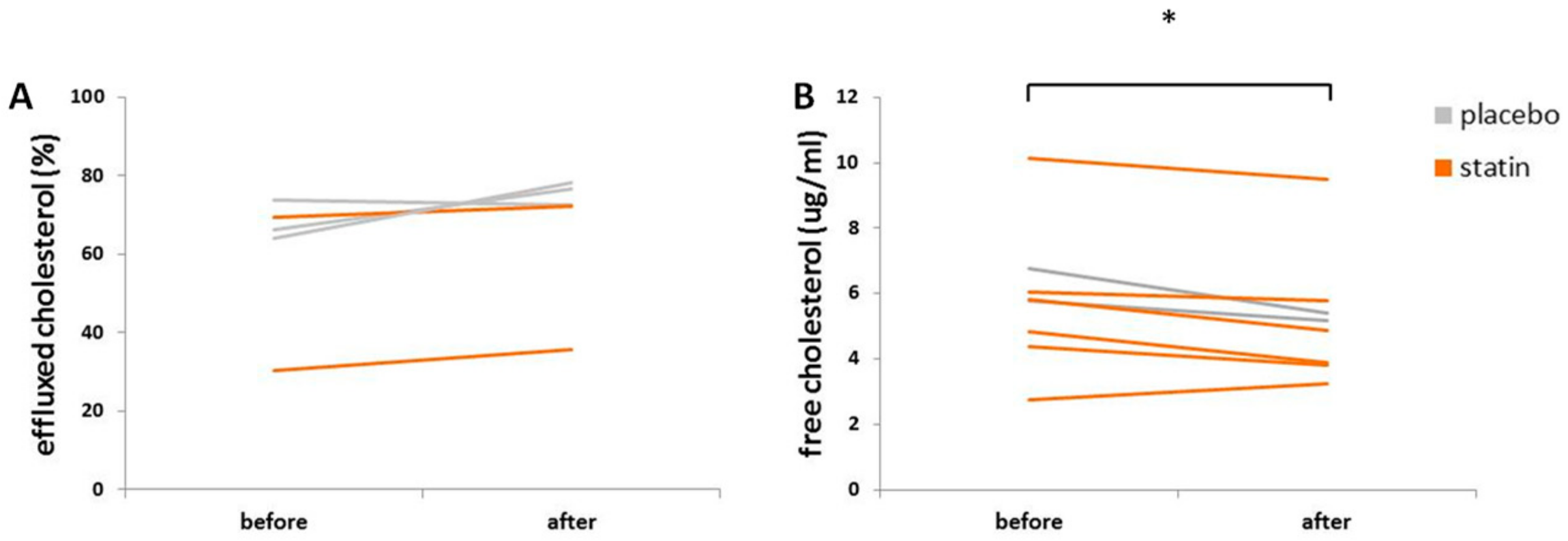
2.5. Cholesterol Efflux Analysis
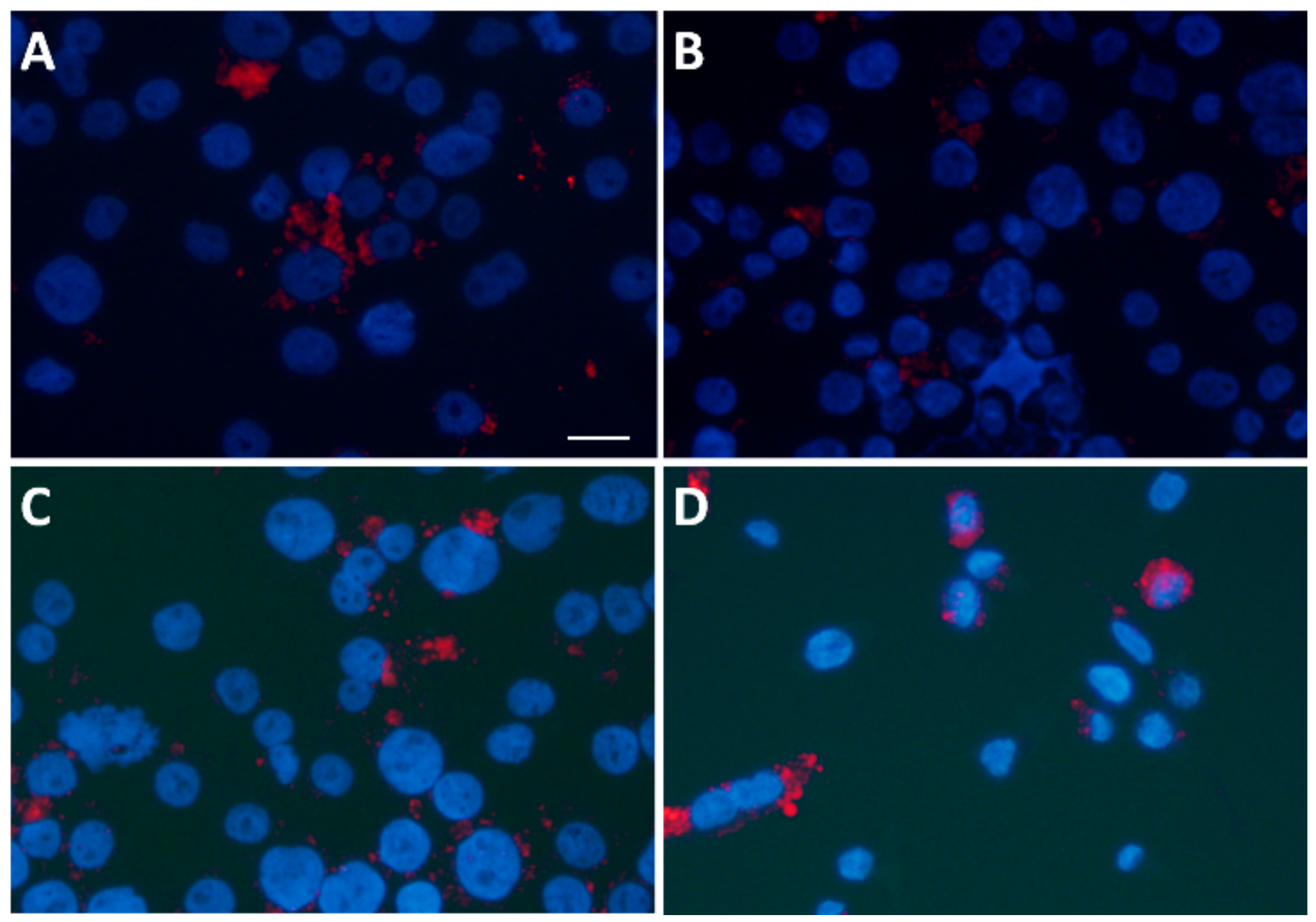
2.6. Oxidized Cholesterol Uptake Analysis
2.7. Data Analysis
3. Results
3.1. Demographics, Clinical Characteristics, and Immunological Status of SLE Patients
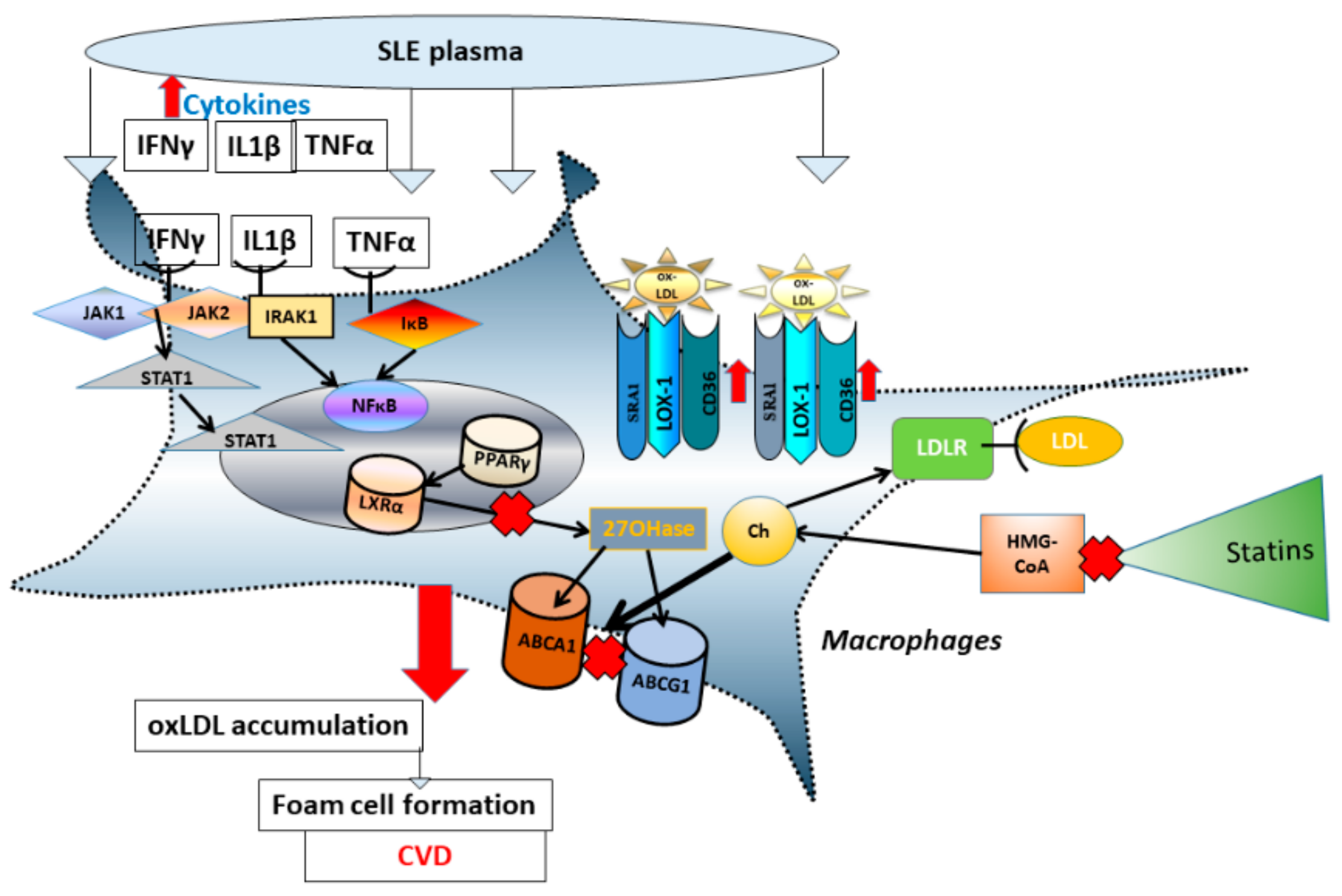
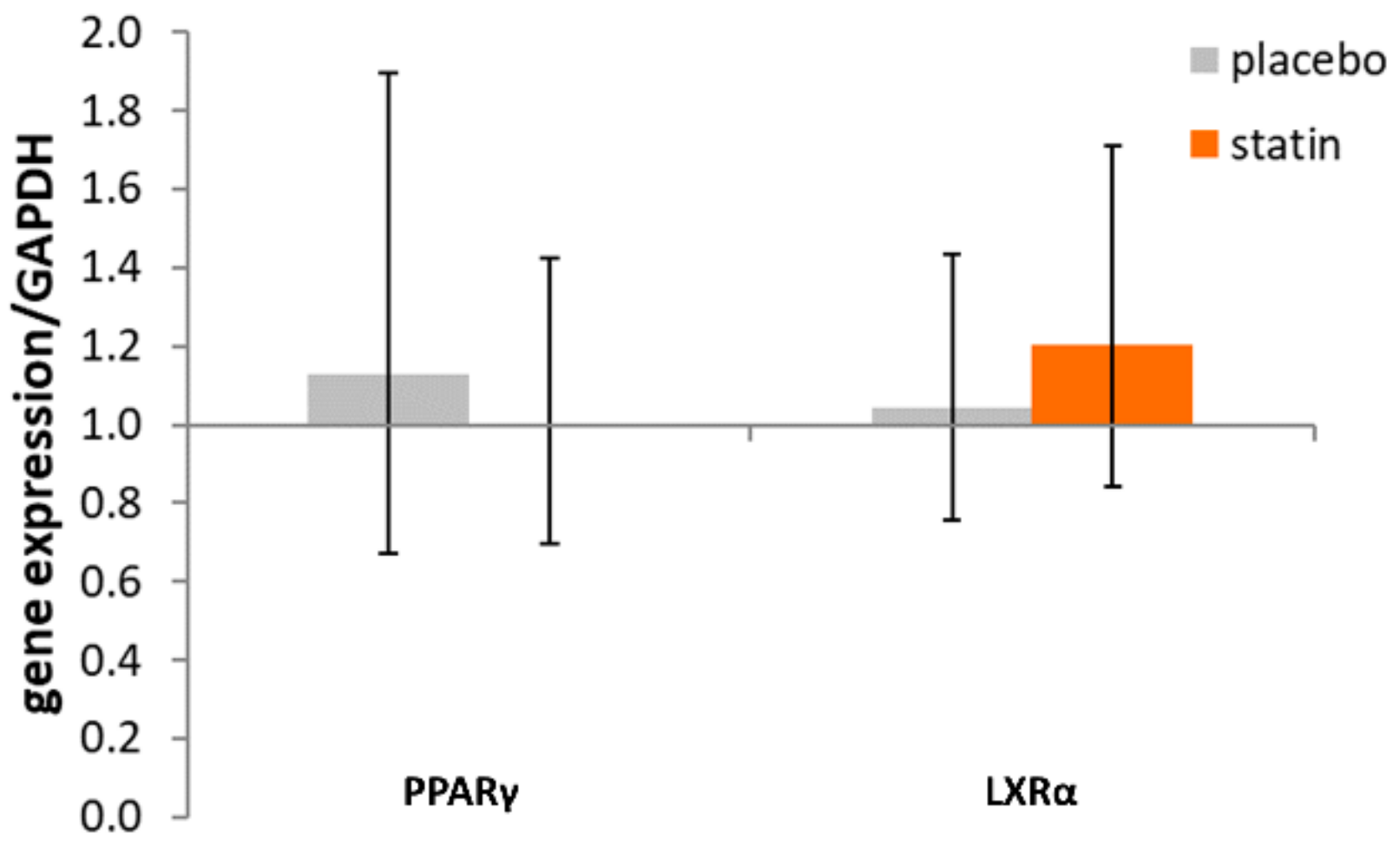
3.2. Comparison of Nuclear Receptor Expression (PPARγ and LXRα) in THP-1 Macrophages Exposed to SLE Plasma Obtained Pre- and Post-Placebo or Statin Therapy
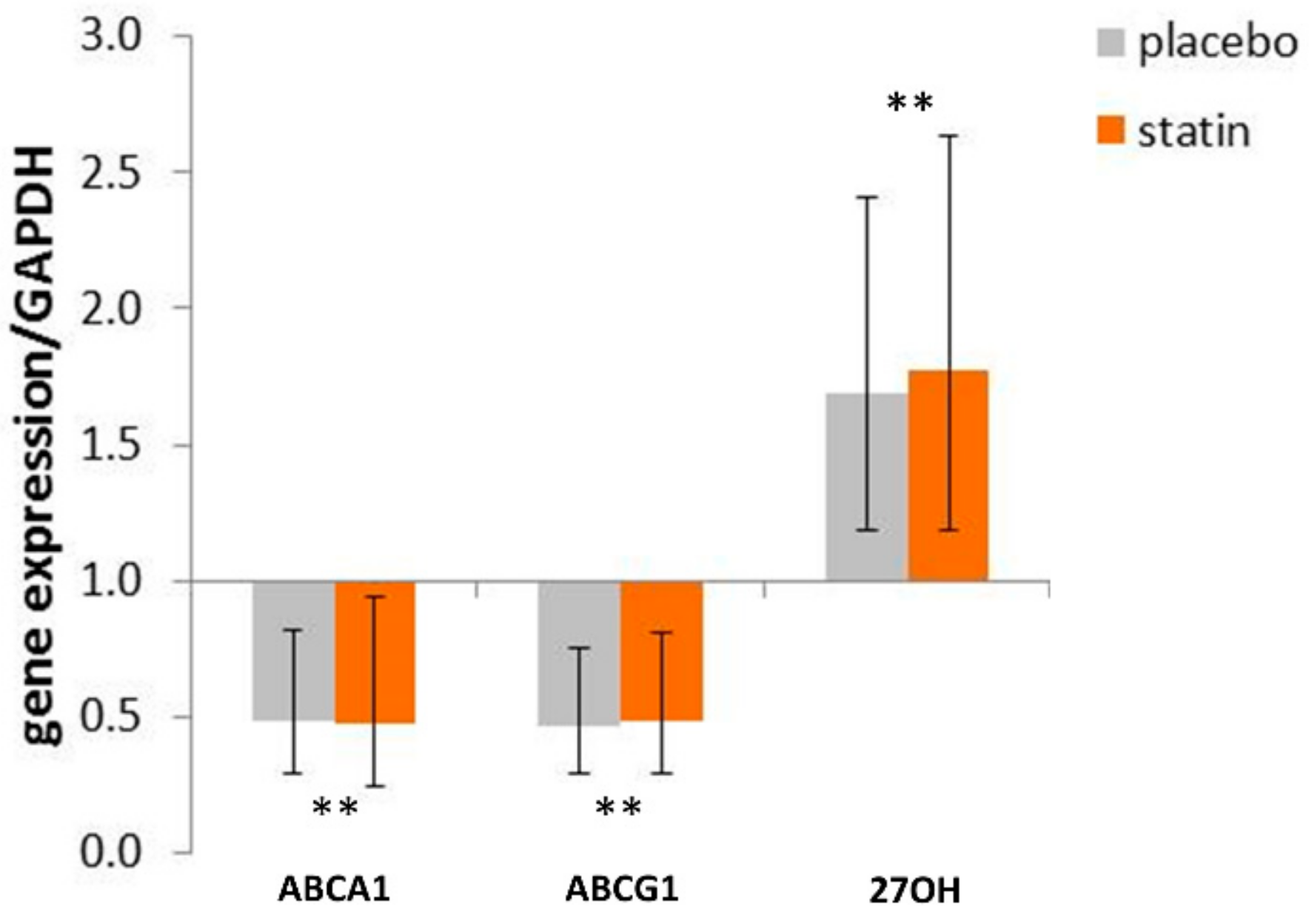
3.3. Comparison of mRNA Level of Reverse Cholesterol Transport Proteins in THP-1 Macrophages Exposed to SLE Plasma Obtained Pre- and Post—Placebo or Statin Therapy
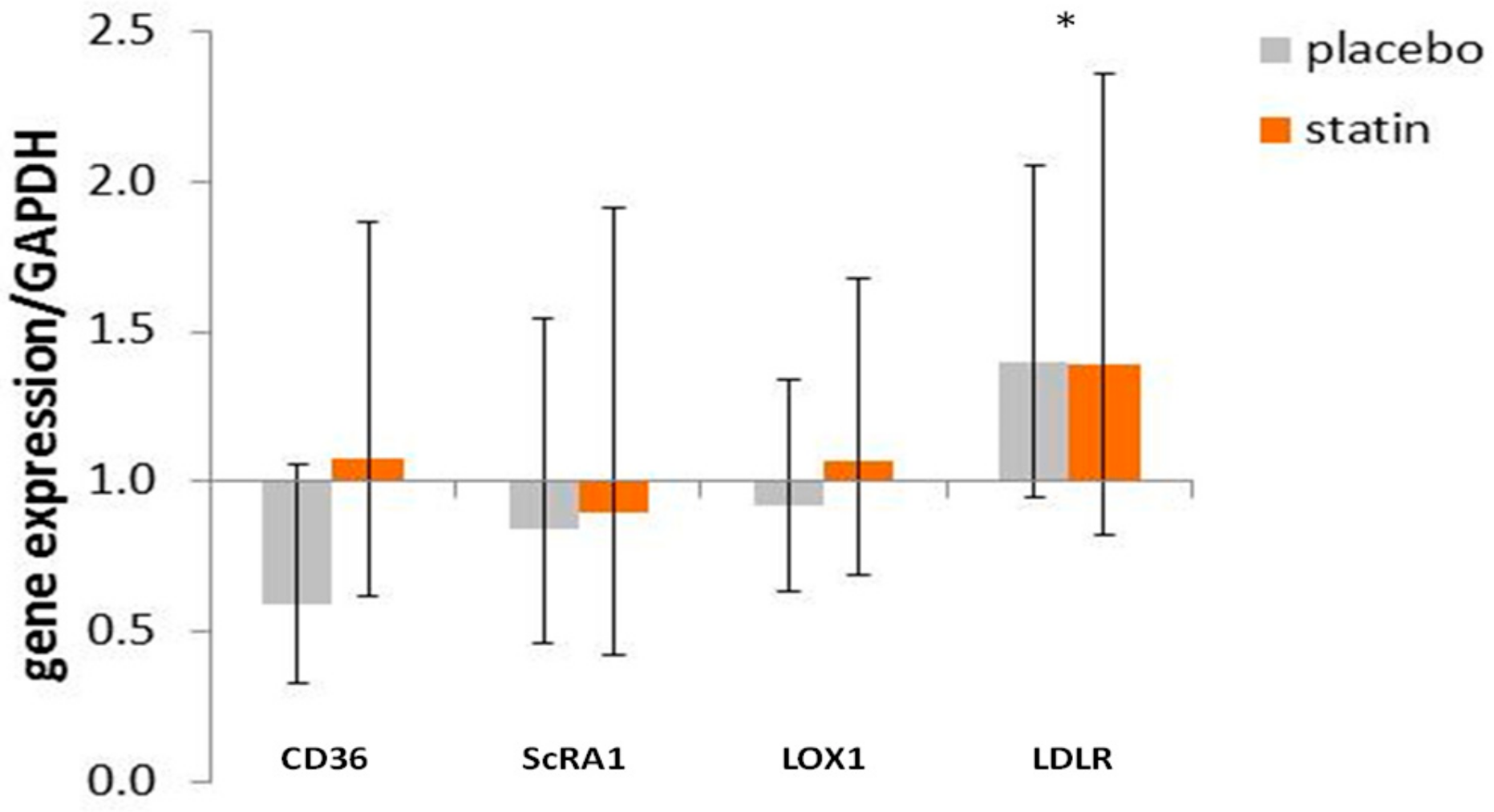
3.4. Comparison of mRNA Level for Scavenger Receptors and Ldl- Receptor Message in THP-1 Macrophages Exposed to Sle Plasma Obtained Pre- and Post-Placebo or Statin Therapy
3.5. Statin Therapy Does Not Improve Cholesterol Efflux in Macrophages Exposed to SLE Plasma
3.6. Statin Therapy Does Not Reduce Oxidized Cholesterol Uptake in Macrophages Exposed to SLE Plasma
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bernatsky, S.; Boivin, J.F.; Joseph, L.; Manzi, S.; Ginzler, E.; Gladman, D.D.; Urowitz, M.; Fortin, P.R.; Petri, M.; Barr, S.; et al. Mortality in systemic lupus erythematosus. Arthritis Rheum. 2006, 54, 2550–2557. [Google Scholar] [CrossRef]
- Crowson, C.S.; Matteson, E.L.; Myasoedova, E.; Michet, C.J.; Ernste, F.C.; Warrington, K.J.; Davis, J.M., 3rd; Hunder, G.G.; Therneau, T.M.; Gabriel, S.E. The lifetime risk of adult-onset rheumatoid arthritis and other inflammatory autoimmune rheumatic diseases. Arthritis Rheum. 2011, 63, 633–639. [Google Scholar] [CrossRef] [PubMed]
- Prasad, M.; Hermann, J.; Gabriel, S.E.; Weyand, C.M.; Mulvagh, S.; Mankad, R.; Oh, J.K.; Matteson, E.L.; Lerman, A. Cardiorheumatology: Cardiac involvement in systemic rheumatic disease. Nat. Rev. Cardiol. 2014, 12, 168–176. [Google Scholar]
- Stone, N.J.; Robinson, J.G.; Lichtenstein, A.H.; Bairey Merz, C.N.; Blum, C.B.; Eckel, R.H.; Goldberg, A.C.; Gordon, D.; Levy, D.; Lloyd-Jones, D.M.; et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J. Am. Coll. Cardiol. 2014, 63, 2889–2934. [Google Scholar] [CrossRef]
- Thavendiranathan, P.; Bagai, A.; Brookhart, M.A.; Choudhry, N.K. Primary prevention of cardiovascular diseases with statin therapy: A meta-analysis of randomized controlled trials. Arch. Intern. Med. 2006, 166, 2307–2313. [Google Scholar] [CrossRef]
- Bruzzone, G.; Corbelli, G.; Belci, P.; Cremonini, A.; Pende, A.; Pisciotta, L. Cholesterol Lowering Therapy: Treat to Target or Reduce the Global Risk? The Unresolved Problem of Residual Risk. Curr. Pharm. Des. 2016, 22, 5676–5686. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Zhao, X.; Xie, H.; Tian, Z.; Zhang, S. Efficacy and safety of statins in the prevention of atherosclerosis in patients with systemic lupus erythematosus—A meta-analysis of randomized controlled trials. Int. J. Cardiol. 2013, 167, 301–303. [Google Scholar] [CrossRef] [PubMed]
- Petri, M.A.; Kiani, A.N.; Post, W.; Christopher-Stine, L.; Magder, L.S. Lupus Atherosclerosis Prevention Study (LAPS). Ann. Rheum. Dis. 2011, 70, 760–765. [Google Scholar] [CrossRef]
- Reiss, A.B.; Wan, D.W.; Anwar, K.; Merrill, J.T.; Wirkowski, P.A.; Shah, N.; Cronstein, B.N.; Chan, E.S.; Carsons, S.E. Enhanced CD36 scavenger receptor expression in THP-1 human monocytes in the presence of lupus plasma: Linking autoimmunity and atherosclerosis. Exp. Biol. Med. (Maywood) 2009, 234, 354–360. [Google Scholar] [CrossRef]
- Voloshyna, I.; Modayil, S.; Littlefield, M.J.; Belilos, E.; Belostocki, K.; Bonetti, L.; Rosenblum, G.; Carsons, S.E.; Reiss, A.B. Plasma from Rheumatoid Arthritis Patients Promotes Pro-atherogenic Cholesterol Transport Gene Expression in THP-1 Human Macrophages. Exp. Biol. Med. (Maywood) 2013, 238, 1192–1197. [Google Scholar] [CrossRef]
- Voloshyna, I.; Teboul, I.; Littlefield, M.J.; Siegart, N.M.; Turi, G.K.; Fazzari, M.J.; Carsons, S.E.; DeLeon, J.; Reiss, A.B. Resveratrol counters systemic lupus erythematosus-associated atherogenicity by normalizing cholesterol efflux. Exp. Biol. Med. (Maywood) 2016, 241, 1611–1619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiss, A.B.; Anwar, K.; Merrill, J.T.; Chan, E.S.; Awadallah, N.W.; Cronstein, B.N.; Michael Belmont, H.; Belilos, E.; Rosenblum, G.; Belostocki, K.; et al. Plasma from systemic lupus patients compromises cholesterol homeostasis: A potential mechanism linking autoimmunity to atherosclerotic cardiovascular disease. Rheumatol. Int. 2010, 30, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Kielar, D.; Dietmaier, W.; Langmann, T.; Aslanidis, C.; Probst, M.; Naruszewicz, M.; Schmitz, G. Rapid Quantification of Human ABCA1 mRNA in Various Cell Types and Tissues by Real-Time Reverse Transcription-PCR. Clin. Chem. 2001, 47, 2089–2097. [Google Scholar] [PubMed]
- Peeters, S.D.; Van der Kolk, D.M.; De Haan, G.; Bystrykh, L.; Kuipers, F.; De Vries, E.G.; Vellenga, E. Selective expression of cholesterol metabolism genes in normal CD34+CD38- cells with a heterogeneous expression pattern in AML cells. Exp. Hematol. 2006, 34, 622–630. [Google Scholar] [CrossRef] [PubMed]
- Reiss, A.B.; Martin, K.O.; Rojer, D.E.; Iyer, S.; Grossi, E.A.; Galloway, A.C.; Javitt, N.B. Sterol 27-hydroxylase: Expression in human arterial endothelium. J. Lipid Res. 1997, 38, 1254–1260. [Google Scholar] [PubMed]
- Mahajan, N.; Dhawan, V. In vitro modulation of peroxisome proliferator-activated receptor-gamma and its genes by C-reactive protein. Role of atorvastatin. Arch. Med. Res. 2010, 41, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Draude, G.; Lorenz, R.L. TGF-beta1 downregulates CD36 and scavenger receptor A but upregulates LOX-1 in human macrophages. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H1042–H1048. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.M.; Doyle, M.V.; Mark, D.F. Quantitation of mRNA by the polymerase chain reaction. Proc. Natl. Acad. Sci. USA 1989, 86, 9717–9721. [Google Scholar] [CrossRef]
- McCloy, R.A.; Rogers, S.; Caldon, C.E.; Lorca, T.; Castro, A.; Burgess, A. Partial inhibition of Cdk1 in G 2 phase overrides the SAC and decouples mitotic events. Cell Cycle 2014, 13, 1400–1412. [Google Scholar] [CrossRef]
- Qin, Z. The use of THP-1 cells as a model for mimicking the function and regulation of monocytes and macrophages in the vasculature. Atherosclerosis 2012, 221, 2–11. [Google Scholar] [CrossRef]
- Moore, K.J.; Tabas, I. Macrophages in the pathogenesis of atherosclerosis. Cell 2011, 145, 341–355. [Google Scholar] [CrossRef]
- Glass, C.K.; Witztum, J.L. Atherosclerosis: The road ahead. Cell 2001, 104, 503–516. [Google Scholar] [CrossRef]
- Lu, H.; Talbot, S.; Robertson, K.A.; Watterson, S.; Forster, T.; Roy, D.; Ghazal, P. Rapid proteasomal elimination of 3-hydroxy-3-methylglutaryl-CoA reductase by interferon-γ in primary macrophages requires endogenous 25-hydroxycholesterol synthesis. Steroids 2015, 99, 219–229. [Google Scholar] [CrossRef]
- Reiss, A.B.; Glass, A.D. CD36 and ScR-A: Scavenger Receptors that Mediate Uptake of Oxidized Low-Density Lipoprotein and Foam Cell Formation. In Proteins Involved in the Pathogenesis of Atherosclerosis, 1st ed.; Reiss, A.B., Carsons, S., Cronstein, B.N., Eds.; Research Signpost: Kerala, India, 2006; pp. 1–12. [Google Scholar]
- Voloshyna, I.; Reiss, A.B. The ABC transporters in lipid flux and atherosclerosis. Prog. Lipid Res. 2011, 50, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Voloshyna, I.; Reiss, A.B. Cytochrome P450 Enzymes in Atherosclerosis. In Cytochrome P450 Enzymes: Biochemistry, Pharmacology and Health Implications; Wu, J., Ed.; Nova Science Publishers: New York, NY, USA, 2014; pp. 87–107. [Google Scholar]
- Fu, X.; Menke, J.G.; Chen, Y.; Zhou, G.; MacNaul, K.L.; Wright, S.D.; Sparrow, C.P.; Lund, E.G. 27-hydroxycholesterol is an endogenous ligand for liver X receptor in cholesterol-loaded cells. J. Biol. Chem. 2001, 276, 38378–38387. [Google Scholar] [CrossRef] [PubMed]
- Rohatgi, A.; Khera, A.; Berry, J.D.; Givens, E.G.; Ayers, C.R.; Wedin, K.E.; Neeland, I.J.; Yuhanna, I.S.; Rader, D.R.; De Lemos, J.A.; et al. HDL cholesterol efflux capacity and incident cardiovascular events. N. Engl. J. Med. 2014, 371, 2383–2393. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhang, Y.; Ding, D.; Li, X.; Yang, Y.; Li, Q.; Zheng, Y.; Wang, D.; Ling, W. Cholesterol efflux capacity is an independent predictor of all-cause and cardiovascular mortality in patients with coronary artery disease: A prospective cohort study. Atherosclerosis 2016, 249, 116–124. [Google Scholar] [CrossRef]
- Sone, H.; Shimano, H.; Shu, M.; Nakakuki, M.; Takahashi, A.; Sakai, M.; Sakamoto, Y.; Yokoo, T.; Matsuzaka, K.; Okazaki, H.; et al. Statins downregulate ATP-binding-cassette transporter A1 gene expression in macrophages. Biochem. Biophys. Res. Commun. 2004, 316, 790–794. [Google Scholar] [CrossRef]
- Wang, W.; Song, W.; Wang, Y.; Chan, L.; Yan, X. HMG- CoA reductase inhibitors, simvastatin and atorvastatin, downregulate ABCG1- mediated cholesterol efflux in human macrophages. J. Cardiovasc. Pharm. 2013, 62, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Keidar, S.; Aviram, M.; Maor, I.; Oiknine, J.; Brook, J.G. Pravastatin inhibits cellular cholesterol synthesis and increases low density lipoprotein receptor activity in macrophages: In vitro and in vivo studies. Br. J. Clin. Pharm. 1994, 38, 513–519. [Google Scholar] [CrossRef]
- Györi, N.; Giannakou, I.; Chatzidionysiou, K.; Magder, L.; Van Vollenhoven, R.F.; Petri, M. Disease activity patterns over time in patients with SLE: Analysis of the Hopkins Lupus Cohort. Lupus Sci. Med. 2017, 4, e000192. [Google Scholar] [CrossRef] [PubMed]
- Labos, C.; Brophy, J.M.; Smith, G.D.; Sniderman, A.D.; Thanassoulis, G. Evaluation of the Pleiotropic Effects of Statins: A Reanalysis of the Randomized Trial Evidence Using Egger Regression-Brief Report. Arter. Thromb. Vasc. Biol. 2018, 38, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Fatemi, A.; Moosavi, M.; Sayedbonakdar, Z.; Farajzadegan, Z.; Kazemi, M.; Smiley, A. Atorvastatin effect on systemic lupus erythematosus disease activity: A double-blind randomized clinical trial. Clin. Rheumatol. 2014, 33, 1273–1278. [Google Scholar] [CrossRef]
- Yamamoto, S.; Narita, I.; Kotani, K. The macrophage and its related cholesterol efflux as a HDL function index in atherosclerosis. Clin. Chim. Acta 2016, 457, 117–122. [Google Scholar] [CrossRef]
- Muñoz-Hernandez, L.; Ortiz-Bautista, R.J.; Brito-Córdova, G.; Lozano-Arvizu, F.; Saucedo, S.; Pérez-Méndez, O.; Zentella-Dehesa, A.; Dauteuille, C.; Lhomme, M.; Lesnik, P.; et al. Cholesterol efflux capacity of large, small and total HDL particles is unaltered by atorvastatin in patients with type 2 diabetes. Atherosclerosis 2018, 277, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Liu, C.; Zhang, L.; Liu, Y.; Guo, A.; Shi, H.; Liu, X.; Cheng, Y. Intensive Atorvastatin Therapy Attenuates the Inflammatory Responses in Monocytes of Patients with Unstable Angina Undergoing Percutaneous Coronary Intervention via Peroxisome Proliferator-Activated Receptor γ Activation. Inflammation 2015, 38, 1415–1423. [Google Scholar] [CrossRef]
- Schanberg, L.E.; Sandborg, C.; Barnhart, H.X.; Ardoin, S.P.; Yow, E.; Evans, G.W.; Mieszkalski, K.L.; Ilowite, N.T.; Eberhard, A.; Imundo, L.F.; et al. Atherosclerosis Prevention in Pediatric Lupus Erythematosus Investigators. Use of atorvastatin in systemic lupus erythematosus in children and adolescents. Arthritis Rheum. 2012, 64, 285–296. [Google Scholar] [PubMed]
- Brownell, N.; Rohatgi, A. Modulating cholesterol efflux capacity to improve cardiovascular disease. Curr. Opin. Lipidol. 2016, 27, 398–407. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Zhao, B.; Liu, G.A. Rosuvastatin promotes the differentiation of peripheral blood monocytes into M2 macrophages in patients with atherosclerosis by activating PPAR-γ. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 238–245. [Google Scholar]
- Mendez-Fernandez, Y.; Major, A. Sizing up stability: Combination therapy with Apo-AI peptide mimetics and statins in systemic lupus erythematosus-mediated atherosclerosis. Arthritis Res. 2010, 12, 139. [Google Scholar] [CrossRef]
- Yurkovich, M.; Vostretsova, K.; Chen, W.; Aviña-Zubieta, J.A. Overall and cause-specific mortality in patients with systemic lupus erythematosus: A meta-analysis of observational studies. Arthritis Care Res. (Hoboken) 2014, 66, 608–616. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Placebo | Atorvastatin | |
|---|---|---|---|
| n | 21 | 23 | |
| gender (%) | female | 100 | 87 |
| male | 0 | 13 | |
| race (%) | African American | 48 | 65 |
| Caucasian | 52 | 35 | |
| age (mean ± SD) | 32 ± 12 | 34 ± 10 | |
| disease duration years (median (range)) | 2 (0–20) | 2 (0–22) | |
| anti-ANA antibodies present (%) | 100 | 100 | |
| anti-ANA titer (median (range)) | 1280 (40–20,480) | 640 (160–2560) | |
| anti-dsDNA antibodies present (%) | 67 | 74 | |
| anti-Sm antibodies present (%) | 10 | 18 | |
| RNP antibodies present (%) | 30 | 30 | |
| low C3 or C4 (%) | 69 | 67 | |
| lupus nephritis (%) | 17 | 32 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reiss, A.B.; Arain, H.A.; Kasselman, L.J.; Renna, H.A.; Zhen, J.; Voloshyna, I.; DeLeon, J.; Carsons, S.E.; Petri, M. Human Lupus Plasma Pro-Atherogenic Effects on Cultured Macrophages Are Not Mitigated by Statin Therapy: A Mechanistic LAPS Substudy. Medicina 2019, 55, 514. https://0-doi-org.brum.beds.ac.uk/10.3390/medicina55090514
Reiss AB, Arain HA, Kasselman LJ, Renna HA, Zhen J, Voloshyna I, DeLeon J, Carsons SE, Petri M. Human Lupus Plasma Pro-Atherogenic Effects on Cultured Macrophages Are Not Mitigated by Statin Therapy: A Mechanistic LAPS Substudy. Medicina. 2019; 55(9):514. https://0-doi-org.brum.beds.ac.uk/10.3390/medicina55090514
Chicago/Turabian StyleReiss, Allison B., Hirra A. Arain, Lora J. Kasselman, Heather A. Renna, Juan Zhen, Iryna Voloshyna, Joshua DeLeon, Steven E. Carsons, and Michelle Petri. 2019. "Human Lupus Plasma Pro-Atherogenic Effects on Cultured Macrophages Are Not Mitigated by Statin Therapy: A Mechanistic LAPS Substudy" Medicina 55, no. 9: 514. https://0-doi-org.brum.beds.ac.uk/10.3390/medicina55090514