
Clinical and Histopathological Features of Scleroderma-like Disorders: An Update

Abstract
:1. Introduction
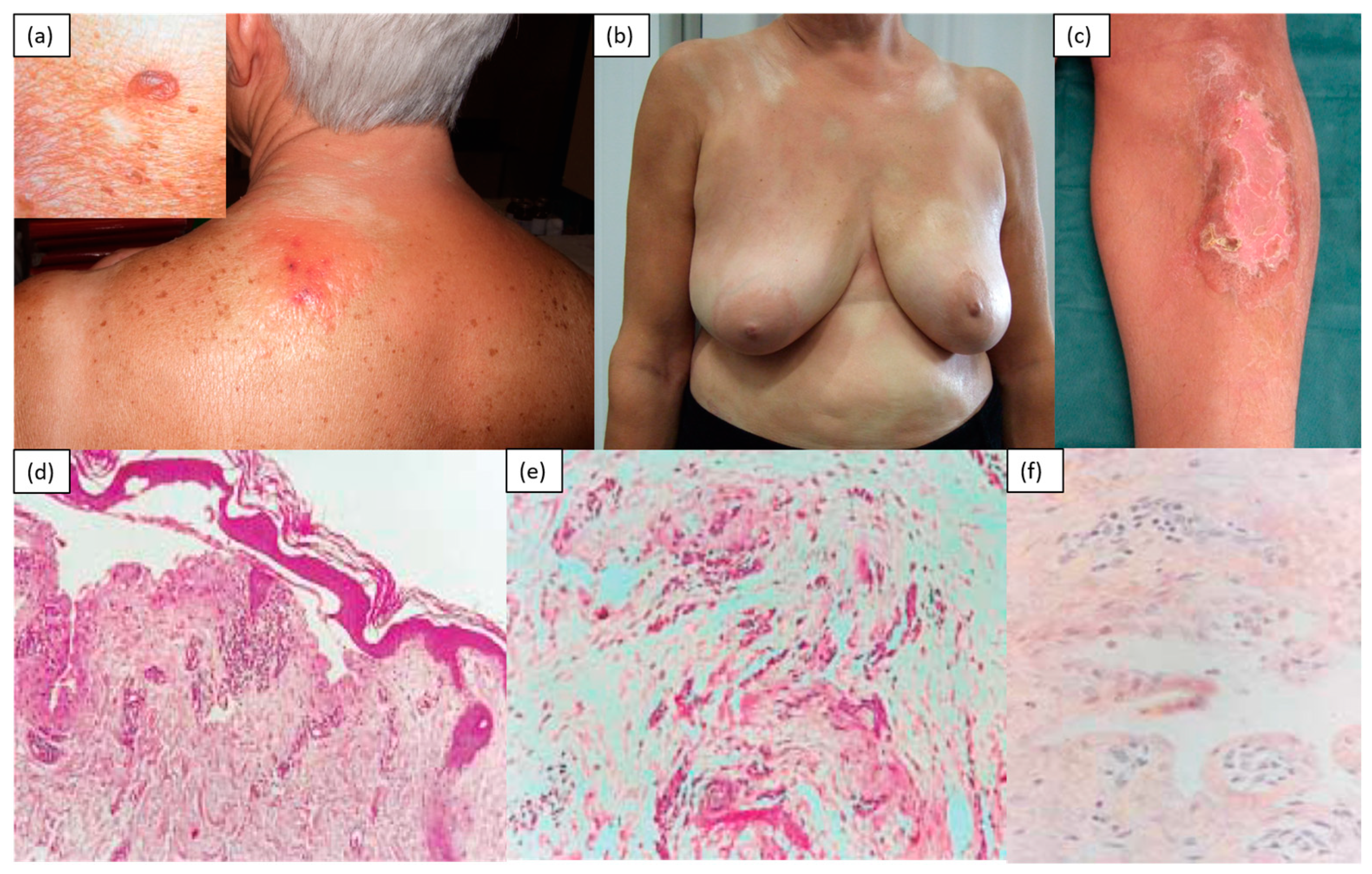
2. Inflammatory/Immunomediated
3. Chronic Graft-versus-Host Disease (Gvhd)
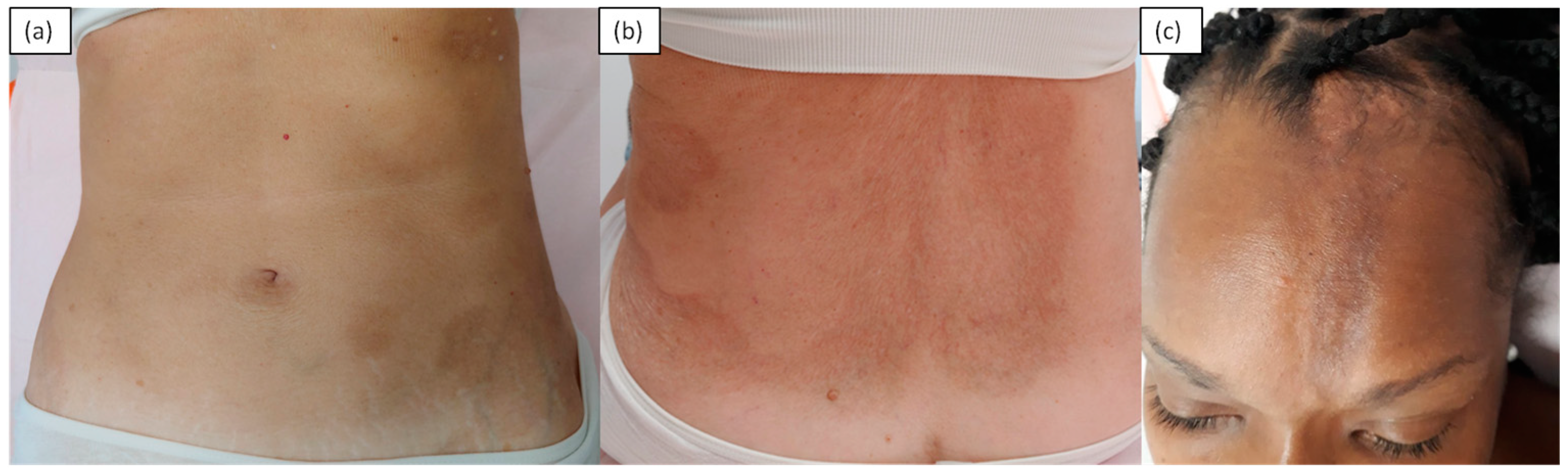
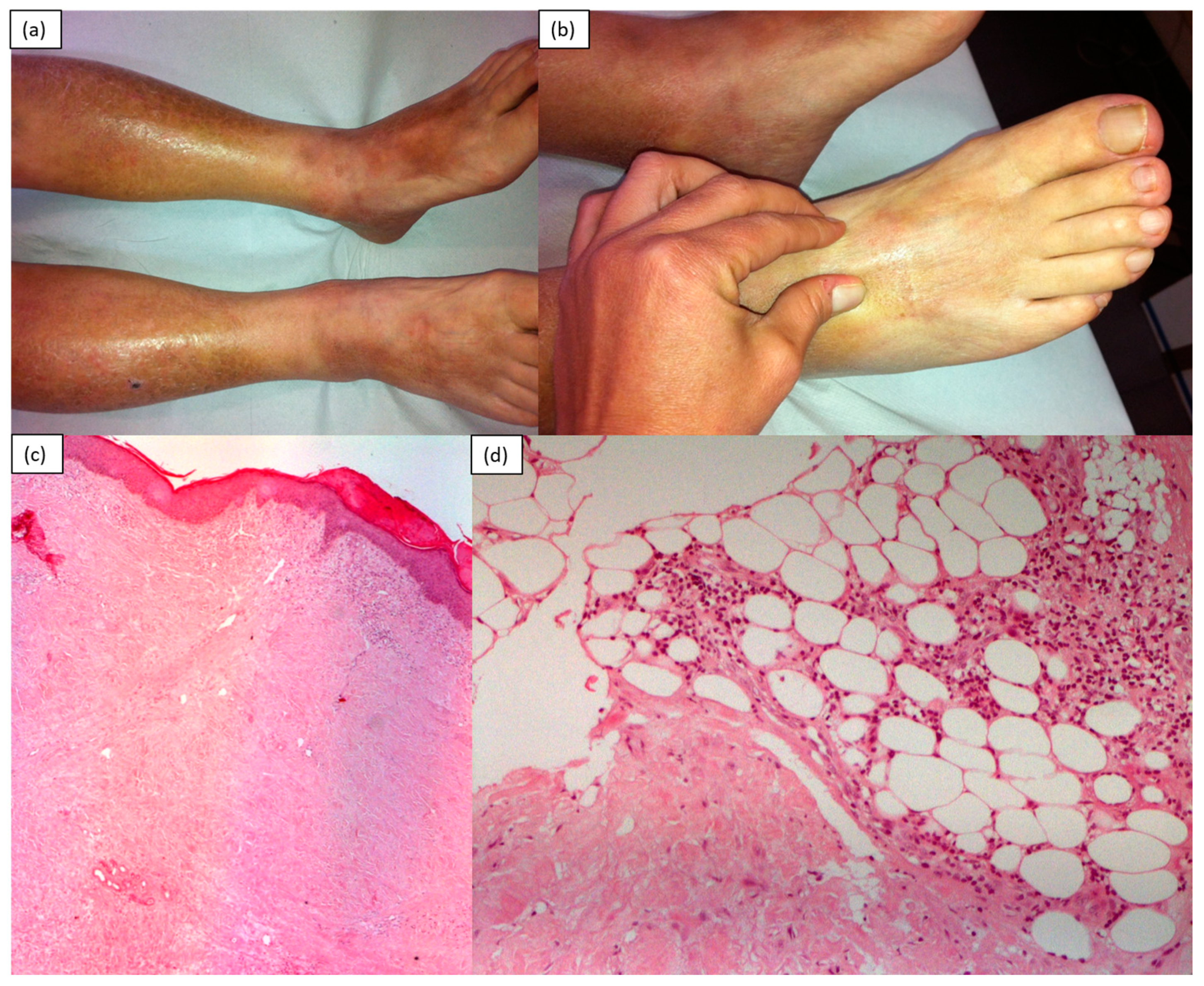
4. Sclerodermiform Mucinoses
5. Genetic
6. Drug-Induced Scleroderma-like Illnesses
7. Toxic
8. Metabolic
9. Paraneoplastic
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Chung, L.; Lin, J.; Furst, D.E.; Fiorentino, D. Systemic and localized scleroderma. Clin. Dermatol. Ottobre 2006, 24, 374–392. [Google Scholar] [CrossRef] [PubMed]
- Ferreli, C.; Gasparini, G.; Parodi, A.; Cozzani, E.; Rongioletti, F.; Atzori, L. Cutaneous Manifestations of Scleroderma and Scleroderma-Like Disorders, a Comprehensive Review. Clin. Rev. Allergy Immunol. 2017, 53, 306–336. [Google Scholar] [CrossRef]
- Mori, Y.; Kahari, V.-M.; Varga, J. Scleroderma-like cutaneous syndromes. Curr. Rheumatol. Rep. 2002, 4, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Cokonis Georgakis, C.-D.; Falasca, G.; Georgakis, A.; Heymann, W.R. Scleromyxedema. Clin. Dermatol. 2006, 24, 493–497. [Google Scholar] [CrossRef]
- Dinneen, A.M.; Dicken, C.H. Scleromyxedema. J. Am. Acad. Dermatol. 1995, 33, 37–43. [Google Scholar] [CrossRef]
- Rongioletti, F. Lichen myxedematosus (papular mucinosis), new concepts and perspectives for an old disease. Semin. Cutan. Med. Surg. 2006, 25, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Torok, K.S. Pediatric scleroderma, systemic or localized forms. Pediatr. Clin. N. Am. 2012, 59, 381–405. [Google Scholar] [CrossRef] [Green Version]
- Careta, M.F.; Romiti, R. Localized scleroderma, clinical spectrum and therapeutic update. An. Bras. Dermatol. 2015, 90, 62–73. [Google Scholar] [CrossRef] [Green Version]
- Jablonska, S.; Blaszczyk, M. Scleroderma-like disorders. Semin. Cutan. Med. Surg. 1998, 17, 65–76. [Google Scholar] [CrossRef]
- Jinnin, M.; Yamamoto, T.; Asano, Y.; Ishikawa, O.; Sato, S.; Takehara, K.; Hasegawa, M.; Fujimoto, M.; Ihn, H. Diagnostic criteria, severity classification and guidelines of eosinophilic fasciitis. J. Dermatol. 2018, 45, 881–890. [Google Scholar] [CrossRef] [Green Version]
- Fujimoto, M.; Sato, S.; Ihn, H.; Kikuchi, K.; Yamada, N.; Takehara, K. Serum aldolase level is a useful indicator of disease activity in eosinophilic fasciitis. J. Rheumatol. 1995, 22, 563–565. [Google Scholar]
- Ihn, H. Eosinophilic fasciitis, From pathophysiology to treatment. Allergol. Int. 2019, 68, 437–439. [Google Scholar] [CrossRef]
- Jinnin, M.; Ihn, H.; Yamane, K.; Asano, Y.; Yazawa, N.; Tamaki, K. Serum levels of tissue inhibitor of metalloproteinase-1 and 2 in patients with eosinophilic fasciitis. Br. J. Dermatol. 2004, 151, 407–412. [Google Scholar] [CrossRef]
- Nishiya, K.; Tanimoto, N.; Hashimoto, K.; Hashizume, M.; Tominaga, A. Serum and synovial fluid levels of interleukin-5 in a patient with eosinophilic fasciitis. Ann. Rheum. Dis. 1996, 55, 935–936. [Google Scholar] [CrossRef] [Green Version]
- Wasserman, S.I.; Seibold, J.R.; Medsger, T.A.; Rodnan, G.P. Serum eosinophilotactic activity in eosinophilic fasciitis. Arthritis Rheum. 1982, 25, 1352–1356. [Google Scholar] [CrossRef]
- Baumann, F.; Brühlmann, P.; Andreisek, G.; Michel, B.A.; Marincek, B.; Weishaupt, D. MRI for diagnosis and monitoring of patients with eosinophilic fasciitis. AJR Am. J. Roentgenol. 2005, 184, 169–174. [Google Scholar] [CrossRef]
- Schaffer, J.V.; McNiff, J.M.; Seropian, S.; Cooper, D.L.; Bolognia, J.L. Lichen sclerosus and eosinophilic fasciitis as manifestations of chronic graft-versus-host disease, expanding the sclerodermoid spectrum. J. Am. Acad. Dermatol. 2005, 53, 591–601. [Google Scholar] [CrossRef]
- Ratanatharathorn, V.; Ayash, L.; Lazarus, H.M.; Fu, J.; Uberti, J.P. Chronic graft-versus-host disease, clinical manifestation and therapy. Bone Marrow Transpl. 2001, 28, 121–129. [Google Scholar] [CrossRef] [Green Version]
- Cepeda, E.J.; Reveille, J.D. Autoantibodies in systemic sclerosis and fibrosing syndromes, clinical indications and relevance. Curr. Opin. Rheumatol. 2004, 16, 723–732. [Google Scholar] [CrossRef]
- Hummers, L.K. Scleromyxedema. Curr. Opin. Rheumatol. 2014, 26, 658–662. [Google Scholar] [CrossRef]
- Shrestha, B.; Neopane, A.K.; Panth, R. Scleredema—An uncommon cause of swelling in a child--a case report and review of the literature. BMC Res. Notes 2014, 7, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Rongioletti, F.; Kaiser, F.; Cinotti, E.; Metze, D.; Battistella, M.; Calzavara-Pinton, P.G.; Damevska, K.; Girolomoni, G.; André, J.; Perrot, J.L.; et al. Scleredema. A multicentre study of characteristics, comorbidities, course and therapy in 44 patients. J. Eur. Acad. Dermatol. Venereol. 2015, 29, 2399–2404. [Google Scholar] [CrossRef]
- Murphy-Chutorian, B.; Han, G.; Cohen, S.R. Dermatologic manifestations of diabetes mellitus, a review. Endocrinol. Metab. Clin. N. Am. 2013, 42, 869–898. [Google Scholar] [CrossRef]
- Woolen, S.A.; Shankar, P.R.; Gagnier, J.J.; MacEachern, M.P.; Singer, L.; Davenport, M.S. Risk of Nephrogenic Systemic Fibrosis in Patients with Stage 4 or 5 Chronic Kidney Disease Receiving a Group II Gadolinium-Based Contrast Agent, A Systematic Review and Meta-analysis. JAMA Intern. Med. 2020, 180, 223–230. [Google Scholar] [CrossRef]
- Grobner, T.; Prischl, F.C. Gadolinium and nephrogenic systemic fibrosis. Kidney Int. 2007, 72, 260–264. [Google Scholar] [CrossRef] [Green Version]
- Mendoza, F.A.; Artlett, C.M.; Sandorfi, N.; Latinis, K.; Piera-Velazquez, S.; Jimenez, S.A. Description of 12 cases of nephrogenic fibrosing dermopathy and review of the literature. Semin. Arthritis Rheum. 2006, 35, 238–249. [Google Scholar] [CrossRef] [Green Version]
- Wechalekar, A.D.; Gillmore, J.D.; Hawkins, P.N. Systemic amyloidosis. Lancet 2016, 387, 2641–2654. [Google Scholar] [CrossRef]
- Borowicz, J.; Gillespie, M.; Miller, R. Cutaneous amyloidosis. Skinmed 2011, 9, 96–101. [Google Scholar]
- Cho, S.B.; Park, J.S.; Kim, H.O.; Chung, K.Y. Scleroderma-like manifestation in a patient with primary systemic amyloidosis, response to high-dose intravenous immunoglobulin and plasma exchange. Yonsei Med. J. 2006, 47, 737–740. [Google Scholar] [CrossRef]
- Sun, L.; Zhang, L.; Hu, W.; Li, T.-F.; Liu, S. Case report, One case of primary AL amyloidosis repeatedly misdiagnosed as scleroderma. Medicine 2017, 96, e8771. [Google Scholar] [CrossRef]
- Kikuchi, N.; Sakai, E.; Nishibu, A.; Ohtsuka, M.; Yamamoto, T. Primary localized cutaneous amyloidosis in patients with scleroderma. Acta Derm. Venereol. 2010, 90, 326–327. [Google Scholar] [CrossRef]
- Keijzers, G.; Maynard, S.; Shamanna, R.A.; Rasmussen, L.J.; Croteau, D.L.; Bohr, V.A. The role of RecQ helicases in non-homologous end-joining. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 463–472. [Google Scholar] [CrossRef] [Green Version]
- Oshima, J.; Sidorova, J.M.; Monnat, R.J. Werner syndrome, Clinical features, pathogenesis and potential therapeutic interventions. Ageing Res. Rev. 2017, 33, 105–114. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Lee, L.; Hanson, N.B.; Lenaerts, C.; Hoehn, H.; Poot, M.; Rubin, C.D.; Chen, D.F.; Yang, C.C.; Juch, H.; et al. The spectrum of WRN mutations in Werner syndrome patients. Hum. Mutat. 2006, 27, 558–567. [Google Scholar] [CrossRef] [Green Version]
- Takemoto, M.; Mori, S.; Kuzuya, M.; Yoshimoto, S.; Shimamoto, A.; Igarashi, M.; Tanaka, Y.; Miki, T.; Yokote, K. Diagnostic criteria for Werner syndrome based on Japanese nationwide epidemiological survey. Geriatr. Gerontol. Int. 2013, 13, 475–481. [Google Scholar] [CrossRef]
- Hamm, H.; Traupe, H.; Bröcker, E.B.; Schubert, H.; Kolde, G. The scleroatrophic syndrome of Huriez, a cancer-prone genodermatosis. Br. J. Dermatol. 1996, 134, 512–518. [Google Scholar] [CrossRef]
- Lee, Y.A.; Stevens, H.P.; Delaporte, E.; Wahn, U.; Reis, A. A gene for an autosomal dominant scleroatrophic syndrome predisposing to skin cancer (Huriez syndrome) maps to chromosome 4q23. Am. J. Hum. Genet. 2000, 66, 326–330. [Google Scholar] [CrossRef] [Green Version]
- Çelik, N.S.; Yaşar, Ş.; Aytekin, S.; Güneş, P. A Rare Syndrome Resembling Scleroderma, Huriez Syndrome. Ski. Appendage Disord. 2018, 4, 82–85. [Google Scholar] [CrossRef]
- Kotwal, A.; Clarke, B.L. Melorheostosis, a Rare Sclerosing Bone Dysplasia. Curr Osteoporos Rep. agosto 2017, 15, 335–342. [Google Scholar] [CrossRef]
- Shivanand, G.; Srivastava, D.N. Melorheostosis with scleroderma. Clin. Imaging 2004, 28, 214–215. [Google Scholar] [CrossRef]
- Greenspan, A.; Azouz, E.M. Bone dysplasia series. Melorheostosis, review and update. Can. Assoc. Radiol. J. 1999, 50, 324–330. [Google Scholar]
- Kessler, H.B.; Recht, M.P.; Dalinka, M.K. Vascular anomalies in association with osteodystrophies—A spectrum. Skelet. Radiol. 1983, 10, 95–101. [Google Scholar] [CrossRef]
- Morris, J.M.; Samilson, R.L.; Corley, C.L. Melorheostosis. Review of the literature and report of an interesting case with a nineteen-year follow-up. J. Bone Jt. Surg. Am. 1963, 45, 1191–1206. [Google Scholar] [CrossRef]
- Rhys, R.; Davies, A.M.; Mangham, D.C.; Grimer, R.J. Sclerotome distribution of melorheostosis and multicentric fibromatosis. Skeletal Radiol. 1998, 27, 633–636. [Google Scholar] [CrossRef]
- Woolridge, B.; Stone, N.C.; Denic, N. Melorheostosis isolated to the calcaneus, a case report and review of the literature. Foot Ankle Int. 2005, 26, 660–663. [Google Scholar] [CrossRef]
- Geng, S.; Lei, X.; Toyohara, J.P.; Zhan, P.; Wang, J.; Tan, S. Stiff skin syndrome. J. Eur. Acad. Dermatol. Venereol. 2006, 20, 729–732. [Google Scholar] [CrossRef]
- Wang, T.; Yang, Y.; Dong, Q.; Zhu, H.; Liu, Y. Acromicric dysplasia with stiff skin syndrome-like severe cutaneous presentation in an 8-year-old boy with a missense FBN1 mutation, Case report and literature review. Mol. Genet. Genomic. Med. 2020, 8, e1282. [Google Scholar] [CrossRef]
- Nijsten, T.E.C.; De Moor, A.; Colpaert, C.G.; Robert, K.; Mahieu, L.M.; Lambert, J. Restrictive dermopathy, a case report and a critical review of all hypotheses of its origin. Pediatr. Dermatol. 2002, 19, 67–72. [Google Scholar] [CrossRef]
- Bidier, M.; Salz, M.; Meyburg, J.; Elbe-Bürger, A.; Lasitzschka, F.; Hausser, I.; Schäkel, K. Restrictive Dermopathy, Four Case Reports and Structural Skin Changes. Acta Derm. Venereol. 2018, 98, 807–808. [Google Scholar] [CrossRef] [Green Version]
- Ho, J.; Rothchild, Y.H.; Sengelmann, R. Vitamin B12-associated localized scleroderma and its treatment. Dermatol. Surg. 2004, 30, 1252–1255. [Google Scholar]
- Asano, Y.; Ihn, H.; Shikada, J.; Kadono, T.; Kikuchi, K.; Tamaki, K. A case of peplomycin-induced scleroderma. Br. J. Dermatol. 2004, 150, 1213–1214. [Google Scholar] [CrossRef]
- Ozden, M.G.; Erel, A.; Erdem, O.; Oztas, M.O. Dermal fibrosis and cutaneous necrosis after recombinant interferon-beta1a injection in a multiple sclerosis patient. J. Eur. Acad. Dermatol. Venereol. 2005, 19, 112–113. [Google Scholar] [CrossRef]
- Kono, T.; Ishii, M.; Negoro, N.; Taniguchi, S. Scleroderma-like reaction induced by uracil-tegafur (UFT), a second-generation anticancer agent. J. Am. Acad. Dermatol. 2000, 42, 519–520. [Google Scholar] [CrossRef]
- Kupfer, I.; Balguerie, X.; Courville, P.; Chinet, P.; Joly, P. Scleroderma-like cutaneous lesions induced by paclitaxel, a case study. J. Am. Acad. Dermatol. 2003, 48, 279–281. [Google Scholar] [CrossRef]
- Kluger, N.; Girard, C.; Bessis, D.; Guillot, B. Methysergide-induced scleroderma-like changes of the legs. Br. J. Dermatol. 2005, 153, 224–225. [Google Scholar] [CrossRef]
- Bessis, D.; Guillot, B.; Legouffe, E.; Guilhou, J.-J. Gemcitabine-associated scleroderma-like changes of the lower extremities. J. Am. Acad. Dermatol. 2004, 51, 73–76. [Google Scholar] [CrossRef] [PubMed]
- De Angelis, R.; Bugatti, L.; Cerioni, A.; Del Medico, P.; Filosa, G. Diffuse scleroderma occurring after the use of paclitaxel for ovarian cancer. Clin. Rheumatol. 2003, 22, 49–52. [Google Scholar] [CrossRef]
- D’Cruz, D. Autoimmune diseases associated with drugs, chemicals and environmental factors. Toxicol Lett. 2000, 112, 421–432. [Google Scholar] [CrossRef]
- Ward, A.M.; Udnoon, S.; Watkins, J.; Walker, A.E.; Darke, C.S. Immunological mechanisms in the pathogenesis of vinyl chloride disease. Br Med J 1976, 1, 936–938. [Google Scholar] [CrossRef] [Green Version]
- Czernielewski, A.; Kieć-swierczyńska, M.; Gluszcz, M.; Woźniak, L. Dermatological aspects of so called vinyl chloride monomer disease. Derm. Beruf. Umwelt. 1979, 27, 108–112. [Google Scholar]
- Rich, M.W. Porphyria cutanea tarda. Don’t forget to look at the urine. Postgrad. Med. 1999, 105, 208–214. [Google Scholar] [CrossRef]
- Bonkovsky, H.L.; Lambrecht, R.W.; Shan, Y. Iron as a co-morbid factor in nonhemochromatotic liver disease. Alcohol 2003, 30, 137–144. [Google Scholar] [CrossRef]
- Egger, N.G.; Goeger, D.E.; Payne, D.A.; Miskovsky, E.P.; Weinman, S.A.; Anderson, K.E. Porphyria cutanea tarda, multiplicity of risk factors including HFE mutations, hepatitis C.; and inherited uroporphyrinogen decarboxylase deficiency. Dig. Dis. Sci. 2002, 47, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Maynard, B.; Peters, M.S. Histologic and immunofluorescence study of cutaneous porphyrias. J. Cutan. Pathol. 1992, 19, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Scholnick, P.L.; Epstein, J.; Marver, H.S. The molecular basis of the action of chloroquine in porphyria cutanea tarda. J. Investig. Dermatol. 1973, 61, 226–332. [Google Scholar] [CrossRef] [Green Version]
- Grossman, M.E.; Bickers, D.R.; Poh-Fitzpatrick, M.B.; Deleo, V.A.; Harber, L.C. Porphyria cutanea tarda. Clinical features and laboratory findings in 40 patients. Am. J. Med. 1979, 67, 277–286. [Google Scholar] [CrossRef]
- Varigos, G.; Schiltz, J.R.; Bickers, D.R. Uroporphyrin I stimulation of collagen biosynthesis in human skin fibroblasts. A unique dark effect of porphyrin. J. Clin. Investig. 1982, 69, 129–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Mayouf, S.M.; Al-Owain, M.A. Progressive sclerodermatous skin changes in a child with phenylketonuria. Pediatr. Dermatol. 2006, 23, 136–138. [Google Scholar] [CrossRef]
- Toussaint, P.; Sibaud, V.; Labbe, L.; Geniaux, M. POEMS syndrome revealed by a scleroderma-like skin thickening. Ann. Dermatol. Venereol. 2000, 127, 73–76. [Google Scholar] [PubMed]
- Dispenzieri, A.; Kyle, R.A.; Lacy, M.Q.; Rajkumar, S.V.; Therneau, T.M.; Larson, D.R.; Greipp, P.R.; Witzig, T.E.; Basu, R.; Suarez, G.A.; et al. POEMS syndrome, definitions and long-term outcome. Blood 2003, 101, 2496–2506. [Google Scholar] [CrossRef]
- Dispenzieri, A. POEMS Syndrome. Hematol. Am. Soc. Hematol. Educ. Program. 2005, 2005, 360–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paredes-Suárez, C.; Fernández-Redondo, V.; Blanco, M.V.; Sánchez-Aguilar, D.; Toribio, J. Multiple myeloma with scleroderma-like changes. J. Eur. Acad. Dermatol. Venereol. 2005, 19, 500–502. [Google Scholar] [CrossRef] [PubMed]
- Bell, H.K.; Poston, G.J.; Vora, J.; Wilson, N.J.E. Cutaneous manifestations of the malignant carcinoid syndrome. Br. J. Dermatol. 2005, 152, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Plötz, S.G.; Hüttig, B.; Aigner, B.; Merkel, C.; Brockow, K.; Akdis, C.; Darsow, U.; Ring, J. Clinical overview of cutaneous features in hypereosinophilic syndrome. Curr. Allergy Asthma. Rep. 2012, 12, 85–98. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Classification of Scleroderma-like Disorders | |
|---|---|
| Inflammatory | Morphea, atrophoderma of Pasini and Pierini, eosinophilic fasciitis, graft-versus-host disease |
| Sclerodermiform mucinoses | Scleromyxedema, Buschke scleredema, nefrogenic systemic fibrosis, cutaneous amyloidosis |
| Genetic | Werner’s syndrome, scleroatrophic Huriez syndrome, stiff-skin syndrome, melhoreostosis |
| Drug-induced | Pentazocine, bleomycin, vitamin K, interferon-β1a, peplomycin, cocaine, D-penicillamine, vitamin B12, uracil–tegafur, methysergide, gemcitabine, paclitaxel |
| Toxic | Toxic-oil syndrome, eosinophilia–myalgia syndrome, vinyl-chloride syndrome |
| Metabolic | Porphyria cutanea tarda, diabetes mellitus, phenylketonuria |
| Paraneoplastic | POEMS syndrome, myeloma with scleroderma-like changes, carcinoid syndrome, scleroderma-like manifestations in myeloproliferative hypereosinophilic syndrome |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Foti, R.; De Pasquale, R.; Dal Bosco, Y.; Visalli, E.; Amato, G.; Gangemi, P.; Foti, R.; Ramondetta, A. Clinical and Histopathological Features of Scleroderma-like Disorders: An Update. Medicina 2021, 57, 1275. https://0-doi-org.brum.beds.ac.uk/10.3390/medicina57111275
Foti R, De Pasquale R, Dal Bosco Y, Visalli E, Amato G, Gangemi P, Foti R, Ramondetta A. Clinical and Histopathological Features of Scleroderma-like Disorders: An Update. Medicina. 2021; 57(11):1275. https://0-doi-org.brum.beds.ac.uk/10.3390/medicina57111275
Chicago/Turabian StyleFoti, Rosario, Rocco De Pasquale, Ylenia Dal Bosco, Elisa Visalli, Giorgio Amato, Pietro Gangemi, Riccardo Foti, and Alice Ramondetta. 2021. "Clinical and Histopathological Features of Scleroderma-like Disorders: An Update" Medicina 57, no. 11: 1275. https://0-doi-org.brum.beds.ac.uk/10.3390/medicina57111275