
Central TSH Dysregulation in a Patient with Familial Non-Autoimmune Autosomal Dominant Hyperthyroidism Due to a Novel Thyroid-Stimulating Hormone Receptor Disease-Causing Variant

 , , ,
, , , 
Abstract
:1. Introduction
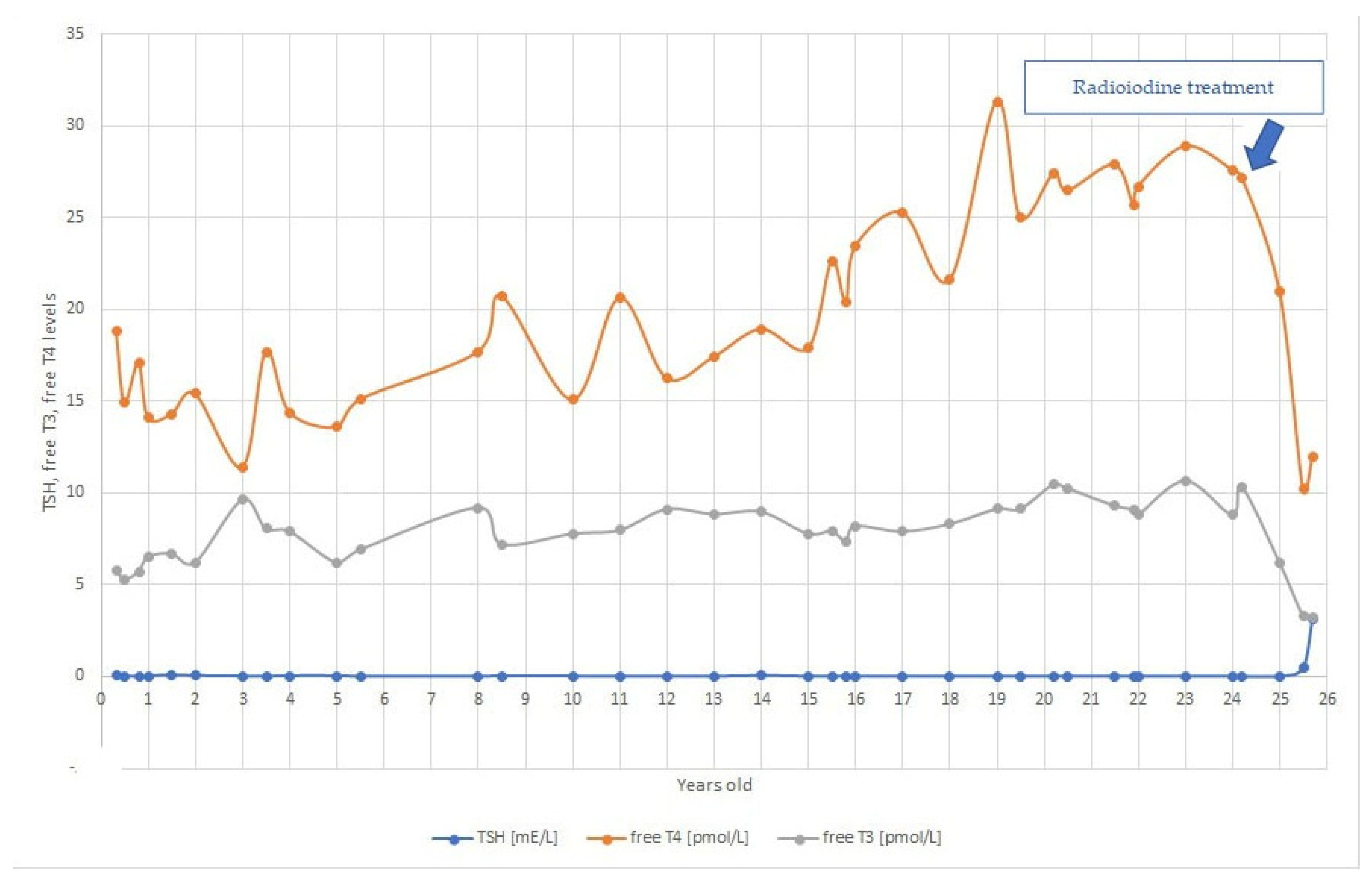
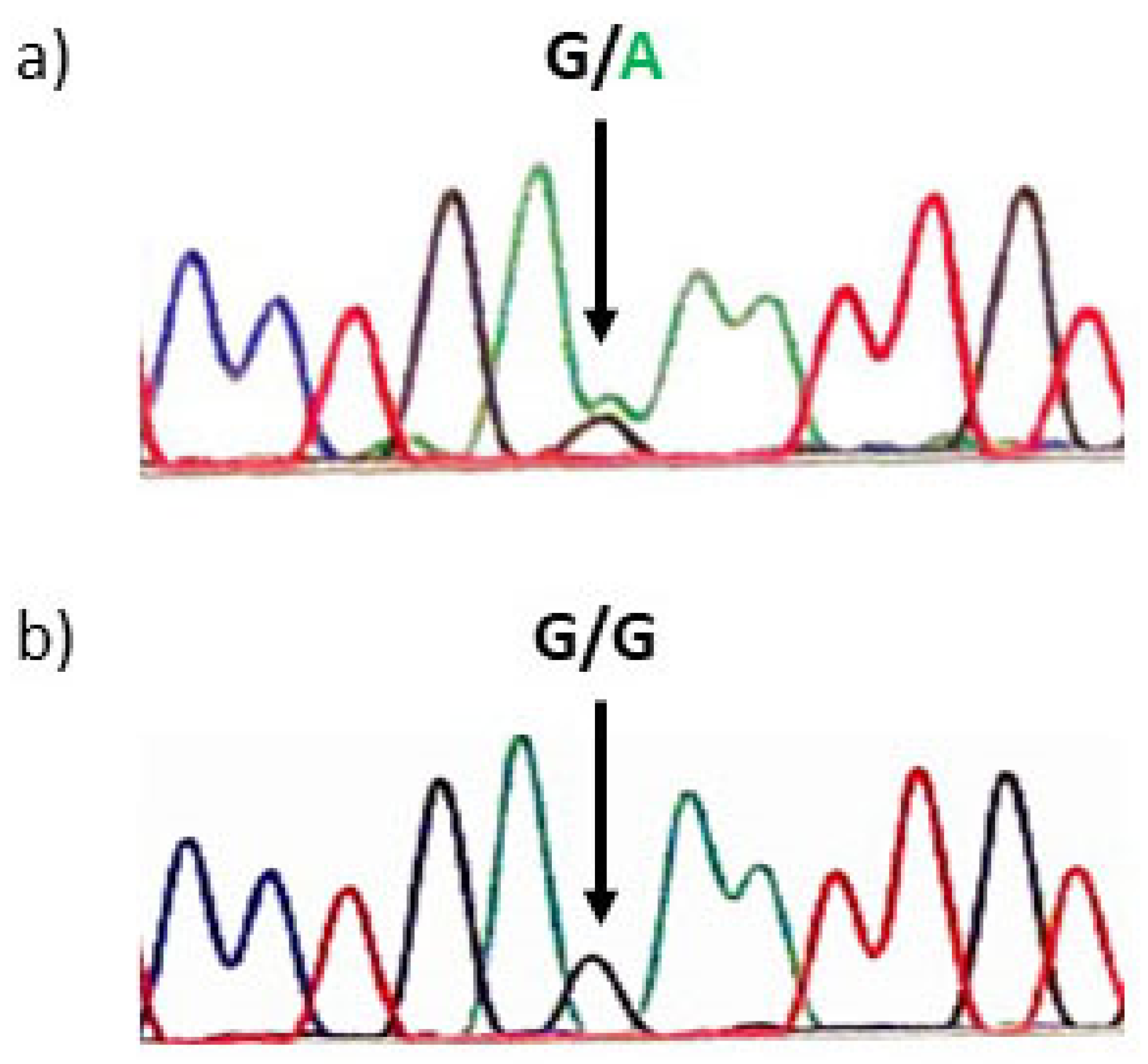
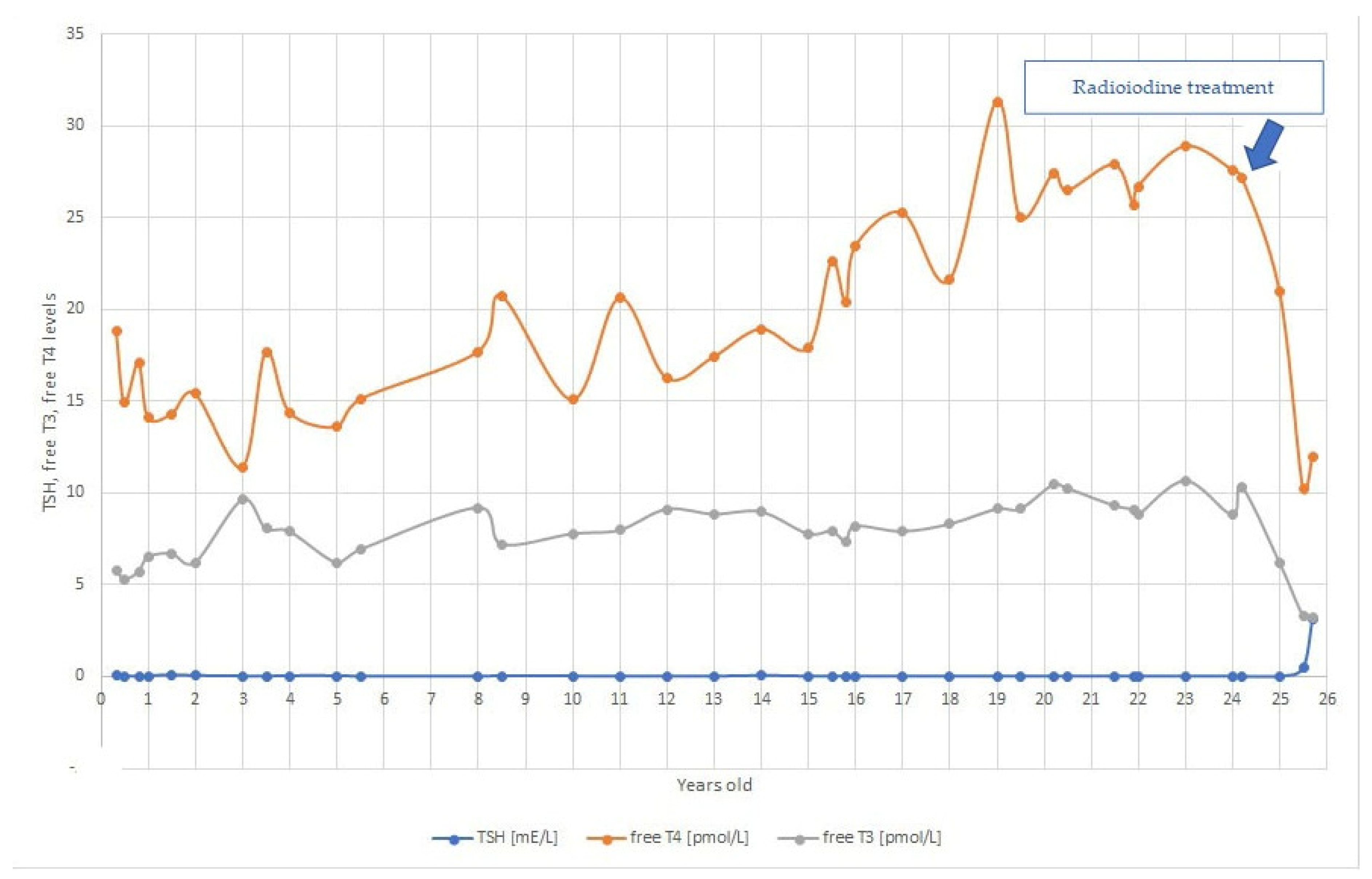
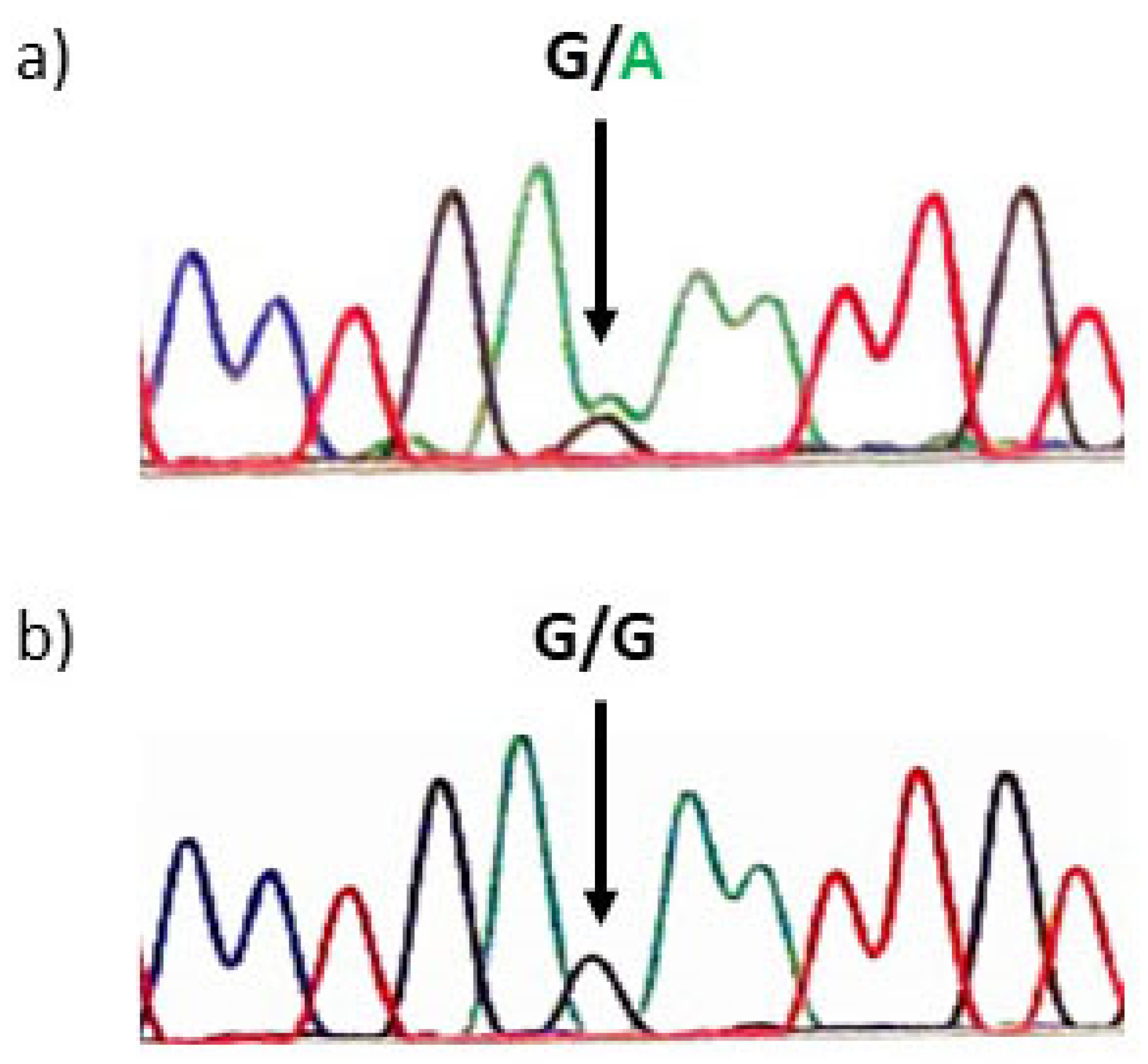
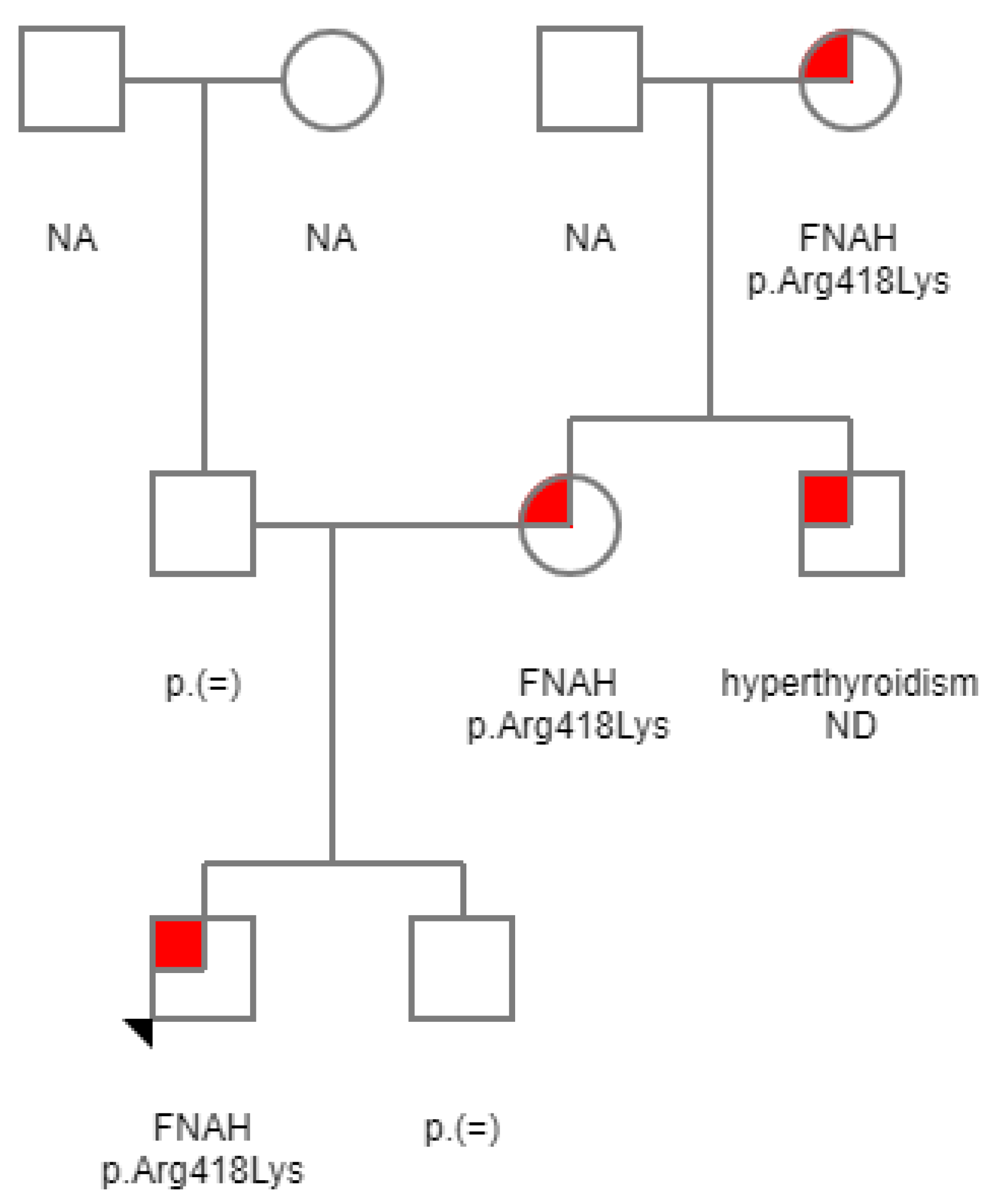
2. Case Report
3. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Léger, J.; Carel, J. Hyperthyroidism in Childhood: Causes, when and how to treat. J. Clin. Res. Pediatric Endocrinol. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Hébrant, A.; van Staveren, W.; Maenhaut, C.; Dumont, J.; Leclère, J. Genetic hyperthyroidism: Hyperthyroidism due to activating TSHR mutations. Eur. J. Endocrinol. 2011, 164, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akcurin, S.; Turkkahraman, D.; Tysoe, C.; Ellard, S.; De Leener, A.; Vassart, G.; Costagliola, S. A family with a novel TSH receptor activating germline mutation (p.Ala485Val). Eur. J. Pediatrics 2008, 167, 1231–1237. [Google Scholar] [CrossRef] [PubMed]
- Börgel, K.; Pohlenz, J.; Koch, H.; Bramswig, J. Long-Term Carbimazole Treatment of Neonatal Nonautoimmune Hyperthyroidism due to a New Activating TSH Receptor Gene Mutation (Ala428Val). Horm. Res. Paediatr. 2005, 64, 203–208. [Google Scholar] [CrossRef]
- Singer, K.; Menon, R.; Lesperance, M.; McHugh, J.; Gebarski, S.; Avram, A. Residual Thyroid Tissue After Thyroidectomy in a Patient With TSH Receptor-Activating Mutation Presenting as a Neck Mass. J. Clin. Endocrinol. Metab. 2013, 98, 448–452. [Google Scholar] [CrossRef] [Green Version]
- Weiss, R.; Dumitrescu, A.; Refetoff, S. Approach to the Patient with Resistance to Thyroid Hormone and Pregnancy. J. Clin. Endocrinol. Metab. 2010, 95, 3094–3102. [Google Scholar] [CrossRef] [Green Version]
- Gozu, H.; Lublinghoff, J.; Bircan, R.; Paschke, R. Genetics and phenomics of inherited and sporadic non-autoimmune hyperthyroidism. Mol. Cell. Endocrinol. 2010, 322, 125–134. [Google Scholar] [CrossRef]
- Lee, Y.; Poh, L.; Loke, K. An Activating Mutation of the Thyrotropin Receptor Gene in Hereditary Non-Autoimmune Hyperthyroidism. J. Pediatric Endocrinol. Metab. 2002, 15. [Google Scholar] [CrossRef] [PubMed]
- Biebermann, H.; Winkler, F.; Handke, D.; Grüters, A.; Krude, H.; Kleinau, G. Molecular description of non-autoimmune hyperthyroidism at a neonate caused by a new thyrotropin receptor germline mutation. Thyroid Res. 2011, 4, S8. [Google Scholar] [CrossRef] [Green Version]
- Jaeschke, H.; Schaarschmidt, J.; Eszlinger, M.; Huth, S.; Puttinger, R.; Rittinger, O.; Meiler, J.; Paschke, R. A Newly Discovered TSHR Variant (L665F) Associated with Nonautoimmune Hyperthyroidism in an Austrian Family Induces Constitutive TSHR Activation by Steric Repulsion Between TM1 and TM7. J. Clin. Endocrinol. Metab. 2014, 99, E2051–E2059. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Sun, Y.; Dong, Q.; He, M.; Cheng, C.; Fan, F. A novel TSHR gene mutation (Ile691Phe) in a Chinese family causing autosomal dominant non-autoimmune hyperthyroidism. J. Hum. Genet. 2008, 53, 475–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biebermann, H.; Winkler, F.; Handke, D.; Teichmann, A.; Gerling, B.; Cameron, F.; Eichhorst, J.; Grüters, A.; Wiesner, B.; Kühnen, P.; et al. New Pathogenic Thyrotropin Receptor Mutations Decipher Differentiated Activity Switching at a Conserved Helix 6 Motif of Family A GPCR. J. Clin. Endocrinol. Metab. 2012, 97, E228–E232. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, A.; Morikawa, S.; Aoyagi, H.; Ishizu, K.; Tajima, T. A Japanese family with nonautoimmune hyperthyroidism caused by a novel heterozygous thyrotropin receptor gene mutation. Pediatric Res. 2014, 75, 749–753. [Google Scholar] [CrossRef] [Green Version]
- Pohlenz, J.; Pfarr, N.; Krüger, S.; Hesse, V. Subclinical hyperthyroidism due to a thyrotropin receptor (TSHR) gene mutation (S505R). Acta Paediatr. 2006, 95, 1685–1687. [Google Scholar] [CrossRef]
- Ferrara, A.; Capalbo, D.; Rossi, G.; Capuano, S.; Del Prete, G.; Esposito, V.; Montesano, G.; Zampella, E.; Fenzi, G.; Salerno, M.; et al. A New Case of Familial Nonautoimmune Hyperthyroidism Caused by the M463V Mutation in the TSH Receptor with Anticipation of the Disease Across Generations: A Possible Role of Iodine Supplementation. Thyroid 2007, 17, 677–680. [Google Scholar] [CrossRef]
- Claus, M.; Maier, J.; Paschke, R.; Kujat, C.; Stumvoll, M.; Führer, D. Novel Thyrotropin Receptor Germline Mutation (Ile568Val) in a Saxonian Family with Hereditary Nonautoimmune Hyperthyroidism. Thyroid 2005, 15, 1089–1094. [Google Scholar] [CrossRef] [PubMed]
- Larsen, C.; Karaviti, L.; Seghers, V.; Weiss, R.; Refetoff, S.; Dumitrescu, A. A new family with an activating mutation (G431S) in the TSH receptor gene: A phenotype discussion and review of the literature. Int. J. Pediatric Endocrinol. 2014, 2014, 23. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Ferraz, C.; Paschke, R. Inheritable and sporadic non-autoimmune hyperthyroidism. Best Pract. Res. Clin. Endocrinol. Metab. 2017, 31, 265–275. [Google Scholar] [CrossRef]
- TSH Receptor Mutations. Available online: www.tsh-receptor-mutation-database.org/list.html?view=mutations (accessed on 20 December 2020).
- InterPro Database. Available online: www.ebi.ac.uk/interpro/protein/UniProt/P16473 (accessed on 20 December 2020).
- Biebermann, H.; Schöneberg, T.; Hess, C.; Germak, J.; Gudermann, T.; Grüters, A. The First Activating TSH Receptor Mutation in Transmembrane Domain 1 Identified in a Family with Nonautoimmune Hyperthyroidism. J. Clin. Endocrinol. Metab. 2001, 86, 4429–4433. [Google Scholar] [CrossRef] [PubMed]
- Lueblinghoff, J.; Eszlinger, M.; Jaeschke, H.; Mueller, S.; Bircan, R.; Gozu, H.; Sancak, S.; Akalin, S.; Paschke, R. Shared Sporadic and Somatic Thyrotropin Receptor Mutations Display More Active In Vitro Activities than Familial Thyrotropin Receptor Mutations. Thyroid 2011, 21, 221–229. [Google Scholar] [CrossRef]
- Lueblinghoff, J.; Mueller, S.; Sontheimer, J.; Paschke, R. Lack of consistent association of thyrotropin receptor mutations in vitro activity with the clinical course of patients with sporadic non-autoimmune hyperthyroidism. J. Endocrinol. Investig. 2009, 33, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Paschke, R.; Niedziela, M.; Vaidya, B.; Persani, L.; Rapoport, B.; Leclere, J. 2012 European Thyroid Association Guidelines for the Management of Familial and Persistent Sporadic Non-Autoimmune Hyperthyroidism Caused by Thyroid-Stimulating Hormone Receptor Germline Mutations. Eur. Thyroid J. 2012, 1, 142–147. [Google Scholar] [CrossRef] [Green Version]
- Bircan, R.; Miehle, K.; Mladenova, G.; Ivanova, R.; Ivanova, R.; Sarafova, A.; Borissova, A.; Lüblinghoff, J.; Paschke, R. Multiple Relapses of Hyperthyroidism after Thyroid Surgeries in a Patient with Long Term Follow-up of Sporadic Non-autoimmune Hyperthyroidism. Exp. Clin. Endocrinol. Diabetes 2008, 116, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Nishihara, E.; Fukata, S.; Hishinuma, A.; Kudo, T.; Ohye, H.; Ito, M.; Kubota, S.; Amino, N.; Kuma, K.; Miyauchi, A. Sporadic Congenital Hyperthyroidism due to a Germline Mutation in the Thyrotropin Receptor Gene (Leu 512 Gln) in a Japanese Patient. Endocr. J. 2006, 53, 735–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaeschke, H.; Eszlinger, M.; Lueblinghoff, J.; Coslovsky, R.; Paschke, R. Prolonged Inappropriate TSH Suppression During Hypothyroidism After Thyroid Ablation in a Patient with Nonautoimmune Familial Hyperthyroidism. Horm. Metab. Res. 2011, 43, 500–504. [Google Scholar] [CrossRef]
- Holzapfel, H.; Wonerow, P.; von Petrykowski, W.; Henschen, M.; Scherbaum, W.; Paschke, R. Sporadic Congenital Hyperthyroidism due to a Spontaneous Germline Mutation in the Thyrotropin Receptor Gene. J. Clin. Endocrinol. Metab. 1997, 82, 3879–3884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taha, D.; Adhikari, A.; Flore, L. Familial neonatal nonautoimmune hyperthyroidism due to a gain-of-function (D619G) thyrotropin-receptor mutation. J. Pediatric Endocrinol. Metab. 2020. [Google Scholar] [CrossRef]
- Grüters, A.; Schöneberg, T.; Biebermann, H.; Krude, H.; Krohn, H.; Dralle, H.; Gudermann, T. Severe Congenital Hyperthyroidism Caused by a Germ-LineneoMutation in the Extracellular Portion of the Thyrotropin Receptor1. J. Clin. Endocrinol. Metab. 1998, 83, 1431–1436. [Google Scholar] [CrossRef] [PubMed]
- Watkins, M.; Dejkhamron, P.; Huo, J.; Vazquez, D.; Menon, R. Persistent Neonatal Thyrotoxicosis in a Neonate Secondary to a Rare Thyroid-Stimulating Hormone Receptor Activating Mutation. Endocr. Pract. 2008, 14, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Seo, G.; Oh, S.; Chung, W.; Kim, H.; Kim, Y.; Bae, M.; Park, K.; Kwak, M. An A627V-activating mutation in the thyroid-stimulating hormone receptor gene in familial nonautoimmune hyperthyroidism. Ann. Pediatric Endocrinol. Metab. 2020, 25, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Farahani, P. Thyroid Stimulating Hormone Suppression Post-Therapy in Patients with Graves’ Disease: A Systematic Review of Pathophysiology and Clinical Data. Clin. Investig. Med. 2015, 38, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prummel, M.; Brokken, L.; Wiersinga, W. Ultra Short-Loop Feedback Control of Thyrotropin Secretion. Thyroid 2004, 14, 825–829. [Google Scholar] [CrossRef]
- Brokken, L.; Bakker, O.; Wiersinga, W.; Prummel, M. Functional Thyrotropin Receptor Expression in the Pituitary Folliculo-Stellate Cell Line TtT/GF. Exp. Clin. Endocrinol. Diabetes 2005, 113, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Bockmann, J.; Winter, C.; Wittkowski, W.; Kreutz, M.; Böckers, T. Cloning and Expression of a Brain-Derived TSH Receptor. Biochem. Biophys. Res. Commun. 1997, 238, 173–178. [Google Scholar] [CrossRef]
- Uy, H.; Reasner, C.; Samuels, M. Pattern of recovery of the hypothalamic-pituitary-thyroid axis following radioactive iodine therapy in patients with Graves’ disease. Am. J. Med. 1995, 99, 173–179. [Google Scholar] [CrossRef]
- Albert, S.; Goodgold, H.; Chehade, J.; Kim, J. Delayed Recovery of Thyrotropin Responsiveness after Radioactive Iodine Therapy for Hyperthyroidism. Am. J. Med. Sci. 2000, 319, 376–379. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Time (Minutes) | 0 | 30 | 120 |
|---|---|---|---|
| TSH (mE/L) | 0.03 | 0.79 | ND |
| Free T3 (pmol/L) | 6.5 | ND | 8.0 |
| Free T4 (pmol/L) | 14.1 | ND | 16.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suput Omladic, J.; Pajek, M.; Groselj, U.; Trebusak Podkrajsek, K.; Avbelj Stefanija, M.; Zerjav Tansek, M.; Kotnik, P.; Battelino, T.; Smigoc Schweiger, D. Central TSH Dysregulation in a Patient with Familial Non-Autoimmune Autosomal Dominant Hyperthyroidism Due to a Novel Thyroid-Stimulating Hormone Receptor Disease-Causing Variant. Medicina 2021, 57, 196. https://0-doi-org.brum.beds.ac.uk/10.3390/medicina57030196
Suput Omladic J, Pajek M, Groselj U, Trebusak Podkrajsek K, Avbelj Stefanija M, Zerjav Tansek M, Kotnik P, Battelino T, Smigoc Schweiger D. Central TSH Dysregulation in a Patient with Familial Non-Autoimmune Autosomal Dominant Hyperthyroidism Due to a Novel Thyroid-Stimulating Hormone Receptor Disease-Causing Variant. Medicina. 2021; 57(3):196. https://0-doi-org.brum.beds.ac.uk/10.3390/medicina57030196
Chicago/Turabian StyleSuput Omladic, Jasna, Maja Pajek, Urh Groselj, Katarina Trebusak Podkrajsek, Magdalena Avbelj Stefanija, Mojca Zerjav Tansek, Primoz Kotnik, Tadej Battelino, and Darja Smigoc Schweiger. 2021. "Central TSH Dysregulation in a Patient with Familial Non-Autoimmune Autosomal Dominant Hyperthyroidism Due to a Novel Thyroid-Stimulating Hormone Receptor Disease-Causing Variant" Medicina 57, no. 3: 196. https://0-doi-org.brum.beds.ac.uk/10.3390/medicina57030196