
TGF-β Physiology as a Novel Therapeutic Target Regarding Autoimmune Thyroid Diseases: Where Do We Stand and What to Expect

 , and
, and
Abstract
:1. Introduction
2. TGF-β Homeostasis and Its Interplay with the Immune System
2.1. Physiology of TGF-β Biosynthesis, Activation, and Signaling Pathways
2.2. The Physiological Role of TGF-β Regarding the Immune System
3. Thyroid Gland Homeostasis and TGF-β
3.1. Interplay between Normal Thyroid Physiology/Function and TGF-β
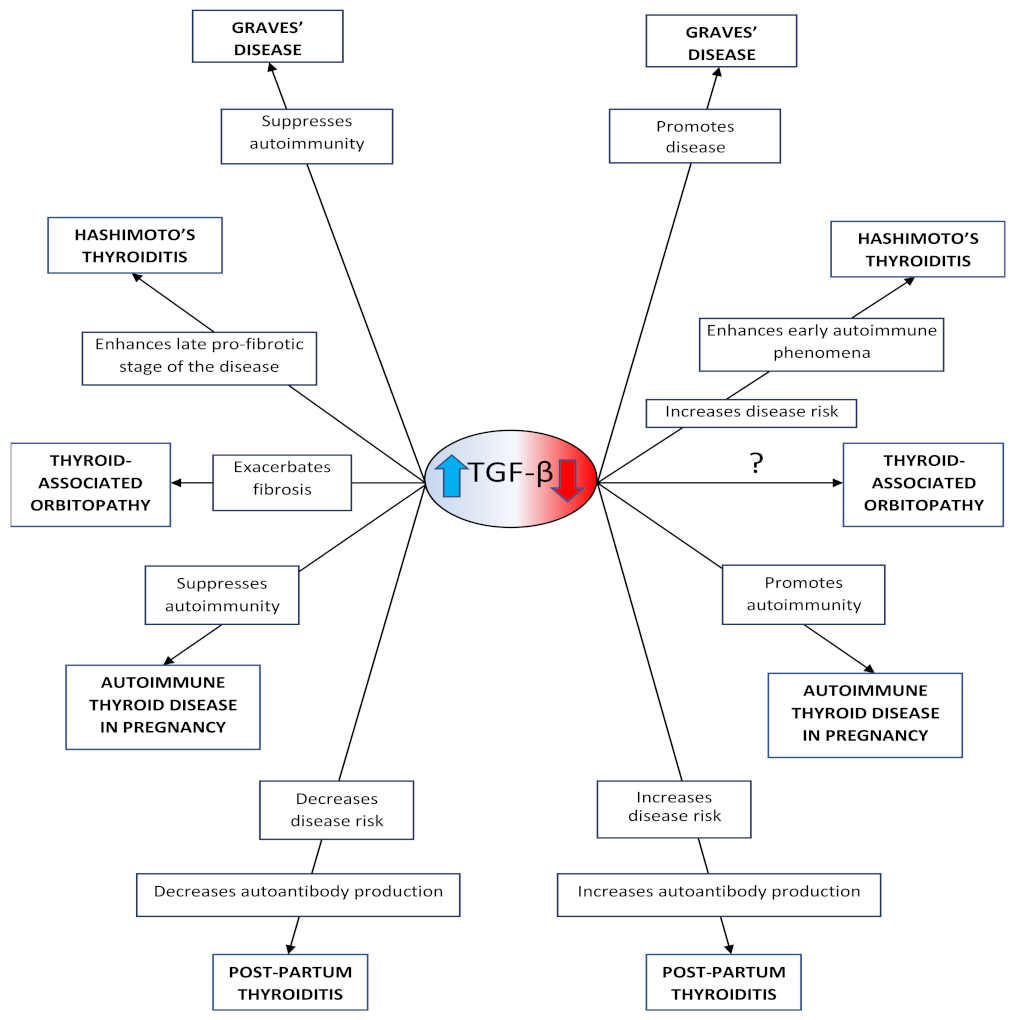
3.2. Association of TGF-β with the Pathophysiology of the Autoimmune Thyroid Diseases
3.2.1. In Graves’ Disease
3.2.2. In Hashimoto’s Thyroiditis
3.2.3. In Thyroid-Associated Orbitopathy
3.2.4. In Autoimmune Thyroid Diseases during Pregnancy
3.2.5. Ιn Post-Partum Thyroiditis
- -
- -
- -
4. TGF-β-Based Therapeutic Interventions in Non-Thyroid Autoimmune, Inflammatory, and Fibrotic Diseases
- -
- Experimental addition of iTregs (generated in healthy subjects) to patients with active multiple sclerosis effectively inhibited the autoimmune phenomena associated with the latter via enhanced TGF-β synthesis [57].
- -
- In arteriosclerotic conditions (especially in atherosclerosis), TGF-β has a highly beneficial and protective role, as it inhibits the migration of macrophages and suppresses their transformation to smooth muscle cells; sustains and maintains the normal function of the endothelium; minimizes the adhesiveness of the latter regarding pro-inflammatory cell types; and protects vascular integrity [58]. Thus, decreased serum TGF-β concentrations correlate with greater cardiovascular death risk. As a result, therapeutic strategies involving TGF-β could be extremely useful toward ischemic heart disease prophylaxis and limitation of cardiovascular mortality.
- -
- Regarding fibrosis, gene transfer of Smad7 (which exerts a suppressive role in TGF-β signaling pathways) has been therapeutically applicated to liver and colon fibrosis, diabetic nephropathy, and vascular smooth cell remodeling [59].
- -
- PPARγ (Peroxisome proliferator-activated receptors-γ) agonists limit fibrosis in liver via mechanisms unknown yet. Eventually, PPARγ agonists inhibit TGF-β-mediated activation of TβRI signaling in hepatic stellate cells, thereby effectively inhibiting Smad3-dependent initiation of ECM gene expression, which leads to substantially decreased PAI-1 and collagen-1α synthesis [60]. The experimental treatment of these cells with PPARγ agonists results in a dose-dependent suppression of TGF-β-mediated functions, preserving at the same time the physiological cell development or survival.
- -
- In many diseases such as alcoholic steatohepatitis, sarcoidosis, post-radiation fibrosis, and various types of neuropathy, the suppressed expression of TGF-β restores the disturbed Th1/Th2 equilibrium and limits Th1-regulated immune responses. Pentoxifylline (PTX) has been therapeutically used to effectively suppress TGF-β activation and prevent the detrimental autoimmune responses associated with this growth factor [61].
- -
- Treatment strategies based on progesterone had been effective in treating preterm infants suffering from bronchopulmonary dysplasia via dose-dependent suppression of TGF-β-mediated activation and ensuing transcription of the Smad2/3 complex [33].
- -
- Suppression of TGF-β activation and/or signaling pathways via small peptides with binding affinity to the LAP region of the TGF-β latent complex can adequately block TGF-β induced fibrosis and thus promote liver regeneration in experimentally hepatectomized rodents [62].
5. Suggested TGF-β-Based Diagnostic, Prognostic, and Therapeutic Interventions in Thyroid Autoimmune Diseases
- -
- Treatment of human cultures of thyroid follicular cells/lymphocytes (from Graves’ disease patients) with hrTGF-β limits autoimmune phenomena in the thyroid gland via substantially diminished target T-cells antigenicity and decreased expression of TPO and MHC class II autoantigens [37].
- -
- Treatment with low-level laser therapy (LLLT) exerts a suppressive role regarding the evolution of autoimmune diseases via the regeneration of various tissues. It has been shown that in case of Hashimoto’s thyroiditis, treatment of patients with LLLT leads to a significant stimulation of TGF-β gene expression [65]. Thus, this therapeutic strategy promotes normal thyroid function, which is ‘mirrored’ in the mean daily levothyroxine dose-reduction. As a result, therapeutically applicated LLLT to Hashimoto’s thyroiditis could be an effective tool of autoimmunity restraint during the initial pathophysiologic stages of this disease via enhanced TGF-β biosynthesis and subsequent cytokine gene expression.
- -
- The limitation of the Smad-regulated TGF-β gene transcription can substantially suppress thyroid gland fibrosis, which is observed in animal models of experimentally induced autoimmune thyroiditis [66]. This limitation can be accomplished following the administration of triiodothyronine (T3), which binds to its thyroid receptors complex and represses specific TGF-β activated gene promoters.
- -
- The fibrotic phenomena observed in the final pathophysiologic stage of Hashimoto’s thyroiditis can be limited in animal models of this disease via monoclonal anti-TGF-β antibodies administration, which neutralizes the excess quantities of extracellular TGF-β [67].
- -
- The application of 17-estradiol (E2) to thyroid follicular cells (rodent models) triggers ER-β-mediated development of EAT [28]. To the contrary, treatment with an E2 antagonist, such as coumestrol, limits ATA production and suppresses TGF-β-induced Th17-type response (which promotes “cell-type” autoimmune response) [29]. It should be noted that ER-α activation can counterbalance the outcomes of TGF-β function in a unique E2-dependent manner [30,31].
- -
- In Hashimoto’s thyroiditis, TGF-β-mediated inflammatory phenomena initiate enhanced cyclooxygenase (COX-2) expression [68]. Thus, suppression of the latter via Celecoxib (an inhibitor of COX-2) might be an effective treatment strategy for the limitations of the autoimmune process in Hashimoto’s thyroiditis.
- -
- The addition of Celecoxib to human myoblasts (cultured from extraocular muscles of patients with thyroid-associated orbitopathy) impedes TGF-β-regulated HA production and disrupts the TGF-β-mediated maturation and differentiation of ocular muscle fibroblasts [68].
- -
- The addition of PPARγ agonists (such as pioglitazone) to above-mentioned human myoblasts can restrain inflammatory phenomena and suppress adipogenesis in orbital fibroblasts [68]. Moreover, PPARγ agonists can inhibit the facilitatory role of TNF regarding TGF-β-biosynthesis via canonical and/or non-canonical TGF-β-related signaling pathways. Consequently, the catalyzing effects of TGF-β regarding (a) myofibroblast formation from fibroblasts and (b) HAS, HA, and TGF-β production in retro-orbital fibroblasts are adequately limited in an indirect, paracrine manner.
- -
- Furthermore, aryl hydrocarbon (AH) can suppress the profibrotic activity of TGF-β in thyroid-associated orbitopathy [69]. Thus, AH agonists or AH receptor ligands inhibit TGF-β mediated myofibroblast formation in patients with thyroid-associated orbitopathy via the blockade/attenuation of the profibrotic Wnt signaling.
- -
- In addition, the stimulatory effect of PAI-1 regarding TGF-β production in patients with thyroid-associated orbitopathy can be reversed via synthetic or natural small molecules, which effectively suppress the function/activity of PAI-1 [70]. The therapeutic application of these molecules regarding PAI-1 has already been initiated with success in fibrotic, malignant, and thromboembolic conditions.
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Yang, L.; Pang, Y.; Moses, H.L. TGF-β and immune cells: An important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 2010, 31, 220–227. [Google Scholar] [CrossRef] [Green Version]
- Widder, J.; Dorfinger, K.; Wilfing, A.; Trieb, K.; Pirich, K.; Loebenstein, R.; Niederle, B.; Gessl, A.; Spitzauer, S.; Grubeck-Loebenstein, B. The immunoregulatory influence of transforming growth factor beta in thyroid autoimmunity: TGF β inhibits autoreactivity in Graves’ disease. J. Autoimmun. 1991, 4, 689–701. [Google Scholar] [CrossRef]
- Ouyang, W.; Filvaroff, E.; Hu, Y.; Grogan, J. Novel therapeutic targets along the Th17 pathway. Eur. J. Immunol. 2009, 39, 670–675. [Google Scholar] [CrossRef] [PubMed]
- Syed, V. TGF-β Signaling in Cancer. J. Cell. Biochem. 2016, 117, 1279–1287. [Google Scholar] [CrossRef]
- Morikawa, M.; Derynck, R.; Miyazono, K. TGF-β and the TGF-β Family: Context-Dependent Roles in Cell and Tissue Physiology. Cold Spring Harb. Perspect. Biol. 2016, 8, a021873. [Google Scholar] [CrossRef] [Green Version]
- Santibañez, J.F.; Quintanilla, M.; Bernabeu, C. TGF-β/TGF-β receptor system and its role in physiological and pathological conditions. Clin. Sci. 2011, 121, 233–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinck, A.; Mueller, T.D.; Springer, T.A. Structural Biology and Evolution of the TGF-β Family. Cold Spring Harb. Perspect. Biol. 2016, 8, a022103. [Google Scholar] [CrossRef] [Green Version]
- Robertson, I.B.; Horiguchi, M.; Zilberberg, L.; Dabovic, B.; Hadjiolova, K.; Rifkin, D.B. Latent TGF-β-binding proteins. Matrix Biol. 2015, 47, 44–53. [Google Scholar] [CrossRef]
- Wipff, P.-J.; Hinz, B. Integrins and the activation of latent transforming growth factor β1—An intimate relationship. Eur. J. Cell Biol. 2008, 87, 601–615. [Google Scholar] [CrossRef]
- Massagué, J.; Blain, S.W.; Lo, R.S. TGFβ Signaling in Growth Control, Cancer, and Heritable Disorders. Cell 2000, 103, 295–309. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Massagué, J. Mechanisms of TGF-β Signaling from Cell Membrane to the Nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef] [Green Version]
- Ark, A.V.; Cao, J.; Li, X. TGF-β receptors: In and beyond TGF-β signaling. Cell. Signal. 2018, 52, 112–120. [Google Scholar] [CrossRef]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nat. Cell Biol. 2003, 425, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Andrieux, G.; Fattet, L.; Le Borgne, M.; Rimokh, R.; Théret, N. Dynamic Regulation of Tgf-B Signaling by Tif1γ: A Computational Approach. PLoS ONE 2012, 7, e33761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zandvoort, A.; Postma, D.S.; Jonker, M.R.; Noordhoek, J.A.; Vos, J.T.W.M.; Van Der Geld, Y.M.; Timens, W. Altered expression of the Smad signalling pathway: Implications for COPD pathogenesis. Eur. Respir. J. 2006, 28, 533–541. [Google Scholar] [CrossRef] [Green Version]
- Colak, S.; Ten Dijke, P. Targeting TGF-β Signaling in Cancer. Trends Cancer 2017, 3, 56–71. [Google Scholar] [CrossRef]
- Zhang, Y.E. Non-Smad Signaling Pathways of the TGF-β Family. Cold Spring Harb. Perspect. Biol. 2016, 9, a022129. [Google Scholar] [CrossRef]
- Ábrigo, J.; Campos, F.; Simon, F.; Riedel, C.; Cabrera, D.; Vilos, C.; Cabello-Verrugio, C. TGF-β requires the activation of canonical and non-canonical signalling pathways to induce skeletal muscle atrophy. Biol. Chem. 2018, 399, 253–264. [Google Scholar] [CrossRef]
- Papageorgis, P. TGFβ Signaling in Tumor Initiation, Epithelial-to-Mesenchymal Transition, and Metastasis. J. Oncol. 2015, 2015, 587193. [Google Scholar] [CrossRef] [Green Version]
- Egesten, A.; Gordon, S.; Herwald, H. Innate immunity—A clinical perspective. J. Intern. Med. 2019, 285, 477–478. [Google Scholar] [CrossRef] [PubMed]
- Bonilla, F.A.; Oettgen, H.C. Adaptive immunity. J. Allergy Clin. Immunol. 2010, 125, S33–S40. [Google Scholar] [CrossRef]
- Kelly, A.; Houston, S.A.; Sherwood, E.; Casulli, J.; Travis, M.A. Regulation of Innate and Adaptive Immunity by TGFβ. Adv. Immunol. 2017, 134, 137–233. [Google Scholar] [CrossRef] [PubMed]
- Travis, M.A.; Sheppard, D. TGF-β Activation and Function in Immunity. Annu. Rev. Immunol. 2014, 32, 51–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, S.A.; Li, M.O. TGF-β: Guardian of T Cell Function. The Journal of Immunology 2013, 191, 3973–3979. [Google Scholar] [CrossRef] [PubMed]
- Rosser, E.C.; Mauri, C. Regulatory B Cells: Origin, Phenotype, and Function. Immunity 2015, 42, 607–612. [Google Scholar] [CrossRef] [Green Version]
- McCarron, M.J.; Marie, J.C. TGF-β prevents T follicular helper cell accumulation and B cell autoreactivity. J. Clin. Investig. 2014, 124, 4375–4386. [Google Scholar] [CrossRef] [Green Version]
- Mincione, G.; Di Marcantonio, M.C.; Tarantelli, C.; D’Inzeo, S.; Nicolussi, A.; Nardi, F.; Donini, C.F.; Coppa, A. EGF and TGF-1 Effects on Thyroid Function. J. Thyroid Res. 2011, 2011, 431718. [Google Scholar] [CrossRef] [Green Version]
- Santin, A.P.; Furlanetto, T.W. Role of estrogen in thyroid function and growth regulation. J. Thyroid Res. 2011, 2011, 875125. [Google Scholar] [CrossRef] [Green Version]
- Gantus, M.; Alves, L.; Stipursky, J.; Souza, E.; Teodoro, A.; Alves, T.; Carvalho, D.; Martinez, A.; Gomes, F.; Nasciutti, L. Estradiol modulates TGF-β1 expression and its signaling pathway in thyroid stromal cells. Mol. Cell. Endocrinol. 2011, 337, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Hanyu, A.; Wayama, M.; Goto, N.; Katsuno, Y.; Kawasaki, S.; Nakajima, Y.; Kajiro, M.; Komatsu, Y.; Fujimura, A.; et al. Estrogen receptor β activation stimulates the development of experimental autoimmune thyroiditis through up-regulation of Th17-type responses. Clin. Immunol. 2018, 190, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Ito, I.; Hanyu, A.; Wayama, M.; Goto, N.; Katsuno, Y.; Kawasaki, S.; Nakajima, Y.; Kajiro, M.; Komatsu, Y.; Fujimura, A.; et al. Estrogen Inhibits Transforming Growth Factor β Signaling by Promoting Smad2/3 Degradation. J. Biol. Chem. 2010, 285, 14747–14755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chipuk, J.E.; Cornelius, S.C.; Pultz, N.J.; Jorgensen, J.S.; Bonham, M.J.; Kim, S.-J.; Danielpour, D. The Androgen Receptor Represses Transforming Growth Factor-β Signaling through Interaction with Smad3. J. Biol. Chem. 2002, 277, 1240–1248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunzmann, S.; Ottensmeier, B.; Speer, C.P.; Fehrholz, M. Effect of progesterone on Smad signaling and TGF-β/Smad-regulated genes in lung epithelial cells. PLoS ONE 2018, 13, e0200661. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, B.B.; Bhattacharya, P.; Gopisetty, A.; Prabhakar, B.S. Role of Cytokines in the Pathogenesis and Suppression of Thyroid Autoimmunity. J. Interferon Cytokine Res. 2011, 31, 721–731. [Google Scholar] [CrossRef]
- Scappaticcio, L.; Castellana, M.; Virili, C.; Bellastella, G.; Centanni, M.; Cannavò, S.; Campennì, A.; Ruggeri, R.M.; Giovanella, L.; Trimboli, P. Alemtuzumab-induced thyroid events in multiple sclerosis: A systematic review and meta-analysis. J. Endocrinol. Investig. 2020, 43, 219–229. [Google Scholar] [CrossRef]
- McLachlan, S.M.; Nagayama, Y.; Pichurin, P.N.; Mizutori, Y.; Chen, C.-R.; Misharin, A.; Aliesky, H.A.; Rapoport, B. The Link between Graves’ Disease and Hashimoto’s Thyroiditis: A Role for Regulatory T Cells. Endocrinology 2007, 148, 5724–5733. [Google Scholar] [CrossRef] [Green Version]
- Saxena, V.; Lienesch, D.W.; Zhou, M.; Bommireddy, R.; Azhar, M.; Doetschman, T.; Singh, R.R. Dual Roles of Immunoregulatory Cytokine TGF-β in the Pathogenesis of Autoimmunity-Mediated Organ Damage. J. Immunol. 2008, 180, 1903–1912. [Google Scholar] [CrossRef] [Green Version]
- Kutluturk, F.; Yarman, S.; Sarvan, F.O.; Kekik, C. Association of Cytokine Gene Polymorphisms (IL6, IL10, TNF-α, TGF-β and IFN-γ) and Graves’ Disease in Turkish Population. Endocr. Metab. Immune Disord. Drug Targets 2013, 13, 163–167. [Google Scholar] [CrossRef]
- Kakudo, K.; Li, Y.; Hirokawa, M.; Ozaki, T. Diagnosis of Hashimoto’s thyroiditis and IgG4-related sclerosing disease. Pathol. Int. 2011, 61, 175–183. [Google Scholar] [CrossRef]
- Chen, K.; Wei, Y.; Sharp, G.C.; Braley-Mullen, H. Characterization of thyroid fibrosis in a murine model of granulomatous experimental autoimmune thyroiditis. J. Leukoc. Biol. 2000, 68, 828–835. [Google Scholar]
- Morris, G.; Brown, N.; Kong, Y.-C.M. Naturally-existing CD4+CD25+Foxp3+ regulatory T cells are required for tolerance to experimental autoimmune thyroiditis induced by either exogenous or endogenous autoantigen. J. Autoimmun. 2009, 33, 68–76. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Wei, Y.; Sharp, G.C.; Braley-Mullen, H. Mechanisms of spontaneous resolution versus fibrosis in granulomatous experimental autoimmune thyroiditis. J. Immunol. 2003, 171, 6236–6243. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Cai, W.; Gu, R.; Zhang, Y.; Zhang, H.; Tang, K.; Xu, P.; Katirai, F.; Shi, W.; Wang, L.; et al. Th17 cell plays a role in the pathogenesis of Hashimoto’s thyroiditis in patients. Clin. Immunol. 2013, 149, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Vural, P.; Degirmencioglu, S.; Doğru-Abbasoğlu, S.; Baki, M.; Ozderya, A.; Karadag, B.; Uysal, M. Arg25Pro (c.915G>C) polymorphism of transforming growth factor β1 gene suggests an association with increased risk for Hashimoto’s thyroiditis. Int. Immunopharmacol. 2015, 28, 521–524. [Google Scholar] [CrossRef] [PubMed]
- Gianoukakis, A.G.; Khadavi, N.; Smith, T.J. Cytokines, Graves’ Disease, and Thyroid-Associated Ophthalmopathy. Thyroid 2008, 18, 953–958. [Google Scholar] [CrossRef] [PubMed]
- Khoo, T.K.; Coenen, M.J.; Schiefer, A.R.; Kumar, S.; Bahn, R.S. Evidence for Enhanced Thy-1 (CD90) Expression in Orbital Fibroblasts of Patients with Graves’ Ophthalmopathy. Thyroid 2008, 18, 1291–1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valyasevi, R.W.; Jyonouchi, S.C.; Dutton, C.M.; Munsakul, N.; Bahn, R.S. Effect of Tumor Necrosis Factor-α, Interferon-γ, and Transforming Growth Factor-β on Adipogenesis and Expression of Thyrotropin Receptor in Human Orbital Preadipocyte Fibroblasts. J. Clin. Endocrinol. Metab. 2001, 86, 903–908. [Google Scholar] [CrossRef] [Green Version]
- Ko, J.; Chae, M.K.; Lee, J.H.; Lee, E.J.; Yoon, J.S. Sphingosine-1-Phosphate Mediates Fibrosis in Orbital Fibroblasts in Graves’ Orbitopathy. Investig. Opthalmol. Vis. Sci. 2017, 58, 2544–2553. [Google Scholar] [CrossRef] [Green Version]
- Fang, S.; Huang, Y.; Zhong, S.; Li, Y.; Zhang, Y.; Li, Y.; Sun, J.; Liu, X.; Wang, Y.; Zhang, S.; et al. Regulation of Orbital Fibrosis and Adipogenesis by Pathogenic Th17 Cells in Graves Orbitopathy. J. Clin. Endocrinol. Metab. 2017, 102, 4273–4283. [Google Scholar] [CrossRef]
- Seo, J.Y.; Park, J.; Yu, M.R.; Kim, Y.S.; Ha, H.; Lee, H.B. Positive Feedback Loop between Plasminogen Activator Inhibitor-1 and Transforming Growth Factor-β1 during Renal Fibrosis in Diabetes. Am. J. Nephrol. 2009, 30, 481–490. [Google Scholar] [CrossRef]
- Khalilzadeh, O.; Anvari, M.; Esteghamati, A.; Momen-Heravi, F.; Rashidi, A.; Amiri, H.M.; Tahvildari, M.; Mahmoudi, M.; Amirzargar, A. Genetic susceptibility to Graves’ ophthalmopathy: The role of polymorphisms in anti-inflammatory cytokine genes. Ophthalmic Genet. 2010, 31, 215–220. [Google Scholar] [CrossRef]
- Singh, M.; Orazulike, N.C.; Ashmore, J.; Konje, J.C. Changes in Maternal Serum Transforming Growth Factor β-1 during Pregnancy: A Cross-Sectional Study. BioMed Res. Int. 2013, 2013, 318464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakkas, E.G.; Paltoglou, G.; Linardi, A.; Gryparis, A.; Nteka, E.; Chalarakis, N.; Mantzou, A.; Vrachnis, N.; Iliodromiti, Z.; Koukkou, E.; et al. Associations of maternal oestradiol, cortisol, and TGF-β1 plasma concentrations with thyroid autoantibodies during pregnancy and postpartum. Clin. Endocrinol. 2018, 89, 789–797. [Google Scholar] [CrossRef]
- Liakos, P.; Lenz, D.; Bernhardt, R.; Feige, J.-J.; Defaye, G. Transforming growth factor β1 inhibits aldosterone and cortisol production in the human adrenocortical cell line NCI-H295R through inhibition of CYP11B1 and CYP11B2 expression. J. Endocrinol. 2003, 176, 69–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olivieri, A.; De Angelis, S.; Vaccari, V.; Valensise, H.; Magnani, F.; Stazi, M.A.; Cotichini, R.; Gilardi, E.; Cordeddu, V.; Sorcini, M.; et al. Postpartum Thyroiditis Is Associated with Fluctuations in Transforming Growth Factor-β1 Serum Levels. J. Clin. Endocrinol. Metab. 2003, 88, 1280–1284. [Google Scholar] [CrossRef]
- Davies, T.F. The Thyroid Immunology of the Postpartum Period. Thyroid 1999, 9, 675–684. [Google Scholar] [CrossRef] [PubMed]
- Duffy, S.S.; Keating, B.A.; Moalem-Taylor, G. Adoptive Transfer of Regulatory T Cells as a Promising Immunotherapy for the Treatment of Multiple Sclerosis. Front. Neurosci. 2019, 13, 1107. [Google Scholar] [CrossRef] [Green Version]
- Stefoni, S.; Cianciolo, G.; Donati, G.; Dormi, A.; Silvestri, M.G.; Colì, L.; De Pascalis, A.; Iannelli, S. Low TGF-β1 serum levels are a risk factor for atherosclerosis disease in ESRD patients. Kidney Int. 2002, 61, 324–335. [Google Scholar] [CrossRef] [Green Version]
- Cutroneo, K.R. Evidence for TGF-β1 and bleomycin intracellular signaling through autocrine regulation of Smad 3 binding to the proximal promoter of theSmad 7 gene. J. Cell. Biochem. 2006, 97, 933–939. [Google Scholar] [CrossRef]
- Zhao, C.; Chen, W.; Yang, L.; Chen, L.; Stimpson, S.A.; Diehl, A.M. PPARγ agonists prevent TGFβ1/Smad3-signaling in human hepatic stellate cells. Biochem. Biophys. Res. Commun. 2006, 350, 385–391. [Google Scholar] [CrossRef] [Green Version]
- Azimi, A. Pentoxifylline Explores New Horizons in Treatment of Hashimoto Thyroiditis. Biol. Med. 2016, 8. [Google Scholar] [CrossRef] [Green Version]
- Kuroki, H.; Hayashi, H.; Nakagawa, S.; Sakamoto, K.; Higashi, T.; Nitta, H.; Hashimoto, D.; Chikamoto, A.; Beppu, T.; Baba, H. Effect of LSKL peptide on thrombospondin 1-mediated transforming growth factor β signal activation and liver regeneration after hepatectomy in an experimental model. Br. J. Surg. 2015, 102, 813–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, L.F.; Jia, H.Y.; Zhang, H.F.; Hu, Y.X. Expression level and clinical significance of IL-2, IL-6 and TGF-β in elderly patients with goiter and hyperthyroidism. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 4680–4686. [Google Scholar]
- Bahn, R.S. Graves’ Ophthalmopathy. N. Engl. J. Med. 2010, 362, 726–738. [Google Scholar] [CrossRef] [Green Version]
- Höfling, D.B.; Chavantes, M.C.; Acencio, M.M.P.; Cerri, G.G.; Marui, S.; Yoshimura, E.M.; Chammas, M.C. Effects of Low-Level Laser Therapy on the Serum TGF-β1 Concentrations in Individuals with Autoimmune Thyroiditis. Photomed. Laser Surg. 2014, 32, 444–449. [Google Scholar] [CrossRef]
- Mastorakos, G.; Doufas, A.G.; Mantzos, E.; Mantzos, J.; Koutras, D.A. T4 but not T3 administration is associated with increased recurrence of Graves’ disease after successful medical therapy. J. Endocrinol. Investig. 2003, 26, 979–984. [Google Scholar] [CrossRef]
- Alonso-Merino, E.; Orozco, R.M.; Ruíz-Llorente, L.; Martínez-Iglesias, O.A.; Velasco-Martín, J.P.; Montero-Pedrazuela, A.; Fanjul-Rodríguez, L.; Contreras-Jurado, C.; Regadera, J.; Aranda, A. Thyroid hormones inhibit TGF-β signaling and attenuate fibrotic responses. Proc. Natl. Acad. Sci. USA 2016, 113, E3451–E3460. [Google Scholar] [CrossRef] [Green Version]
- Cheng, A.M.S.; Yin, H.Y.; Chen, A.; Liu, Y.-W.; Chuang, M.-C.; He, H.; Tighe, S.; Sheha, H.; Liao, S.-L. Celecoxib and Pioglitazone as Potential Therapeutics for Regulating TGF-β–Induced Hyaluronan in Dysthyroid Myopathy. Investig. Opthalmol. Vis. Sci. 2016, 57, 1951–1959. [Google Scholar] [CrossRef] [Green Version]
- Woeller, C.; Roztocil, E.; Hammond, C.L.; Feldon, S.; Phipps, R.P. The Aryl Hydrocarbon Receptor and Its Ligands Inhibit Myofibroblast Formation and Activation: Implications for Thyroid Eye Disease. Am. J. Pathol. 2016, 186, 3189–3202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouch, A.; Vanucci-Bacqué, C.; Bedos-Belval, F.; Baltas, M. Small molecules inhibitors of plasminogen activator inhibitor-1—An overview. Eur. J. Med. Chem. 2015, 92, 619–636. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| TGF-β |
|---|
|
| TGF-β |
|---|
|
| Effect on Synthesis | Effect on Activation | Effect on Action | Experimental Therapeutic Application | |
|---|---|---|---|---|
| Exogenously administrated hr-TGF-β | None | None | None | Cultures of follicular thyroid/lymphocyte cells from Graves’ disease in humans |
| Low-level laser therapy | Increase | None | None | Hashimoto’s thyroiditis in humans |
| Small peptides | None | Inhibit TGF-β disengagement from LAP | None | Cancer animal models |
| Monoclonal anti-TGF-β | None | None | Neutralize excess extracellular TGF-β | Hashimoto’s thyroiditis in animal models [61] |
| Triiodothyronine nuclear receptor ligands | None | None | Limit Smad phosphorylation | Thyroid fibrosis in animal models |
| Estrogen receptor β antagonists | None | None | Inhibit TGF-β-mediated Th17-type response | Experimental autoimmune thyroiditis in animal models |
| Estrogen receptor α agonists | None | None | Suppress TGF-β activity | Experimental autoimmune thyroiditis in animal models |
| COX-2 inhibitors | None | None | Block TGF-β-induced HA synthesis. Decrease TGF-β-induced ocular muscle fibroblasts proliferation | Cultures of extraocular muscle fibroblasts from TAO in humans |
| PPAR-γ agonists | Inhibit TNF- mediated TGF-β synthesis | None | Inhibit TGF-β-induced fibroblast differentiation to myofibroblasts.Decrease HAS and HA synthesis | Cultures of extraocular muscle fibroblasts from TAO in humans |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kardalas, E.; Maraka, S.; Papagianni, M.; Paltoglou, G.; Siristatidis, C.; Mastorakos, G. TGF-β Physiology as a Novel Therapeutic Target Regarding Autoimmune Thyroid Diseases: Where Do We Stand and What to Expect. Medicina 2021, 57, 621. https://0-doi-org.brum.beds.ac.uk/10.3390/medicina57060621
Kardalas E, Maraka S, Papagianni M, Paltoglou G, Siristatidis C, Mastorakos G. TGF-β Physiology as a Novel Therapeutic Target Regarding Autoimmune Thyroid Diseases: Where Do We Stand and What to Expect. Medicina. 2021; 57(6):621. https://0-doi-org.brum.beds.ac.uk/10.3390/medicina57060621
Chicago/Turabian StyleKardalas, Efstratios, Spyridoula Maraka, Maria Papagianni, George Paltoglou, Charalampos Siristatidis, and George Mastorakos. 2021. "TGF-β Physiology as a Novel Therapeutic Target Regarding Autoimmune Thyroid Diseases: Where Do We Stand and What to Expect" Medicina 57, no. 6: 621. https://0-doi-org.brum.beds.ac.uk/10.3390/medicina57060621