New Mammalian Target of Rapamycin (mTOR) Modulators Derived from Natural Product Databases and Marine Extracts by Using Molecular Docking Techniques

 , ,
, ,  , ,
, , 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
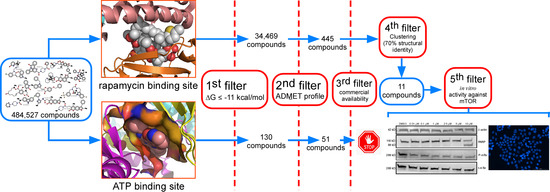
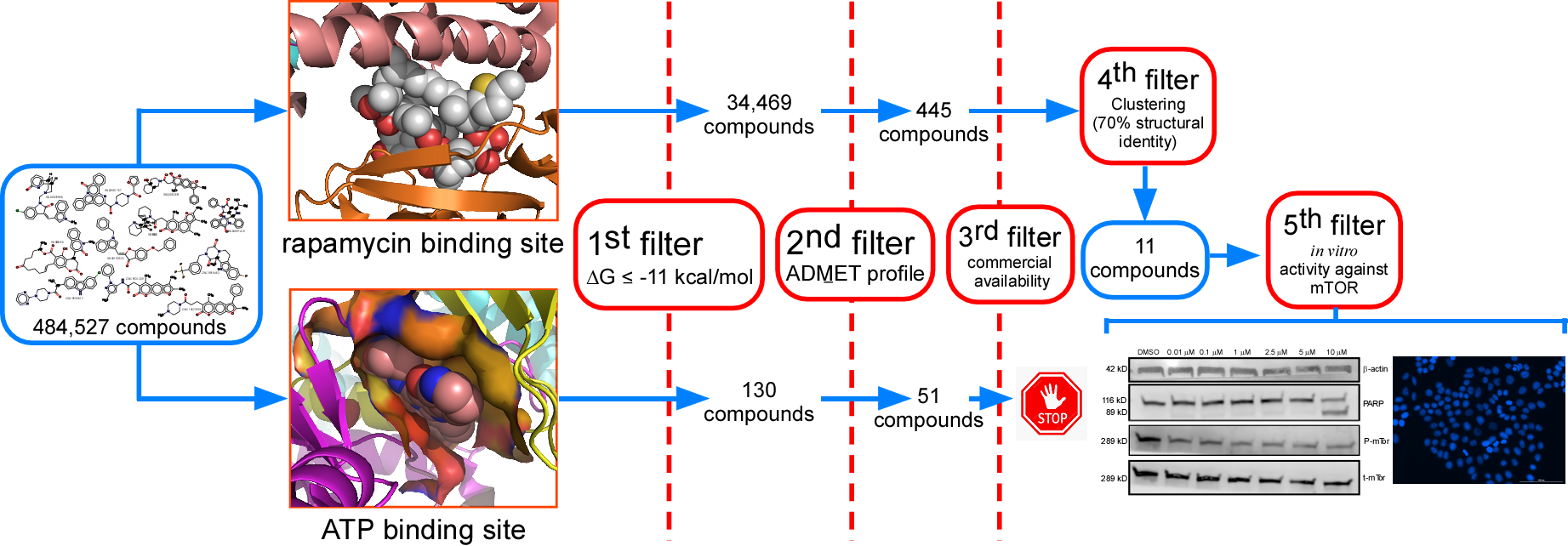
2. Results
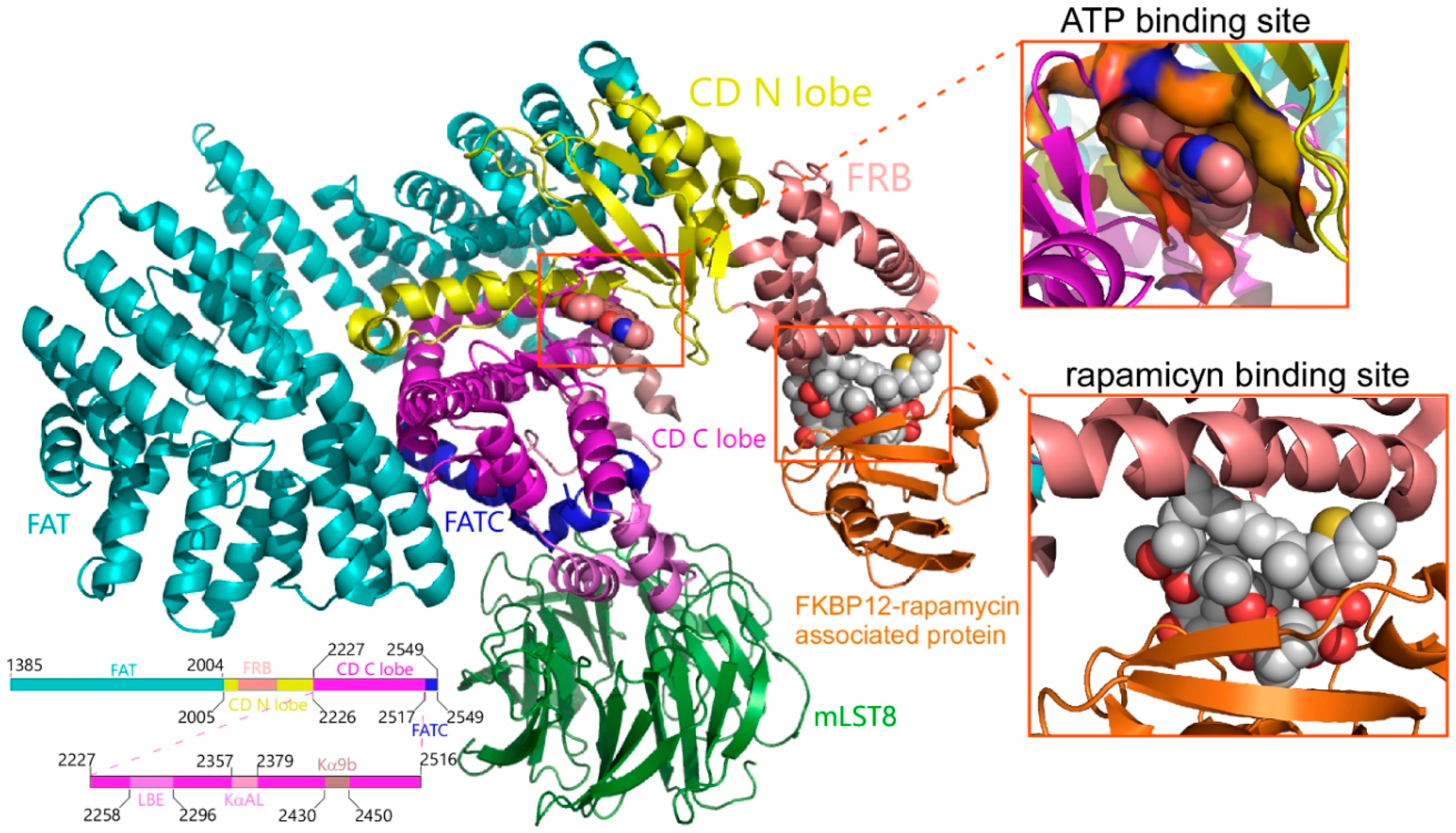
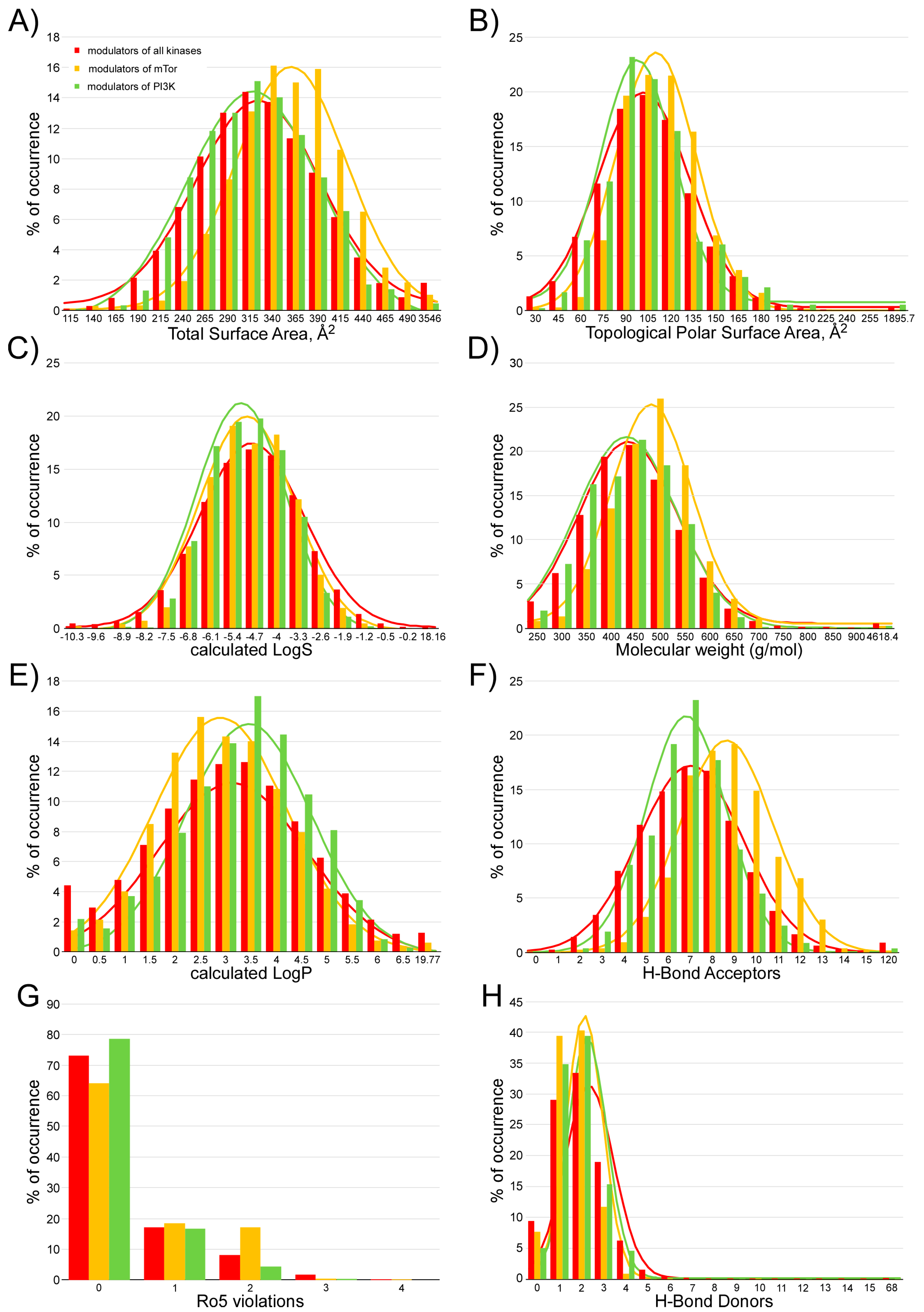
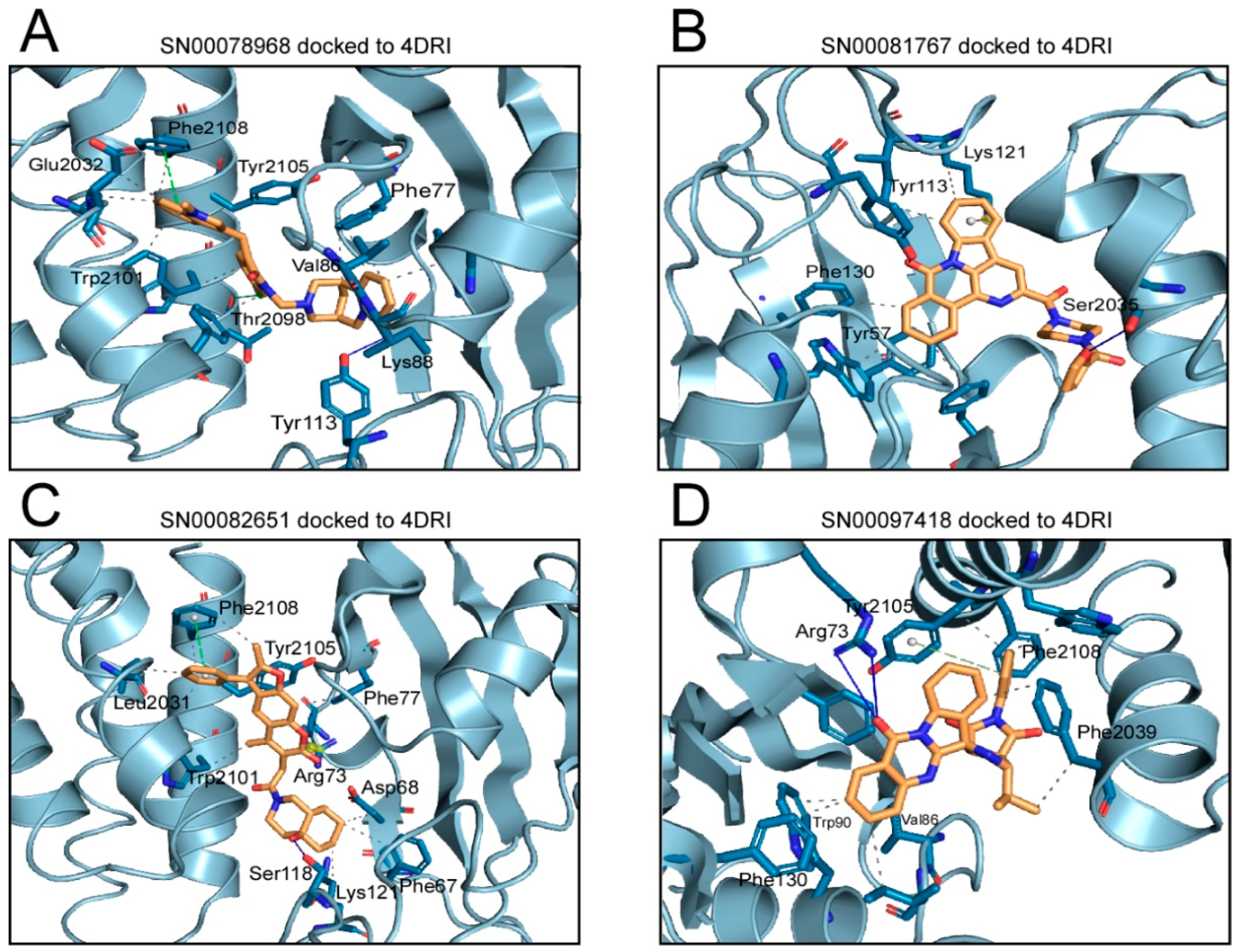
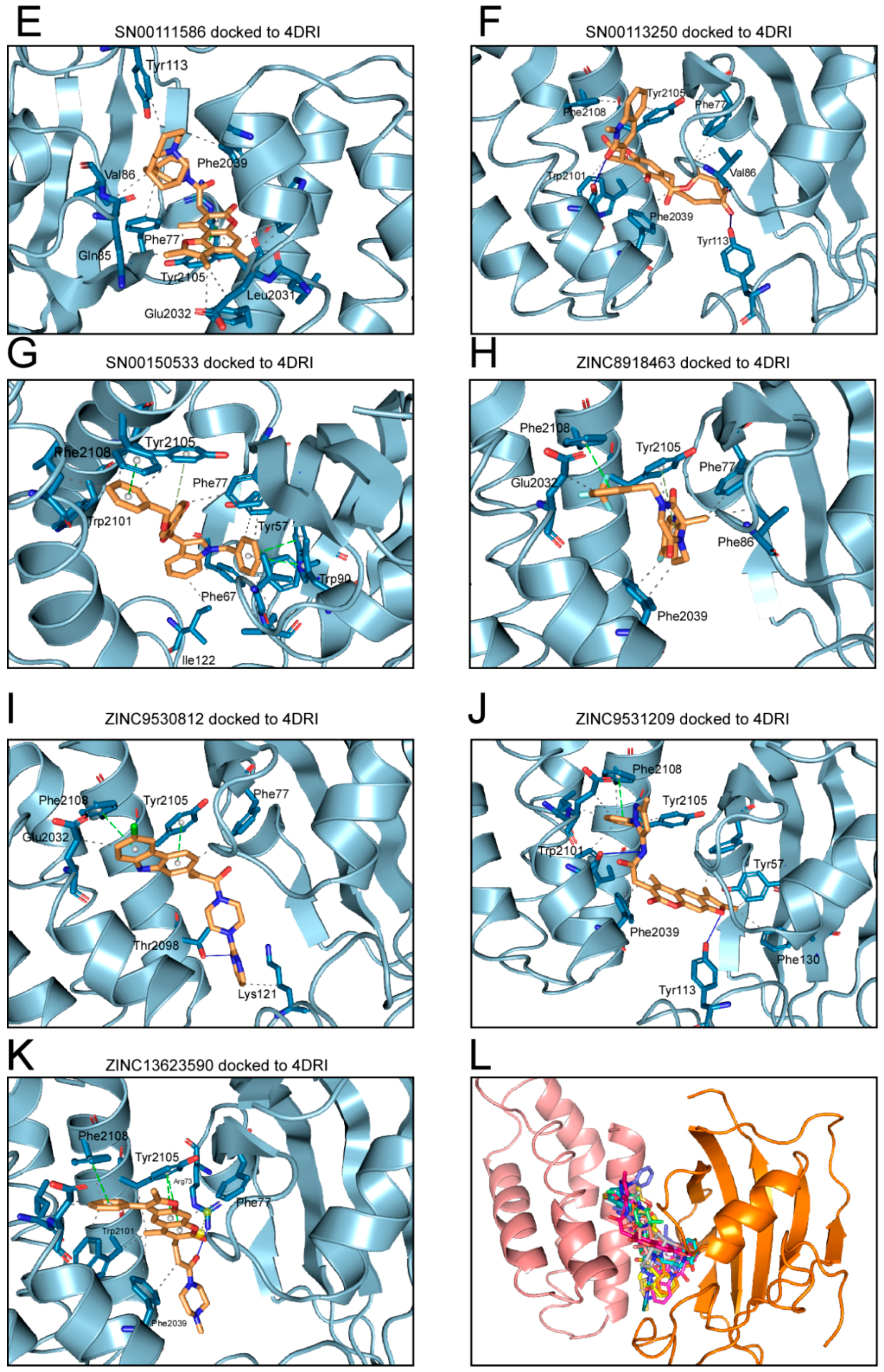
2.1. Analysis of Compounds Docked to ATP and the Rapamycin Binding Sites of the mTOR Catalytic Domain
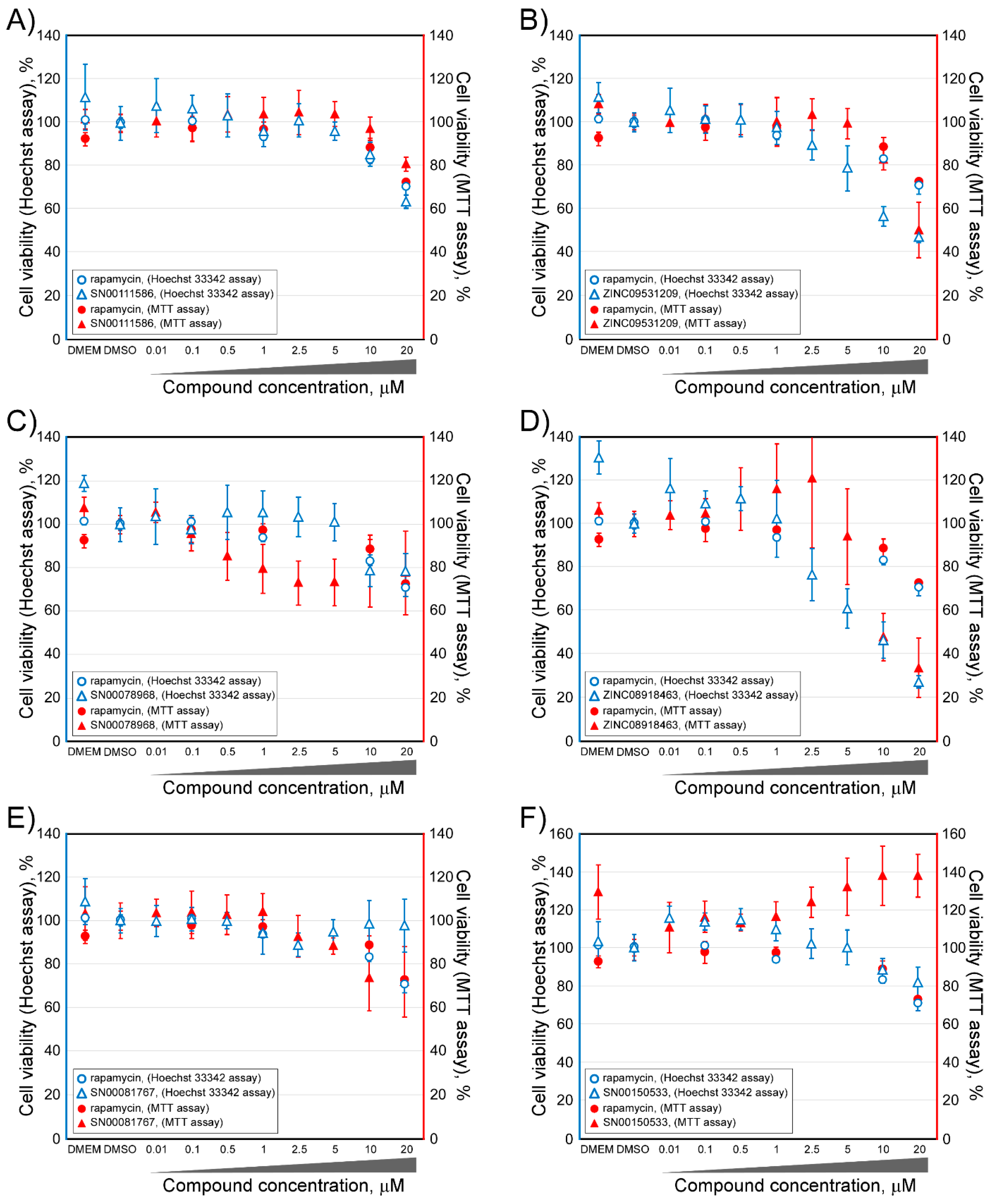
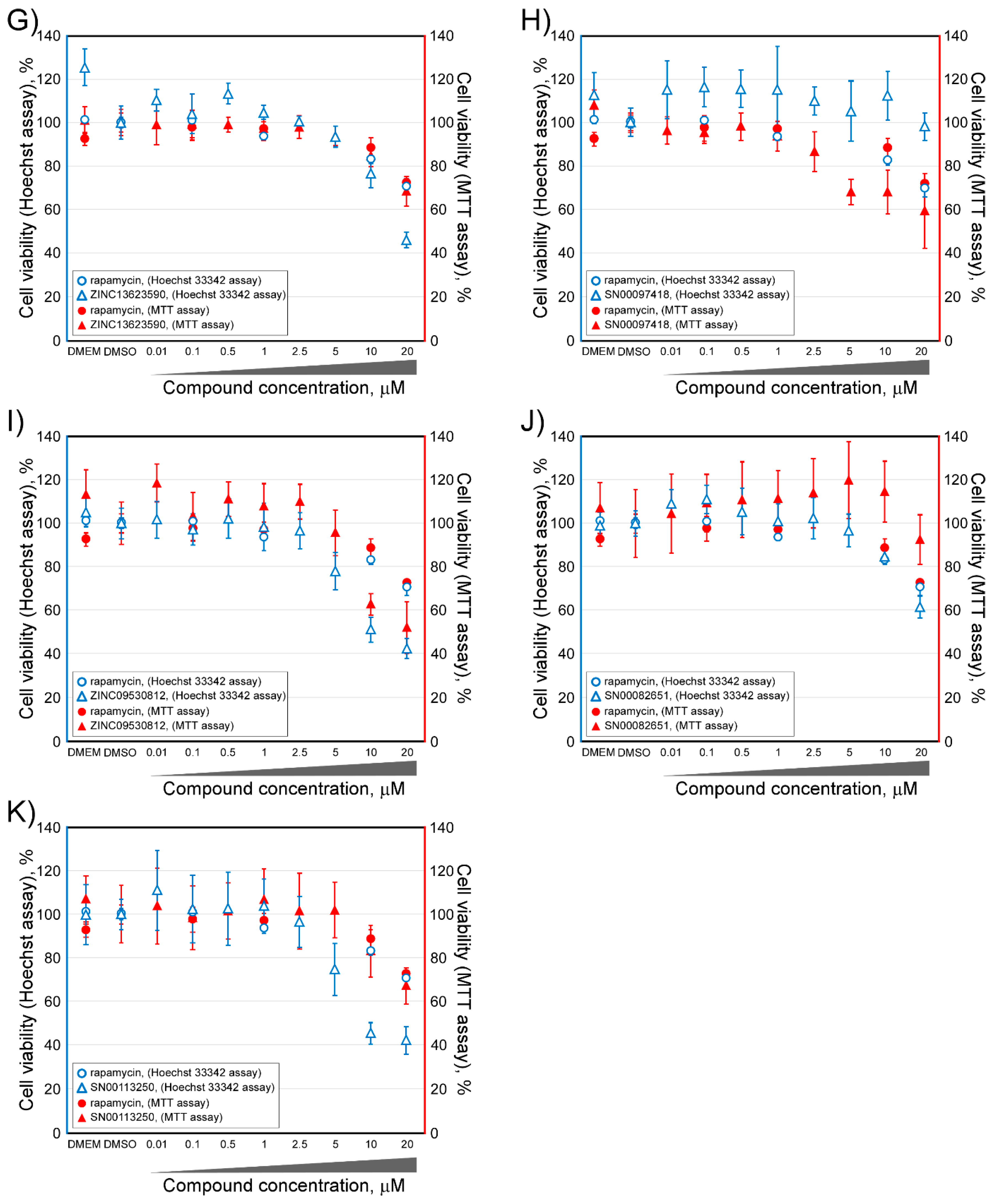
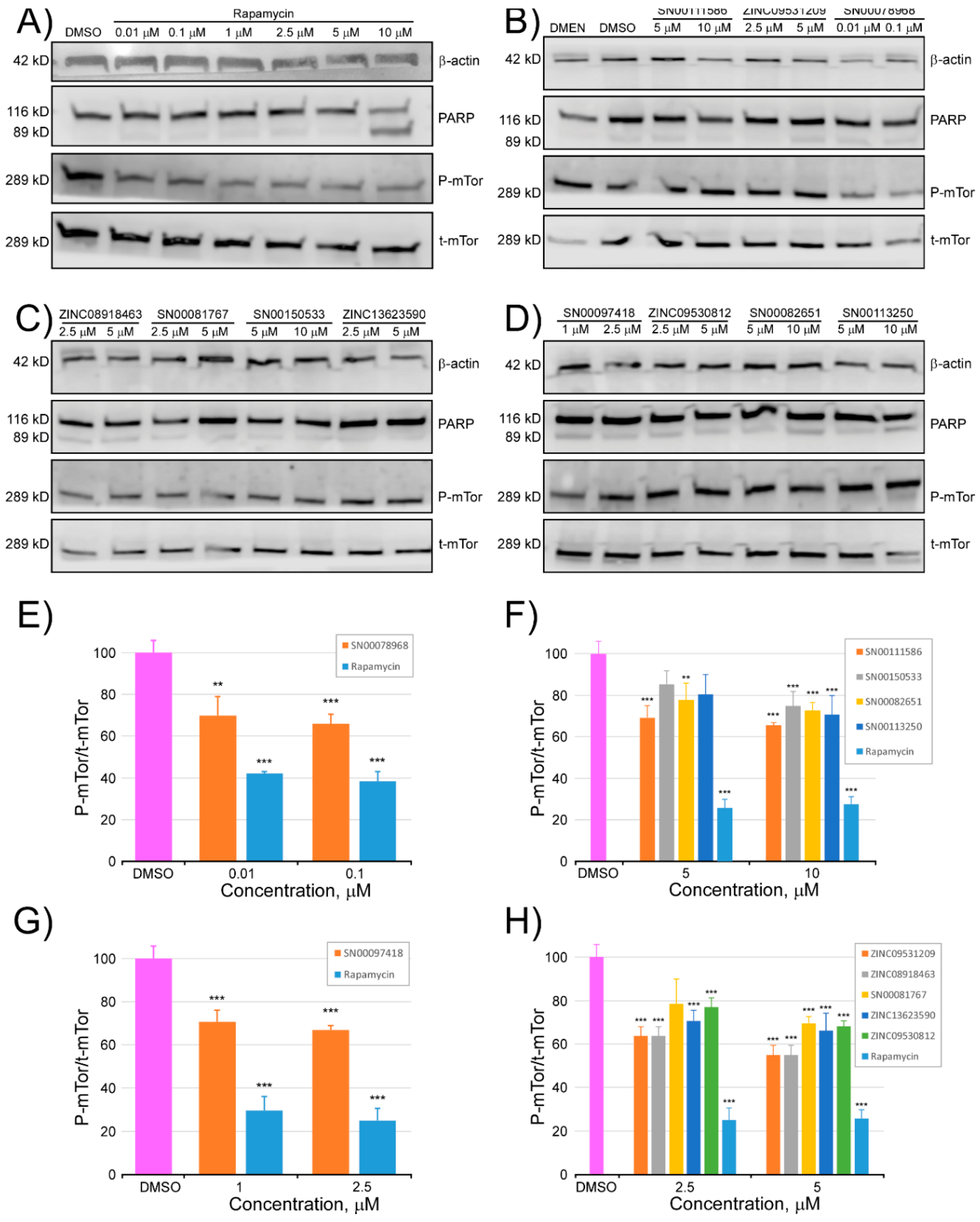
2.2. Determination of the Cell Viability of HCT116 Cells after Treatment with Selected Inhibitor Candidate Compounds
2.3. Determination of the Inhibitory Activity of the Selected Compounds against mTOR
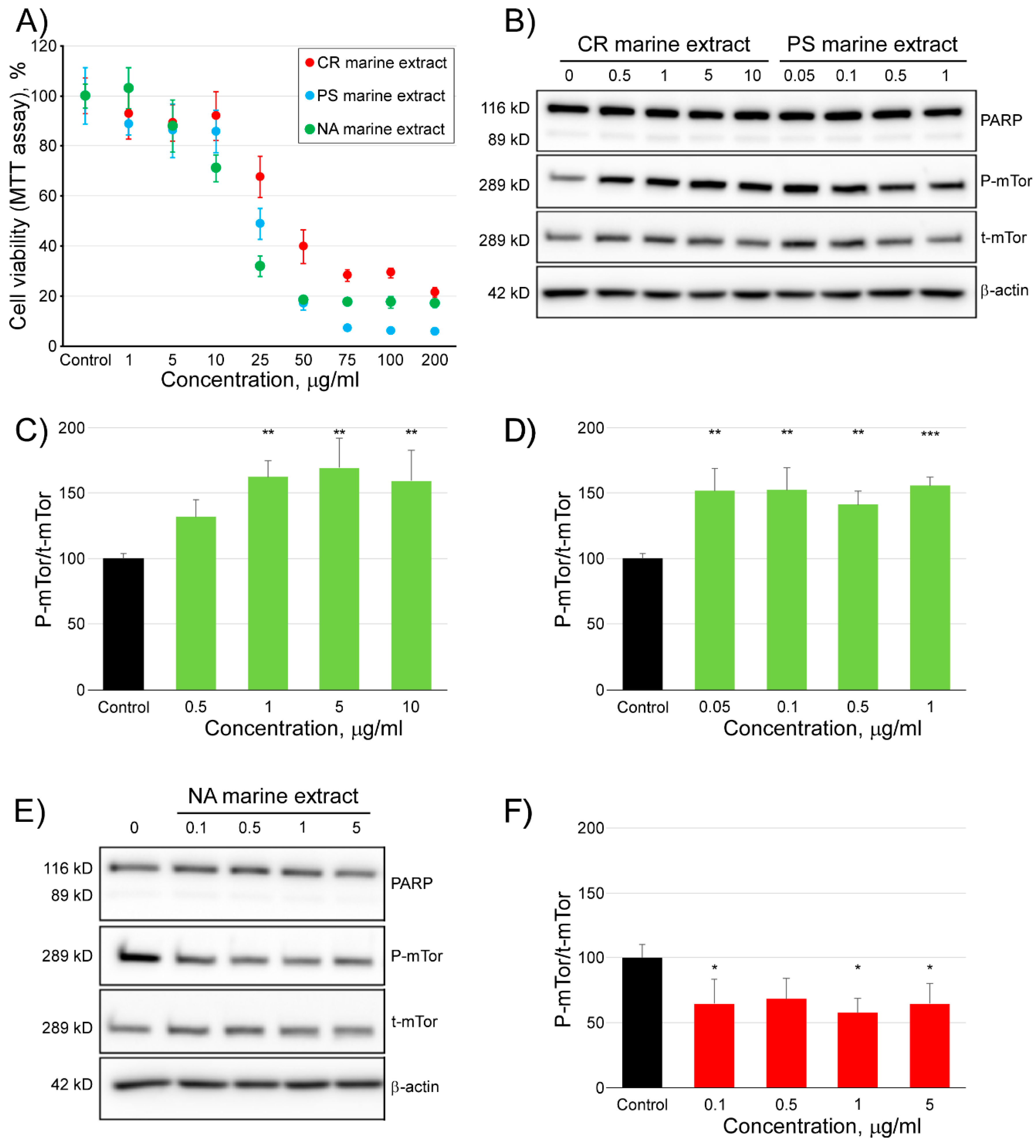
2.4. Modulating Effect of Three Marine Extracts on the Activity of mTOR
3. Discussion
4. Materials and Methods
4.1. Protein Structures of Human mTOR and PI3K Proteins and Chemical Libraries for Molecular Docking
4.2. Molecular Docking Procedure
4.3. Calculation of the Pharmacokinetic Parameters and Potential Toxicity Properties of the Inhibitor Candidates
4.4. Chemical Compounds against mTOR Kinase Activity
4.5. Preparation of Marine Extracts
4.6. Cell Culture and Treatment
4.7. Cytotoxicity Assays
4.8. Quantification of the Total mTOR and Phosphor-mTOR Levels
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chiarini, F.; Evangelisti, C.; McCubrey, J.A.; Martelli, A.M. Current treatment strategies for inhibiting mTOR in cancer. Trends Pharmacol. Sci. 2015, 36, 124–135. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Rudge, D.G.; Koos, J.D.; Vaidialingam, B.; Yang, H.J.; Pavletich, N.P. mTOR kinase structure, mechanism and regulation. Nature 2013, 497, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Hara, K.; Maruki, Y.; Long, X.; Yoshino, K.-I.; Oshiro, N.; Hidayat, S.; Tokunaga, C.; Avruch, J.; Yonezawa, K. Raptor, a Binding Partner of Target of Rapamycin (TOR), Mediates TOR Action. Cell 2002, 110, 177–189. [Google Scholar] [CrossRef] [Green Version]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [PubMed]
- Ieranò, C.; Santagata, S.; Napolitano, M.; Guardia, F.; Grimaldi, A.; Antignani, E.; Botti, G.; Consales, C.; Riccio, A.; Nanayakkara, M.; et al. CXCR4 and CXCR7 transduce through mTOR in human renal cancer cells. Cell Death Dis. 2014, 5, e1310. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.; Chang, F.; Lehmann, B.; Terrian, D.M.; Milella, M.; Tafuri, A.; et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim. Biophys. Acta 2007, 1773, 1263–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laplante, M.; Sabatini, D.M. mTOR Signaling in Growth Control and Disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoki, K.; Ouyang, H.; Zhu, T.; Lindvall, C.; Wang, Y.; Zhang, X.; Yang, Q.; Bennett, C.; Harada, Y.; Stankunas, K.; et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell 2006, 126, 955–968. [Google Scholar] [CrossRef] [PubMed]
- Anastasiou, D. Tumour microenvironment factors shaping the cancer metabolism landscape. Br. J. Cancer 2016, 116, 277. [Google Scholar] [CrossRef] [PubMed]
- Balgi, A.D.; Diering, G.H.; Donohue, E.; Lam, K.K.; Fonseca, B.D.; Zimmerman, C.; Numata, M.; Roberge, M. Regulation of mTORC1 signaling by pH. PLoS ONE 2011, 6, e21549. [Google Scholar] [CrossRef] [PubMed]
- Arsham, A.M.; Howell, J.J.; Simon, M.C. A novel hypoxia-inducible factor-independent hypoxic response regulating mammalian target of rapamycin and its targets. J. Biol. Chem. 2003, 278, 29655–29660. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.H.; Han, J.S. Phospholipase D and Its Essential Role in Cancer. Mol. Cells 2017, 40, 805–813. [Google Scholar] [CrossRef] [PubMed]
- Yoon, M.S.; Rosenberger, C.L.; Wu, C.; Truong, N.; Sweedler, J.V.; Chen, J. Rapid mitogenic regulation of the mTORC1 inhibitor, DEPTOR, by phosphatidic acid. Mol. Cell 2015, 58, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Thoreen, C.C.; Chantranupong, L.; Keys, H.R.; Wang, T.; Gray, N.S.; Sabatini, D.M. A unifying model for mTORC1-mediated regulation of mRNA translation. Nature 2012, 485, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, H.X.; Xiong, Y.; Guan, K.L. Nutrient sensing, metabolism, and cell growth control. Mol. Cell 2013, 49, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell 2009, 20, 1981–1991. [Google Scholar] [CrossRef] [PubMed]
- Dodd, K.M.; Yang, J.; Shen, M.H.; Sampson, J.R.; Tee, A.R. mTORC1 drives HIF-1α and VEGF-A signalling via multiple mechanisms involving 4E-BP1, S6K1 and STAT3. Oncogene 2014, 34, 2239. [Google Scholar] [CrossRef] [PubMed]
- Shimobayashi, M.; Hall, M.N. Making new contacts: The mTOR network in metabolism and signalling crosstalk. Nat. Rev. Mol. Cell Biol. 2014, 15, 155. [Google Scholar] [CrossRef] [PubMed]
- Cornu, M.; Albert, V.; Hall, M.N. mTOR in aging, metabolism, and cancer. Curr. Opin. Genet. Dev. 2013, 23, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Vucenik, I.; Stains, J.P. Obesity and cancer risk: Evidence, mechanisms, and recommendations. Ann. N. Y. Acad. Sci. 2012, 1271, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Dong, L.Q.; Liu, F. Recent Advances in Adipose mTOR Signaling and Function: Therapeutic Prospects. Trends Pharmacol. Sci. 2016, 37, 303–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faes, S.; Demartines, N.; Dormond, O. Resistance to mTORC1 Inhibitors in Cancer Therapy: From Kinase Mutations to Intratumoral Heterogeneity of Kinase Activity. Oxid. Med. Cell. Longev. 2017, 2017, 1726078. [Google Scholar] [CrossRef] [PubMed]
- Malley, C.O.; Pidgeon, G.P. The mTOR pathway in obesity driven gastrointestinal cancers: Potential targets and clinical trials. BBA Clin. 2016, 5, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. A historical overview of protein kinases and their targeted small molecule inhibitors. Pharmacol. Res. 2015, 100, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, D.; Colombi, M.; Moroni, C.; Hall, M.N. Rapamycin passes the torch: A new generation of mTOR inhibitors. Nat. Rev. Drug Discov. 2011, 10, 868. [Google Scholar] [CrossRef] [PubMed]
- Rodrik-Outmezguine, V.S.; Okaniwa, M.; Yao, Z.; Novotny, C.J.; McWhirter, C.; Banaji, A.; Won, H.; Wong, W.; Berger, M.; de Stanchina, E.; et al. Overcoming mTOR resistance mutations with a new-generation mTOR inhibitor. Nature 2016, 534, 272–276. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Torres, V.; Encinar, J.A.; Herranz-Lopez, M.; Perez-Sanchez, A.; Galiano, V.; Barrajon-Catalan, E.; Micol, V. An Updated Review on Marine Anticancer Compounds: The Use of Virtual Screening for the Discovery of Small-Molecule Cancer Drugs. Molecules 2017, 22. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.; Erehman, J.; Gohlke, B.O.; Wilhelm, T.; Preissner, R.; Dunkel, M. Super Natural II--a database of natural products. Nucleic Acids Res. 2015, 43, D935–D939. [Google Scholar] [CrossRef] [PubMed]
- Sterling, T.; Irwin, J.J. ZINC 15—Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef] [PubMed]
- Bello-Perez, M.; Falco, A.; Galiano, V.; Coll, J.; Perez, L.; Encinar, J.A. Discovery of nonnucleoside inhibitors of polymerase from infectious pancreatic necrosis virus (IPNV). Drug Des. Dev. Ther. 2018, 12, 2337–2359. [Google Scholar] [CrossRef] [PubMed]
- Encinar, J.A.; Fernandez-Ballester, G.; Galiano-Ibarra, V.; Micol, V. In silico approach for the discovery of new PPARgamma modulators among plant-derived polyphenols. Drug Des. Dev. Ther. 2015, 9, 5877–5895. [Google Scholar] [CrossRef] [PubMed]
- Galiano, V.; Garcia-Valtanen, P.; Micol, V.; Encinar, J.A. Looking for inhibitors of the dengue virus NS5 RNA-dependent RNA-polymerase using a molecular docking approach. Drug Des. Dev. Ther. 2016, 10, 3163–3181. [Google Scholar] [CrossRef] [PubMed]
- Gilson, M.K.; Liu, T.; Baitaluk, M.; Nicola, G.; Hwang, L.; Chong, J. BindingDB in 2015: A public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 2016, 44, D1045–D1053. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Yu, C.; Zhang, C.; Li, Y.; Wang, T.; Huang, Z.; Wang, X.; Zhou, W.; Li, Y.; Qin, Z.; et al. Highly Selective, Potent, and Oral mTOR Inhibitor for Treatment of Cancer as Autophagy Inducer. J. Med. Chem. 2018, 61, 881–904. [Google Scholar] [CrossRef] [PubMed]
- Salentin, S.; Schreiber, S.; Haupt, V.J.; Adasme, M.F.; Schroeder, M. PLIP: Fully automated protein-ligand interaction profiler. Nucleic Acids Res. 2015, 43, W443–W447. [Google Scholar] [CrossRef] [PubMed]
- Rajput, A.; Dominguez San Martin, I.; Rose, R.; Beko, A.; Levea, C.; Sharratt, E.; Mazurchuk, R.; Hoffman, R.M.; Brattain, M.G.; Wang, J. Characterization of HCT116 human colon cancer cells in an orthotopic model. J. Surg. Res. 2008, 147, 276–281. [Google Scholar] [CrossRef] [PubMed]
- Cádiz-Gurrea, M.d.l.L.; Olivares-Vicente, M.; Herranz-López, M.; Arraez-Roman, D.; Fernández-Arroyo, S.; Micol, V.; Segura-Carretero, A. Bioassay-guided purification of Lippia citriodora polyphenols with AMPK modulatory activity. J. Funct. Foods 2018, 46, 514–520. [Google Scholar] [CrossRef]
- Nave, B.T.; Ouwens, M.; Withers, D.J.; Alessi, D.R.; Shepherd, P.R. Mammalian target of rapamycin is a direct target for protein kinase B: Identification of a convergence point for opposing effects of insulin and amino-acid deficiency on protein translation. Biochem. J. 1999, 344 Pt 2, 427–431. [Google Scholar] [CrossRef]
- Peterson, R.T.; Beal, P.A.; Comb, M.J.; Schreiber, S.L. FKBP12-rapamycin-associated protein (FRAP) autophosphorylates at serine 2481 under translationally repressive conditions. J. Biol. Chem. 2000, 275, 7416–7423. [Google Scholar] [CrossRef] [PubMed]
- Edinger, A.L.; Thompson, C.B. Death by design: Apoptosis, necrosis and autophagy. Curr. Opin. Cell Biol. 2004, 16, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, D.W. Caspase structure, proteolytic substrates, and function during apoptotic cell death. Cell Death Differ. 1999, 6, 1028. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Wang, W.; Fang, G. Targeting protein-protein interaction by small molecules. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 435–456. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.A.; Khuri, F.R.; Fu, H. Targeting protein-protein interactions as an anticancer strategy. Trends Pharmacol. Sci. 2013, 34, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Pereira, F.; Aires-de-Sousa, J. Computational Methodologies in the Exploration of Marine Natural Product Leads. Mar. Drugs 2018, 16. [Google Scholar] [CrossRef] [PubMed]
- Nantasenamat, C.; Prachayasittikul, V. Maximizing computational tools for successful drug discovery. Expert Opin. Drug Discov. 2015, 10, 321–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapahi, P.; Chen, D.; Rogers, A.N.; Katewa, S.D.; Li, P.W.; Thomas, E.L.; Kockel, L. With TOR, less is more: A key role for the conserved nutrient-sensing TOR pathway in aging. Cell Metab. 2010, 11, 453–465. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.C.; Rabinovitch, P.S.; Kaeberlein, M. mTOR is a key modulator of ageing and age-related disease. Nature 2013, 493, 338. [Google Scholar] [CrossRef] [PubMed]
- Sadowski, K.; Kotulska, K.; Jozwiak, S. Management of side effects of mTOR inhibitors in tuberous sclerosis patients. Pharmacol. Rep. 2016, 68, 536–542. [Google Scholar] [CrossRef] [PubMed]
- Pallet, N.; Legendre, C. Adverse events associated with mTOR inhibitors. Expert Opin. Drug Saf. 2013, 12, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Kim, S.G.; Blenis, J. Rapamycin: One drug, many effects. Cell Metab. 2014, 19, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Neves, J.; Sousa-Victor, P.; Jasper, H. Rejuvenating Strategies for Stem Cell-Based Therapies in Aging. Cell Stem Cell 2017, 20, 161–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weichhart, T. mTOR as Regulator of Lifespan, Aging, and Cellular Senescence: A Mini-Review. Gerontology 2018, 64, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Ge, D.; Han, L.; Huang, S.; Peng, N.; Wang, P.; Jiang, Z.; Zhao, J.; Su, L.; Zhang, S.; Zhang, Y.; et al. Identification of a novel MTOR activator and discovery of a competing endogenous RNA regulating autophagy in vascular endothelial cells. Autophagy 2014, 10, 957–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, N.; Meng, N.; Wang, S.; Zhao, F.; Zhao, J.; Su, L.; Zhang, S.; Zhang, Y.; Zhao, B.; Miao, J. An activator of mTOR inhibits oxLDL-induced autophagy and apoptosis in vascular endothelial cells and restricts atherosclerosis in apolipoprotein E-/- mice. Sci. Rep. 2014, 4, 5519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bottegoni, G.; Rocchia, W.; Rueda, M.; Abagyan, R.; Cavalli, A. Systematic exploitation of multiple receptor conformations for virtual ligand screening. PLoS ONE 2011, 6, e18845. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schymkowitz, J.; Borg, J.; Stricher, F.; Nys, R.; Rousseau, F.; Serrano, L. The FoldX web server: An online force field. Nucleic Acids Res. 2005, 33, W382–W388. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Sander, T.; Freyss, J.; von Korff, M.; Rufener, C. DataWarrior: An open-source program for chemistry aware data visualization and analysis. J. Chem. Inf. Model. 2015, 55, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Li, W.; Zhou, Y.; Shen, J.; Wu, Z.; Liu, G.; Lee, P.W.; Tang, Y. admetSAR: A comprehensive source and free tool for assessment of chemical ADMET properties. J. Chem. Inf. Model. 2012, 52, 3099–3105. [Google Scholar] [CrossRef] [PubMed]












© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruiz-Torres, V.; Losada-Echeberría, M.; Herranz-López, M.; Barrajón-Catalán, E.; Galiano, V.; Micol, V.; Encinar, J.A. New Mammalian Target of Rapamycin (mTOR) Modulators Derived from Natural Product Databases and Marine Extracts by Using Molecular Docking Techniques. Mar. Drugs 2018, 16, 385. https://0-doi-org.brum.beds.ac.uk/10.3390/md16100385
Ruiz-Torres V, Losada-Echeberría M, Herranz-López M, Barrajón-Catalán E, Galiano V, Micol V, Encinar JA. New Mammalian Target of Rapamycin (mTOR) Modulators Derived from Natural Product Databases and Marine Extracts by Using Molecular Docking Techniques. Marine Drugs. 2018; 16(10):385. https://0-doi-org.brum.beds.ac.uk/10.3390/md16100385
Chicago/Turabian StyleRuiz-Torres, Verónica, Maria Losada-Echeberría, Maria Herranz-López, Enrique Barrajón-Catalán, Vicente Galiano, Vicente Micol, and José Antonio Encinar. 2018. "New Mammalian Target of Rapamycin (mTOR) Modulators Derived from Natural Product Databases and Marine Extracts by Using Molecular Docking Techniques" Marine Drugs 16, no. 10: 385. https://0-doi-org.brum.beds.ac.uk/10.3390/md16100385