13-Acetoxysarcocrassolide Exhibits Cytotoxic Activity against Oral Cancer Cells through the Interruption of the Keap1/Nrf2/p62/SQSTM1 Pathway: The Need to Move Beyond Classical Concepts

 ,
,
Abstract
:1. Introduction
2. Results
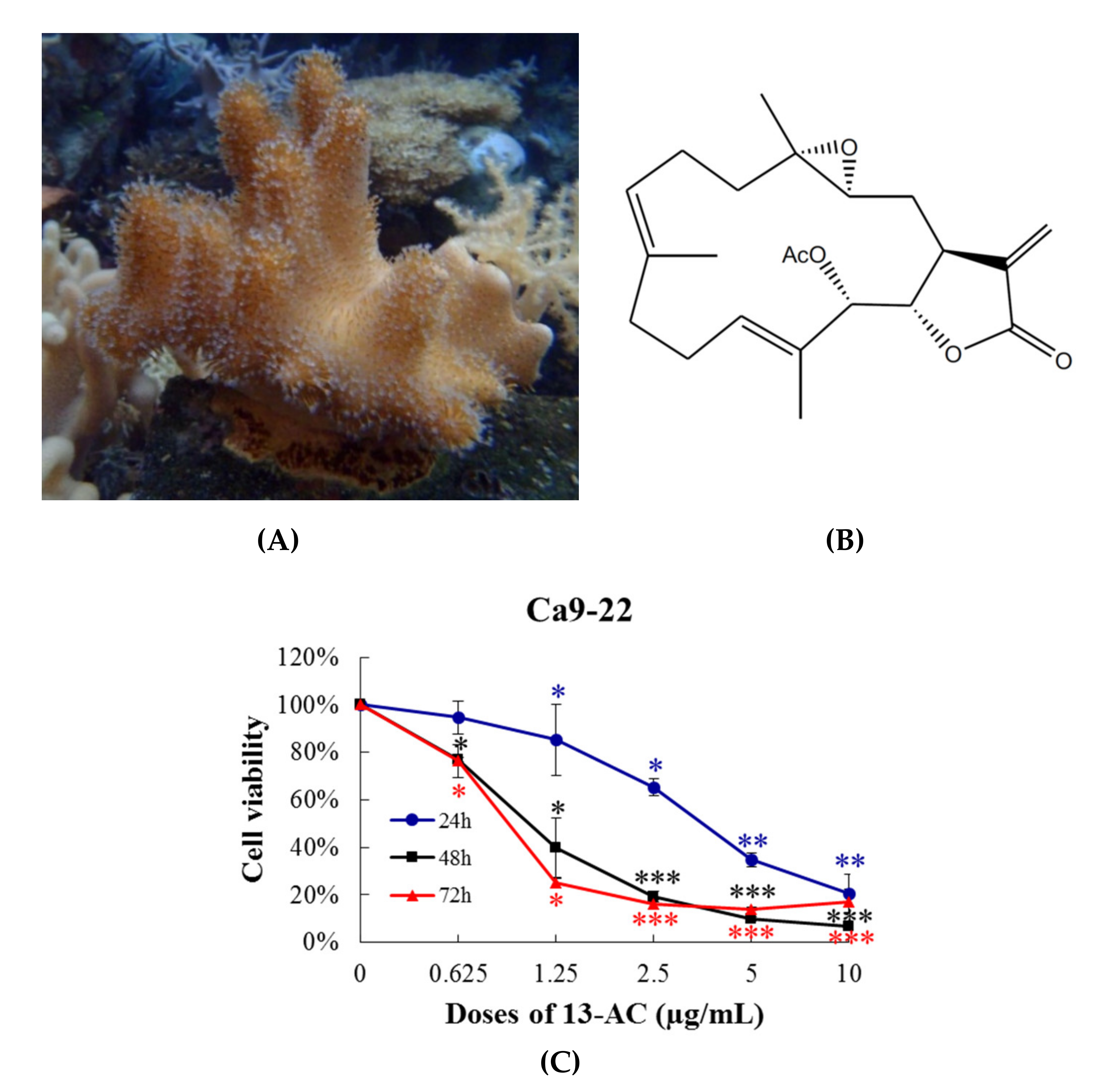
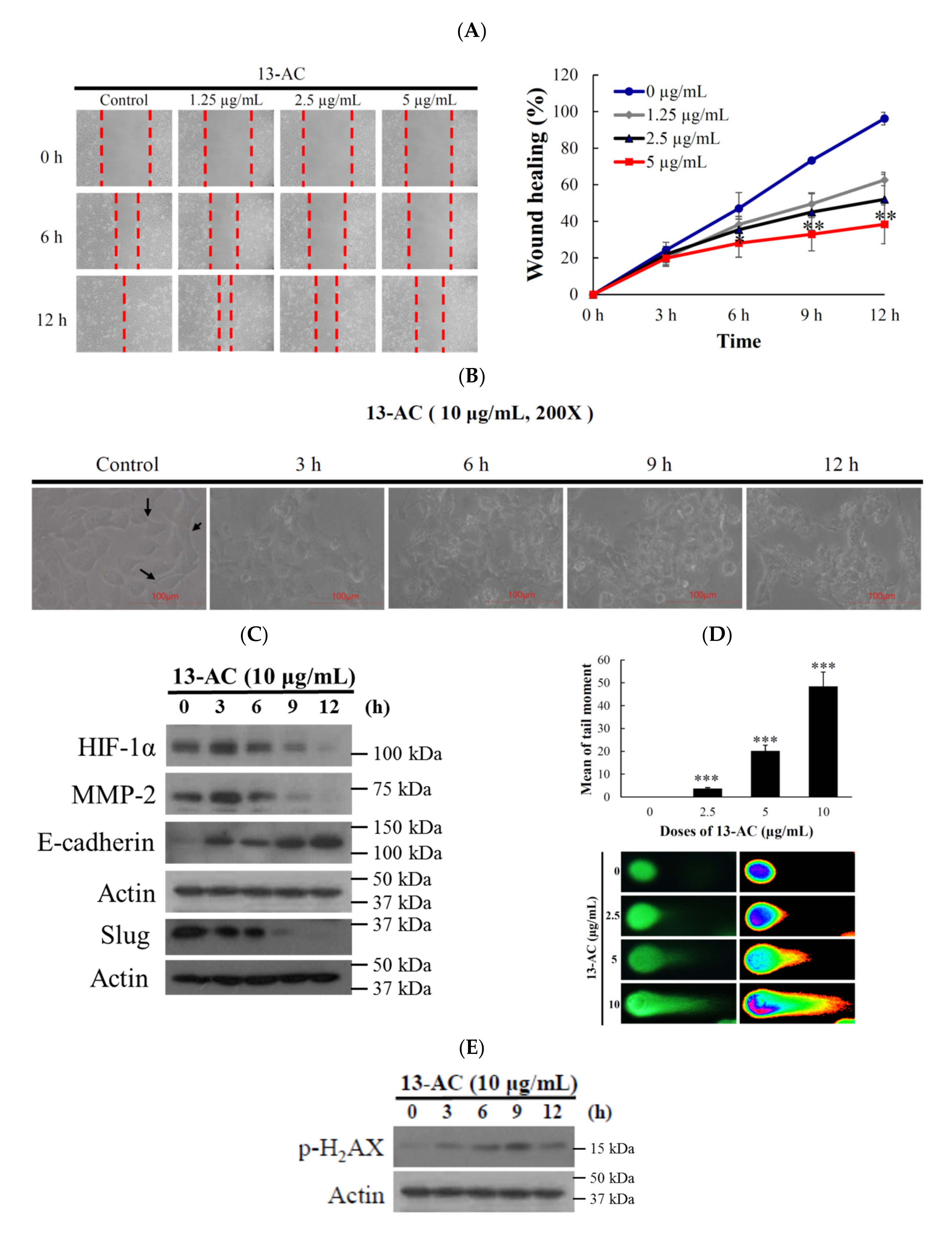
2.1. Effect of 13-AC on Cellular Proliferation, Migration and DNA Damage in Oral Cancer Cells
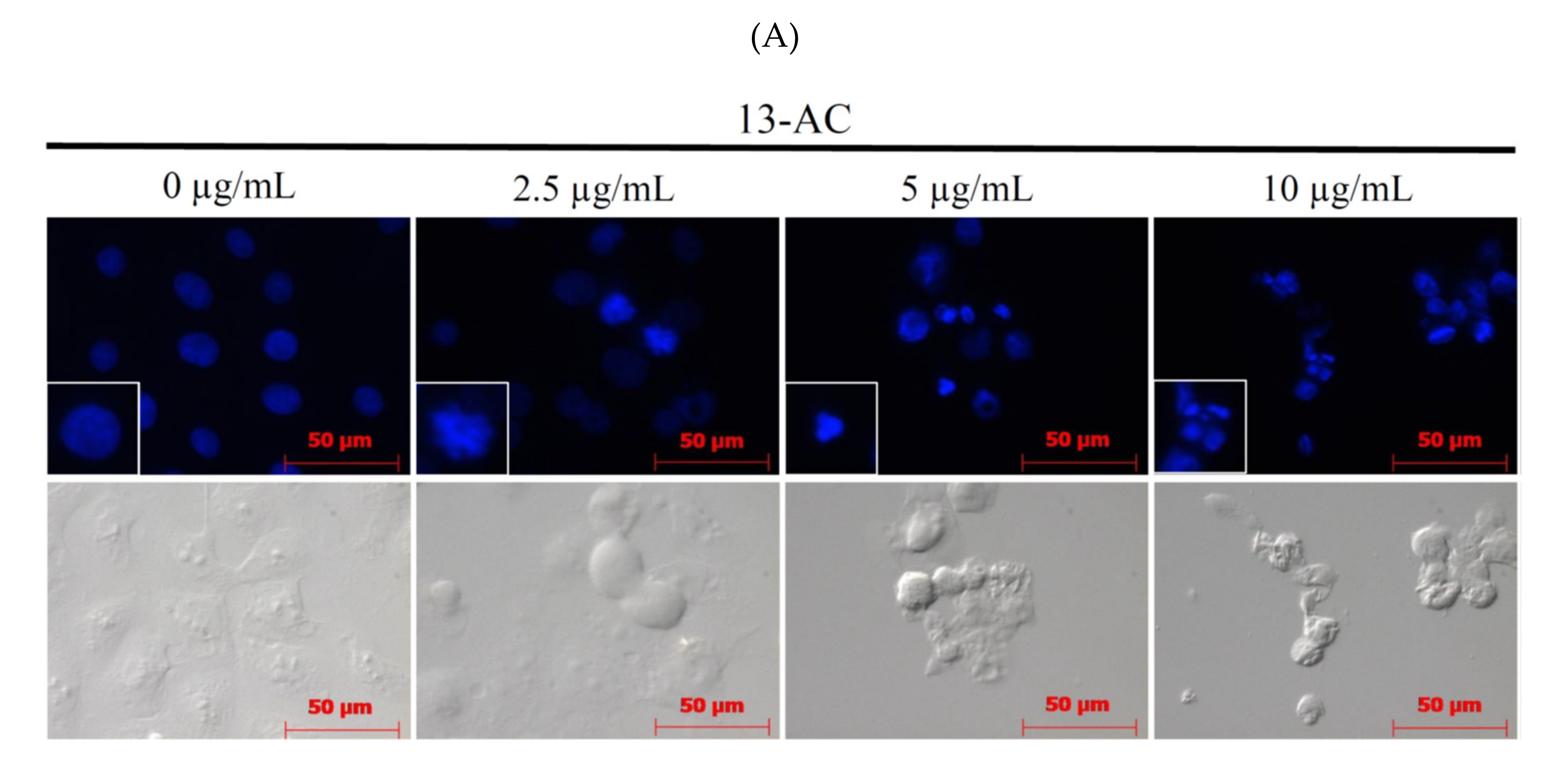
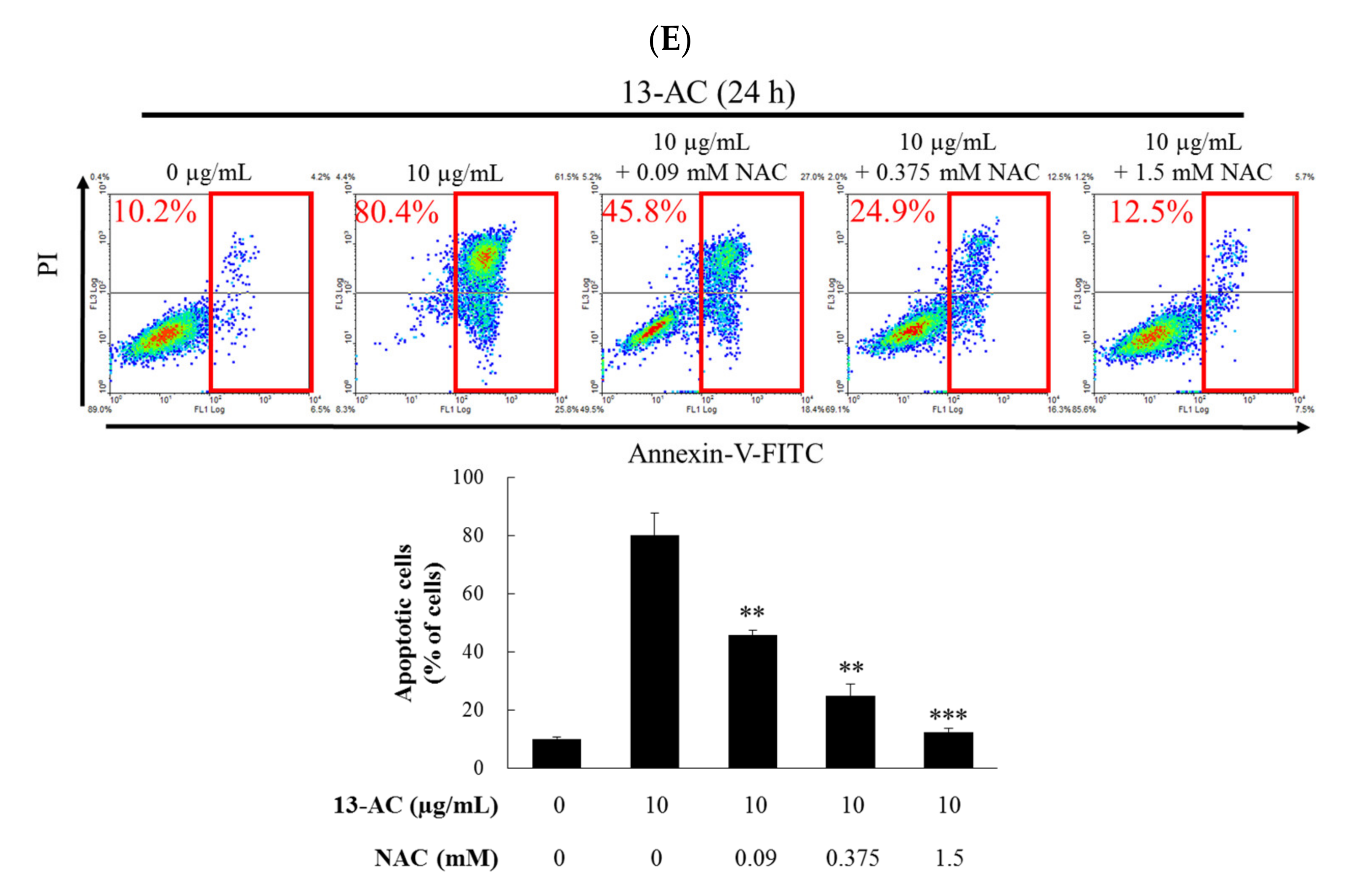
2.2. 13-AC Induces Apoptosis in Ca9-22 Cancer Cells
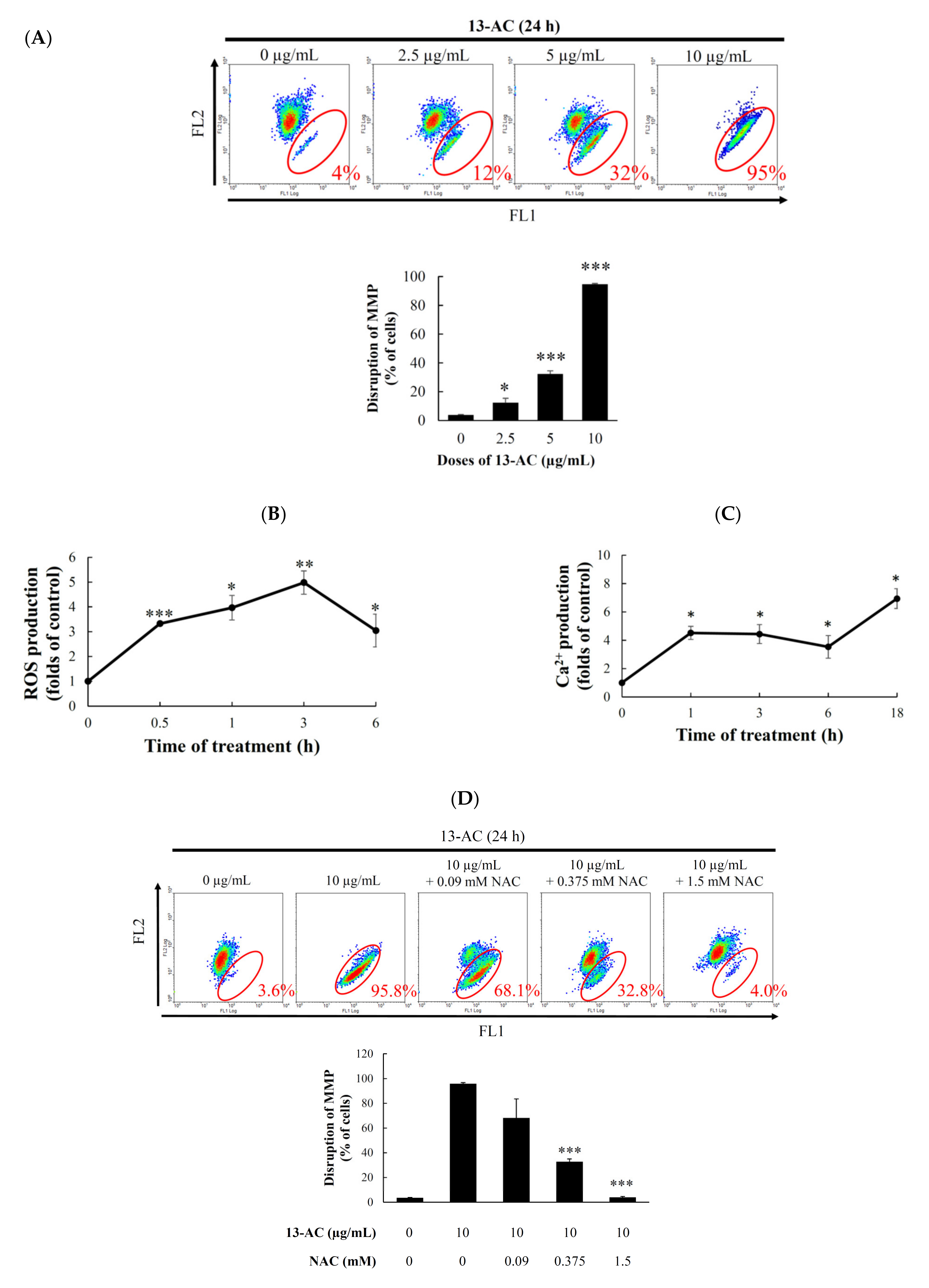
2.3. Effect of 13-AC on Mitochondrial Membrane Potential, ROS Generation, and Calcium Accumulation
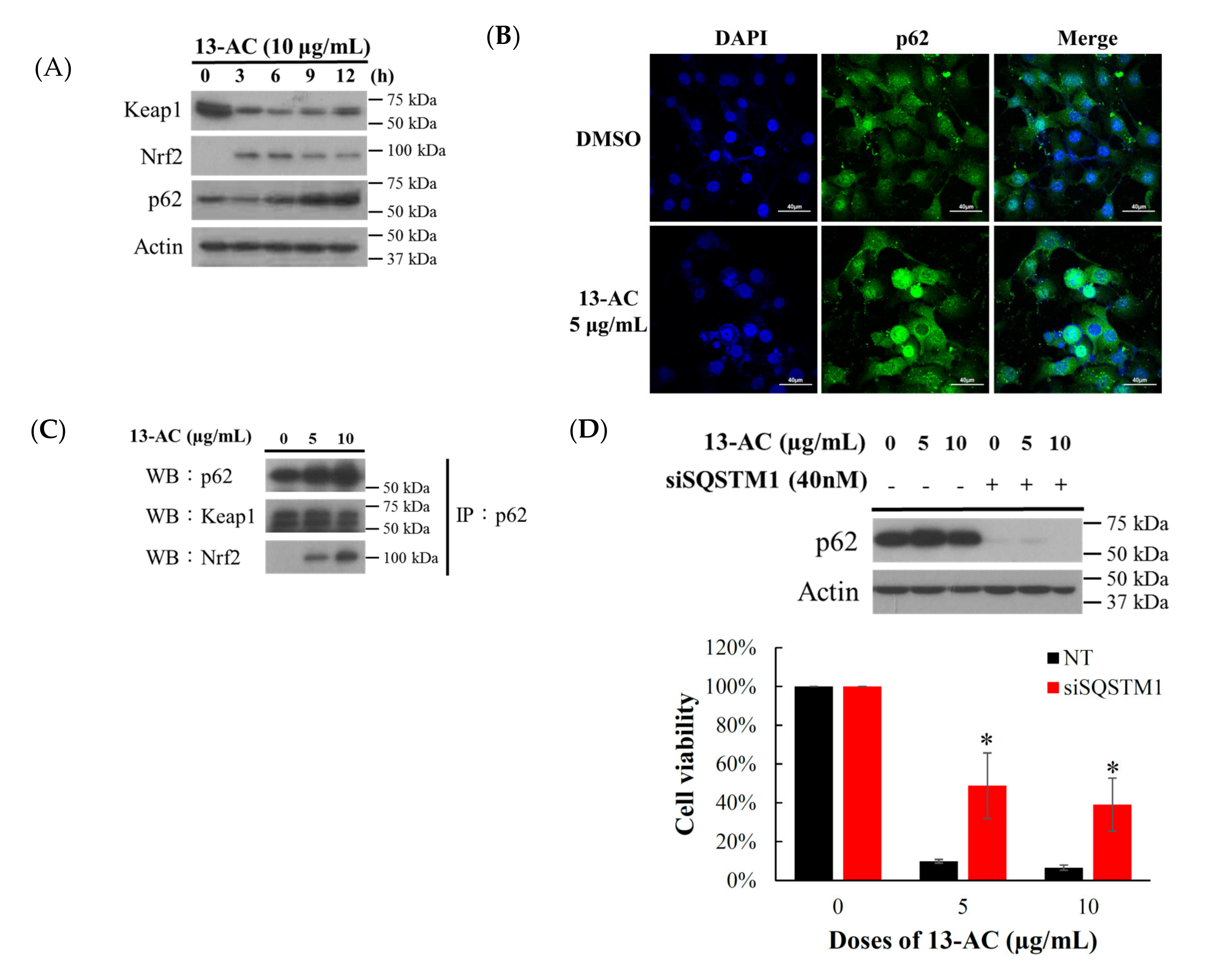
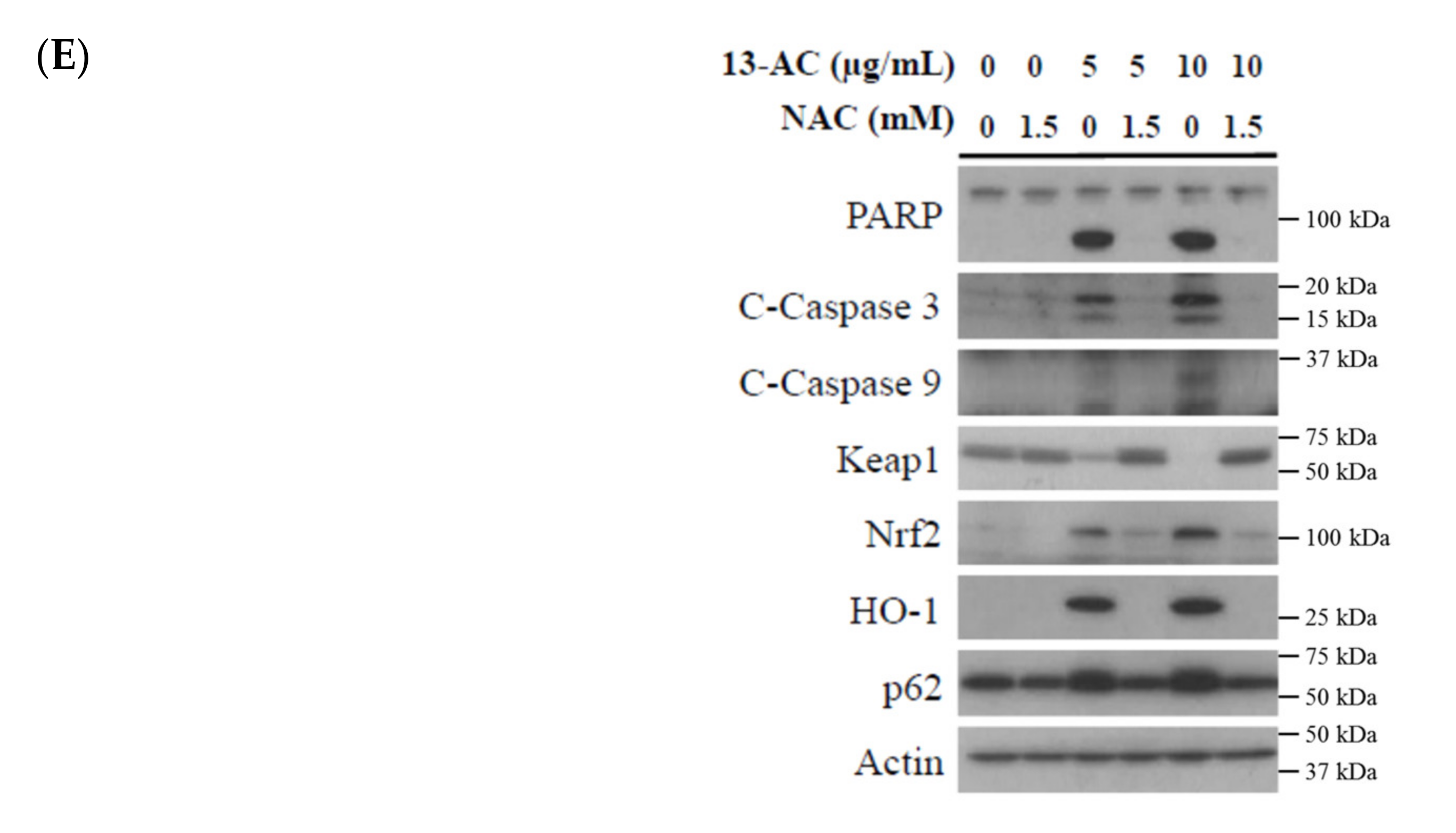
2.4. Oxidative Stress-Induced by 13-AC Involves the Dysregulation of Nrf2/Keap1/p62/SQSTM1 Pathway
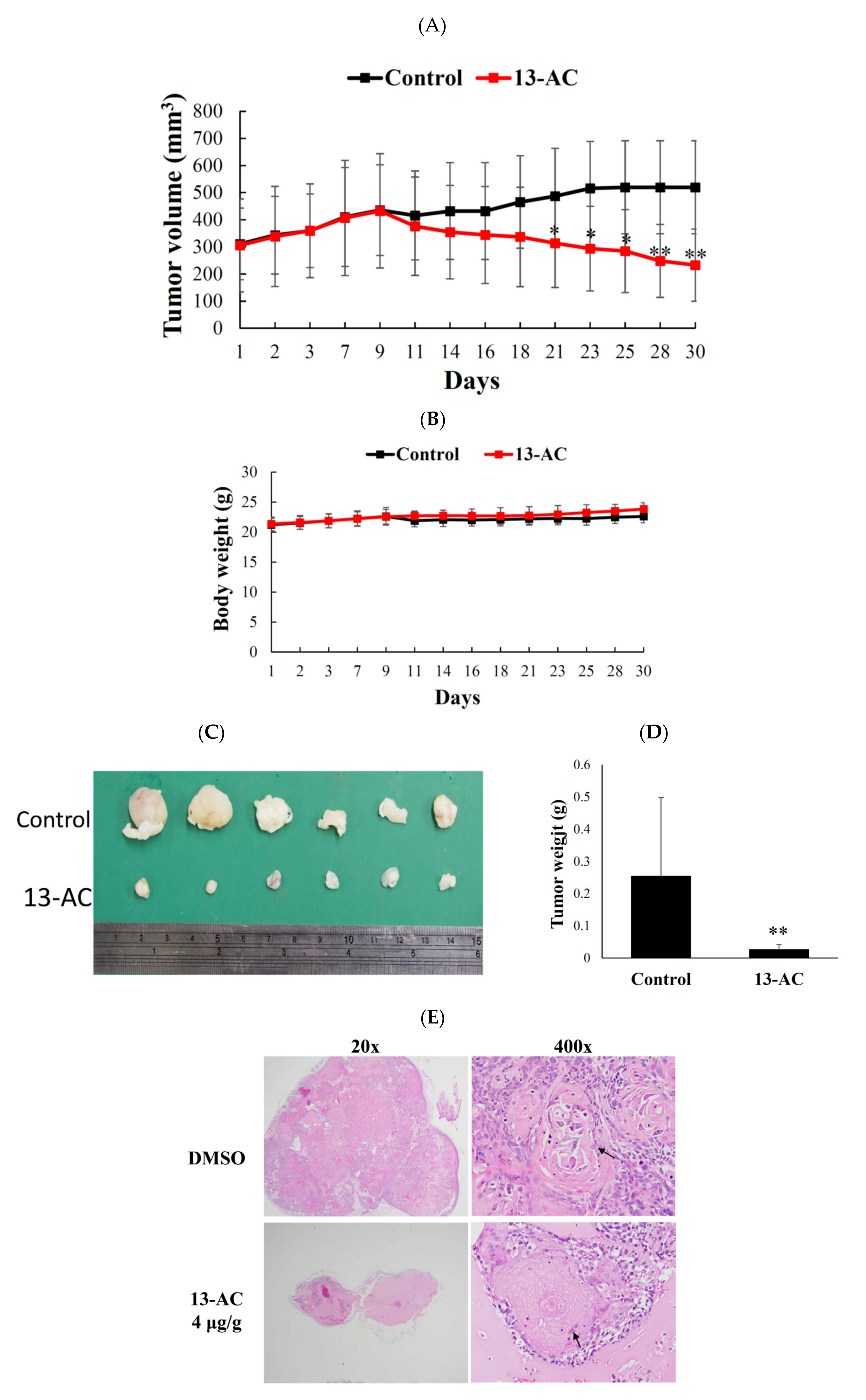
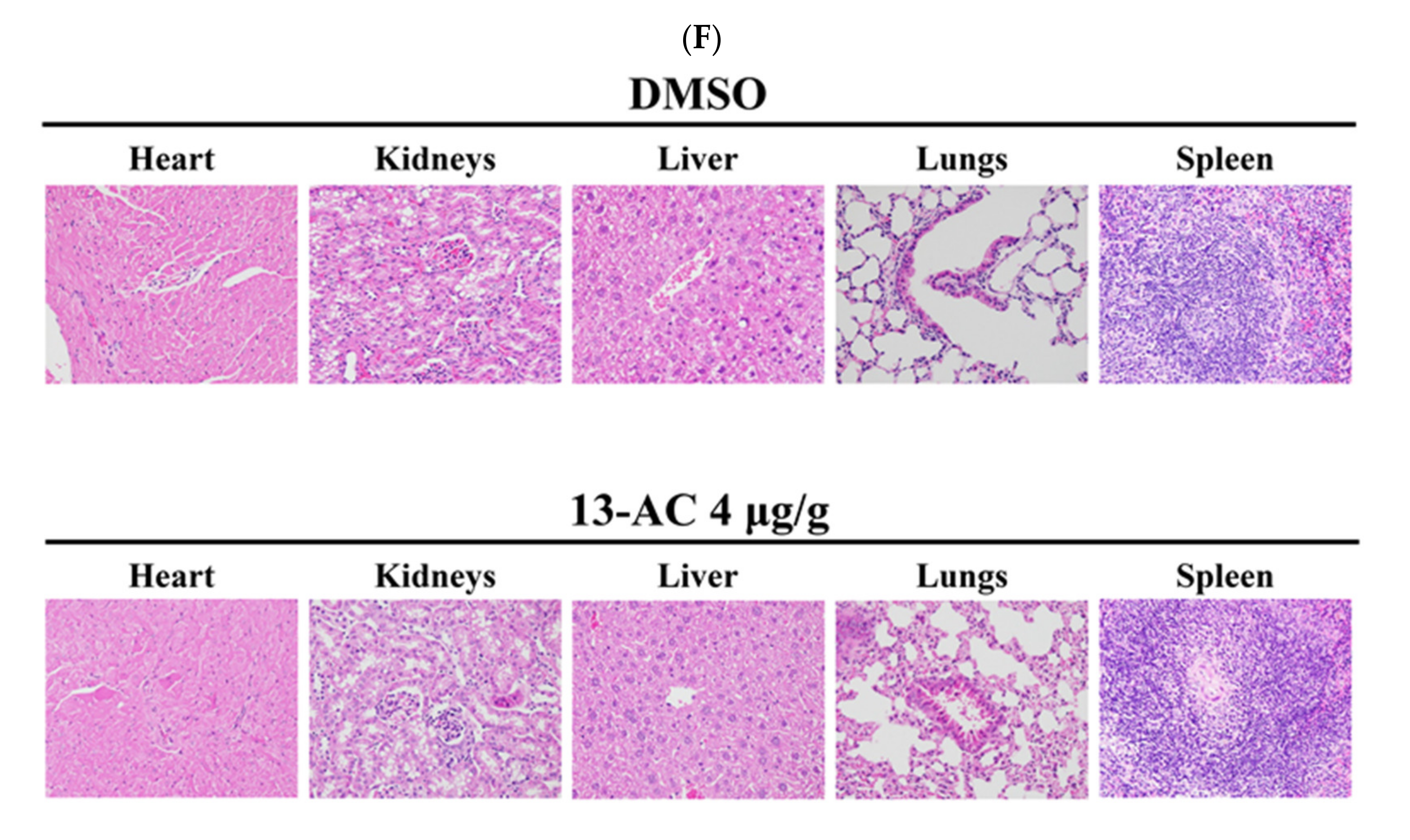
2.5. Antitumor Effect of 13-AC on Human Oral Ca9-22 Cancer Cells Xenograft Animal Model
3. Discussion
4. Materials and Methods
4.1. Cell Lines, Biological Materials and Chemicals
4.2. The Stock Solution of 13-AC
4.3. MTT Cell Proliferation Assay
4.4. Annexin V/PI Apoptotic Assay
4.5. Determination of ROS Generation, MMP Disruption, and Calcium Accumulation
4.6. Protein Immunoprecipitation and Western Blot Analysis
4.7. Immunofluorescence Analysis
4.8. The Detection of DNA Double-Strand Breaks (DSBs) Using Neutral Comet Assay For
4.9. RNA Interference Transfection
4.10. Animal Material and Xenograft Animal Model with Human Oral Cancer Cells
4.11. Statistics
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Oosting, S.F.; Haddad, R.I. Best Practice in Systemic Therapy for Head and Neck Squamous Cell Carcinoma. Front. Oncol. 2019, 9, 815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cramer, J.D.; Burtness, B.; Le, Q.T.; Ferris, R.L. The changing therapeutic landscape of head and neck cancer. Nat. Rev. Clin. Oncol. 2019, 16, 669–983. [Google Scholar] [CrossRef]
- Shah, M.; Jomaa, M.K.; Ferrarotto, R.; Yeung, S.J.; Hanna, E.Y.; Reyes-Gibby, C.C. Serious immune-related adverse events in patients with head and neck cancer after checkpoint blockade: Systematic review. Head Neck 2019, 41, 4036–4050. [Google Scholar] [CrossRef] [PubMed]
- Bossi, P.; Alfieri, S.; Strojan, P.; Takes, R.P.; Lopez, F.; Makitie, A.; Saba, N.F.; Rodrigo, J.P.; Bradford, C.; Suarez, C.; et al. Prognostic and predictive factors in recurrent and/or metastatic head and neck squamous cell carcinoma: A review of the literature. Crit. Rev. Oncol. Hematol. 2019, 137, 84–91. [Google Scholar] [CrossRef]
- Rojo de la Vega, M.; Chapman, E.; Zhang, D.D. Nrf2 and the Hallmarks of Cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.; Glorieux, C.; Liao, J.; Chen, P.; Lu, W.; Liang, Z.; Wen, S.; Hu, Y.; Huang, P. Impact of Nrf2 on tumour growth and drug sensitivity in oncogenic K-ras-transformed cells in vitro and in vivo. Free Radic. Res. 2018, 52, 661–671. [Google Scholar] [CrossRef] [PubMed]
- Roh, J.L.; Kim, E.H.; Jang, H.; Shin, D. Nrf2 inhibition reverses the resistance of cisplatin-resistant head and neck cancer cells to artesunate-induced ferroptosis. Redox Biol. 2017, 11, 254–262. [Google Scholar] [CrossRef]
- Milkovic, L.; Zarkovic, N.; Saso, L. Controversy about pharmacological modulation of Nrf2 for cancer therapy. Redox Biol. 2017, 12, 727–732. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, I.G.; Miguel, M.G.; Mnif, W. A Brief Review on New Naturally Occurring Cembranoid Diterpene Derivatives from the Soft Corals of the Genera Sarcophyton, Sinularia, and Lobophytum Since 2016. Molecules 2019, 24, 781. [Google Scholar] [CrossRef] [Green Version]
- Duh, C.Y.; Wang, S.K.; Chung, S.G.; Chou, G.C.; Dai, C.F. Cytotoxic cembrenolides and steroids from the formosan soft coral Sarcophyton crassocaule. J. Nat. Prod. 2000, 63, 1634–1637. [Google Scholar] [CrossRef]
- Rashid, M.A.; Gustafson, K.R.; Boyd, M.R. HIV-inhibitory cembrane derivatives from a Philippines collection of the soft coral Lobophytum species. J. Nat. Prod. 2000, 63, 531–533. [Google Scholar] [CrossRef] [PubMed]
- Roy, P.K.; Ashimine, R.; Miyazato, H.; Taira, J.; Ueda, K. New Casbane and Cembrane Diterpenoids from an Okinawan Soft Coral, Lobophytum sp. Molecules 2016, 21, 679. [Google Scholar] [CrossRef] [Green Version]
- Lai, K.H.; You, W.J.; Lin, C.C.; El-Shazly, M.; Liao, Z.J.; Su, J.H. Anti-Inflammatory Dembranoids from the Soft Coral Lobophytum crassum. Mar. Drugs 2017, 15, 327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, P.K.; Roy, S.; Ueda, K. New cytotoxic cembranolides from an Okinawan soft coral, Lobophytum sp. Fitoterapia 2019, 136, 104162. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Lu, M.C.; Su, J.H.; Chu, C.L.; Shiuan, D.; Weng, C.F.; Sung, P.J.; Huang, K.J. Immunomodulatory effect of marine cembrane-type diterpenoids on dendritic cells. Mar. Drugs 2013, 11, 1336–1350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, B.R.; Lu, M.C.; El-Shazly, M.; Wu, S.L.; Lai, K.H.; Su, J.H. Aquaculture Soft Coral Lobophytum crassum as a Producer of Anti-Proliferative Cembranoids. Mar. Drugs 2018, 16, 15. [Google Scholar] [CrossRef] [Green Version]
- Su, C.C.; Su, J.H.; Lin, J.J.; Chen, C.C.; Hwang, W.I.; Huang, H.H.; Wu, Y.J. An investigation into the cytotoxic effects of 13-acetoxysarcocrassolide from the soft coral Sarcophyton crassocaule on bladder cancer cells. Mar. Drugs 2011, 9, 2622–2642. [Google Scholar] [CrossRef]
- Su, C.C.; Chen, J.Y.; Din, Z.H.; Su, J.H.; Yang, Z.Y.; Chen, Y.J.; Wang, R.Y.; Wu, Y.J. 13-acetoxysarcocrassolide induces apoptosis on human gastric carcinoma cells through mitochondria-related apoptotic pathways: p38/JNK activation and PI3K/AKT suppression. Mar. Drugs 2014, 12, 5295–5315. [Google Scholar] [CrossRef] [Green Version]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef] [Green Version]
- Al-Lihaibi, S.S.; Alarif, W.M.; Abdel-Lateff, A.; Ayyad, S.E.; Abdel-Naim, A.B.; El-Senduny, F.F.; Badria, F.A. Three new cembranoid-type diterpenes from Red Sea soft coral Sarcophyton glaucum: Isolation and antiproliferative activity against HepG2 cells. Eur. J. Med. Chem. 2014, 81, 314–322. [Google Scholar] [CrossRef]
- Liu, H.; Zhao, S.; Zhang, Y.; Wu, J.; Peng, H.; Fan, J.; Liao, J. Reactive oxygen species-mediated endoplasmic reticulum stress and mitochondrial dysfunction contribute to polydatin-induced apoptosis in human nasopharyngeal carcinoma CNE cells. J. Cell. Biochem. 2011, 112, 3695–3703. [Google Scholar] [CrossRef] [PubMed]
- Awasthi, B.P.; Mitra, K. In vitro leishmanicidal effects of the anti-fungal drug natamycin are mediated through disruption of calcium homeostasis and mitochondrial dysfunction. Apoptosis 2018, 23, 420–435. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Li, R.; Zhao, X.; Ma, C.; Lv, X.; Liu, L.; Liu, P. Morusin induces paraptosis-like cell death through mitochondrial calcium overload and dysfunction in epithelial ovarian cancer. Chem. Biol. Interact. 2018, 283, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.G.; Liu, Y.C.; Lee, Y.L.; El-Shazly, M.; Lai, K.H.; Shih, S.P.; Ke, S.C.; Hong, M.C.; Du, Y.C.; Yang, J.C.; et al. Heteronemin, a Marine Sesterterpenoid-Type Metabolite, Induces Apoptosis in Prostate LNcap Cells via Oxidative and ER Stress Combined with the Inhibition of Topoisomerase II and Hsp90. Mar. Drugs 2018, 16, 204. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.C.; Lu, M.C.; El-Shazly, M.; Lai, K.H.; Wu, T.Y.; Hsu, Y.M.; Lee, Y.L.; Liu, Y.C. Breaking down Leukemia Walls: Heteronemin, a Sesterterpene Derivative, Induces Apoptosis in Leukemia Molt4 Cells through Oxidative Stress, Mitochondrial Dysfunction and Induction of Talin Expression. Mar. Drugs 2018, 16, 212. [Google Scholar] [CrossRef] [Green Version]
- Lu, M.C.; Li, T.Y.; Hsieh, Y.C.; Hsieh, P.C.; Chu, Y.L. Chemical evaluation and cytotoxic mechanism investigation of Clinacanthus nutans extract in lymphoma SUP-T1 cells. Environ. Toxicol. 2018, 33, 1229–1236. [Google Scholar] [CrossRef] [PubMed]
- Gill, J.G.; Piskounova, E.; Morrison, S.J. Cancer, Oxidative Stress, and Metastasis. Cold Spring Harb. Symp. Quant. Biol. 2016, 81, 163–175. [Google Scholar] [CrossRef] [Green Version]
- Tong, L.; Chuang, C.C.; Wu, S.; Zuo, L. Reactive oxygen species in redox cancer therapy. Cancer Lett. 2015, 367, 18–25. [Google Scholar] [CrossRef]
- Ma, Q. Role of Nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [Green Version]
- Rubio-Navarro, A.; Vazquez-Carballo, C.; Guerrero-Hue, M.; Garcia-Caballero, C.; Herencia, C.; Gutierrez, E.; Yuste, C.; Sevillano, A.; Praga, M.; Egea, J.; et al. Nrf2 Plays a Protective Role Against Intravascular Hemolysis-Mediated Acute Kidney Injury. Front. Pharmacol. 2019, 10, 740. [Google Scholar] [CrossRef] [Green Version]
- Furfaro, A.L.; Traverso, N.; Domenicotti, C.; Piras, S.; Moretta, L.; Marinari, U.M.; Pronzato, M.A.; Nitti, M. The Nrf2/HO-1 Axis in Cancer Cell Growth and Chemoresistance. Oxidative Med. Cell Longev. 2016, 2016, 1958174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef]
- Copple, I.M.; Lister, A.; Obeng, A.D.; Kitteringham, N.R.; Jenkins, R.E.; Layfield, R.; Foster, B.J.; Goldring, C.E.; Park, B.K. Physical and functional interaction of sequestosome 1 with Keap1 regulates the Keap1-Nrf2 cell defense pathway. J. Biol. Chem. 2010, 285, 16782–16788. [Google Scholar] [CrossRef] [Green Version]
- Ichimura, Y.; Waguri, S.; Sou, Y.S.; Kageyama, S.; Hasegawa, J.; Ishimura, R.; Saito, T.; Yang, Y.; Kouno, T.; Fukutomi, T.; et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol. Cell 2013, 51, 618–631. [Google Scholar] [CrossRef] [Green Version]
- Kageyama, S.; Saito, T.; Obata, M.; Koide, R.H.; Ichimura, Y.; Komatsu, M. Negative Regulation of the Keap1-Nrf2 Pathway by a p62/Sqstm1 Splicing Variant. Mol. Cell Biol. 2018, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, A.; Wang, X.J.; Zhao, F.; Villeneuve, N.F.; Wu, T.; Jiang, T.; Sun, Z.; White, E.; Zhang, D.D. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: Direct interaction between Keap1 and p62. Mol. Cell Biol. 2010, 30, 3275–3285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, A.; Teknos, T.N.; Pan, Q. Epithelial to mesenchymal transition in head and neck squamous cell carcinoma. Oral Oncol. 2013, 49, 287–292. [Google Scholar] [CrossRef] [Green Version]
- Chung, C.H.; Parker, J.S.; Ely, K.; Carter, J.; Yi, Y.; Murphy, B.A.; Ang, K.K.; El-Naggar, A.K.; Zanation, A.M.; Cmelak, A.J.; et al. Gene expression profiles identify epithelial-to-mesenchymal transition and activation of nuclear factor-κB signaling as characteristics of a high-risk head and neck squamous cell carcinoma. Cancer Res. 2006, 66, 8210–8218. [Google Scholar] [CrossRef] [Green Version]
- Thiery, J.P. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Nijkamp, M.M.; Span, P.N.; Hoogsteen, I.J.; van der Kogel, A.J.; Kaanders, J.H.; Bussink, J. Expression of E-cadherin and vimentin correlates with metastasis formation in head and neck squamous cell carcinoma patients. Radiother Oncol. 2011, 99, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef] [PubMed]
- Montero, A.J.; Diaz-Montero, C.M.; Deutsch, Y.E.; Hurley, J.; Koniaris, L.G.; Rumboldt, T.; Yasir, S.; Jorda, M.; Garret-Mayer, E.; Avisar, E.; et al. Phase 2 study of neoadjuvant treatment with NOV-002 in combination with doxorubicin and cyclophosphamide followed by docetaxel in patients with HER-2 negative clinical stage II-IIIc breast cancer. Breast Cancer Res. Treat. 2012, 132, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Lo, M.; Dockery, P.; Mahon, S.; Karp, C.M.; Buckley, A.R.; Lam, S.; Gout, P.W.; Wang, Y.Z. The xc cystine/glutamate antiporter as a potential therapeutic target for small-cell lung cancer: Use of sulfasalazine. Cancer Chemother Pharmacol. 2009, 64, 463. [Google Scholar] [CrossRef]
- Zhu, J.; Song, X.; Lin, H.P.; Young, D.C.; Yan, S.; Marquez, V.E.; Chen, C.S. Using cyclooxygenase-2 inhibitors as molecular platforms to develop a new class of apoptosis-inducing agents. J. Natl. Cancer Inst. 2002, 94, 1745–1757. [Google Scholar] [CrossRef]
- Gills, J.J.; Lopiccolo, J.; Tsurutani, J.; Shoemaker, R.H.; Best, C.J.; Abu-Asab, M.S.; Borojerdi, J.; Warfel, N.A.; Gardner, E.R.; Danish, M.; et al. Nelfinavir, A lead HIV protease inhibitor, is a broad-spectrum, anticancer agent that induces endoplasmic reticulum stress, autophagy, and apoptosis in vitro and in vivo. Clin. Cancer Res. 2007, 13, 5183–5194. [Google Scholar] [CrossRef] [Green Version]
- Papa, L.; Gomes, E.; Rockwell, P. Reactive oxygen species induced by proteasome inhibition in neuronal cells mediate mitochondrial dysfunction and a caspase-independent cell death. Apoptosis 2007, 12, 1389–1405. [Google Scholar] [CrossRef]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-fluorouracil: Mechanisms of action and clinical strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef]
- Wang, J.; Yuan, Z. Gambogic acid sensitizes ovarian cancer cells to doxorubicin through ROS-mediated apoptosis. Cell Biochem. Biophys. 2013, 67, 199–206. [Google Scholar] [CrossRef]
- Lai, K.H.; Liu, Y.C.; Su, J.H.; El-Shazly, M.; Wu, C.F.; Du, Y.C.; Hsu, Y.M.; Yang, J.C.; Weng, M.K.; Chou, C.H.; et al. Antileukemic Scalarane Sesterterpenoids and Meroditerpenoid from Carteriospongia (Phyllospongia) sp., Induce Apoptosis via Dual Inhibitory Effects on Topoisomerase II and Hsp90. Sci. Rep. 2016, 6, 36170. [Google Scholar] [CrossRef]
- Tao, S.; Park, S.L.; Rojo de la Vega, M.; Zhang, D.D.; Wondrak, G.T. Systemic administration of the apocarotenoid bixin protects skin against solar UV-induced damage through activation of Nrf2. Free Radic. Biol. Med. 2015, 89, 690–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, S.; Rojo de la Vega, M.; Chapman, E.; Ooi, A.; Zhang, D.D. The effects of Nrf2 modulation on the initiation and progression of chemically and genetically induced lung cancer. Mol. Carcinog. 2018, 57, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Satoh, H.; Moriguchi, T.; Takai, J.; Ebina, M.; Yamamoto, M. Nrf2 prevents initiation but accelerates progression through the Kras signaling pathway during lung carcinogenesis. Cancer Res. 2013, 73, 4158–4168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Lamark, T.; Sjottem, E.; Larsen, K.B.; Awuh, J.A.; Overvatn, A.; McMahon, M.; Hayes, J.D.; Johansen, T. p62/SQSTM1 is a target gene for transcription factor Nrf2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Kong, A.N. Molecular mechanisms of Nrf2-mediated antioxidant response. Mol. Carcinog. 2009, 48, 91–104. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, M.; Waguri, S.; Koike, M.; Sou, Y.S.; Ueno, T.; Hara, T.; Mizushima, N.; Iwata, J.; Ezaki, J.; Murata, S.; et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 2007, 131, 1149–1163. [Google Scholar] [CrossRef] [Green Version]
- Raghunath, A.; Sundarraj, K.; Arfuso, F.; Sethi, G.; Perumal, E. Dysregulation of Nrf2 in Hepatocellular Carcinoma: Role in Cancer Progression and Chemoresistance. Cancers 2018, 10, 481. [Google Scholar] [CrossRef] [Green Version]
- Menegon, S.; Columbano, A.; Giordano, S. The Dual Roles of Nrf2 in Cancer. Trends Mol. Med. 2016, 22, 578–593. [Google Scholar] [CrossRef]
- Shen, H.; Yang, Y.; Xia, S.; Rao, B.; Zhang, J.; Wang, J. Blockage of Nrf2 suppresses the migration and invasion of esophageal squamous cell carcinoma cells in hypoxic microenvironment. Dis. Esophagus 2014, 27, 685–692. [Google Scholar] [CrossRef]
- Moscat, J.; Diaz-Meco, M.T. P62 at the crossroads of autophagy, apoptosis, and cancer. Cell 2009, 137, 1001–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsuragi, Y.; Ichimura, Y.; Komatsu, M. P62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J. 2015, 282, 4672–4678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, C.; Mitter, S.K.; Qi, X.; Beli, E.; Rao, H.V.; Ding, J.; Ip, C.S.; Gu, H.; Akin, D.; Dunn, W.A., Jr.; et al. Oxidative stress-mediated NFκB phosphorylation upregulates p62/SQSTM1 and promotes retinal pigmented epithelial cell survival through increased autophagy. PLoS ONE 2017, 12, e0171940. [Google Scholar] [CrossRef] [PubMed]
- Katsuragi, Y.; Ichimura, Y.; Komatsu, M. Regulation of the Keap1–Nrf2 pathway by p62/SQSTM1. Curr. Opin. Toxicol. 2016, 1, 54–61. [Google Scholar] [CrossRef] [Green Version]
- Su, J.H.; Chen, Y.C.; El-Shazly, M.; Du, Y.C.; Su, C.W.; Tsao, C.W.; Liu, L.L.; Chou, Y.; Chang, W.B.; Su, Y.D.; et al. Towards the small and the beautiful: A small dibromotyrosine derivative from Pseudoceratina sp. sponge exhibits potent apoptotic effect through targeting IKK/NFkappaB signaling pathway. Mar. Drugs 2013, 11, 3168–3185. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.C.; Du, Y.C.; Chuu, J.J.; Hwang, S.L.; Hsieh, P.C.; Hung, C.S.; Chang, F.R.; Wu, Y.C. Active extracts of wild fruiting bodies of Antrodia camphorata (EEAC) induce leukemia HL 60 cells apoptosis partially through histone hypoacetylation and synergistically promote anticancer effect of trichostatin A. Arch. Toxicol. 2009, 83, 121–129. [Google Scholar] [CrossRef]
- Wu, C.F.; Lee, M.G.; El-Shazly, M.; Lai, K.H.; Ke, S.C.; Su, C.W.; Shih, S.P.; Sung, P.J.; Hong, M.C.; Wen, Z.H.; et al. Isoaaptamine Induces T-47D Cells Apoptosis and Autophagy via Oxidative Stress. Mar. Drugs 2018, 16, 18. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer Types | Cell Lines | 13-AC IC50 (µg/mL) | Cisplatin IC50 (µg/mL) |
|---|---|---|---|
| Human oral cancer cells | Ca9-22 | 0.94 ± 0.16 | 3.47 ± 0.33 |
| Cal-27 | 1.31 ± 0.38 | 1.78 ± 0.17 | |
| Human colon cancer cells | HCT116 | 1.36 ± 0.27 | 3.62 ± 0.19 |
| LoVo | 1.38 ± 0.37 | NA | |
| DLD1 | 1.64 ± 0.36 | 8.79 ± 0.52 | |
| Human prostate cancer cells | DU145 | 4.85 ± 0.92 | 3.24 ± 0.36 |
| LNcap | 3.93 ± 1.06 | 2.10 ± 0.20 | |
| Human breast cancer cells | MCF-7 | 2.44 ± 0.22 | NA |
| T47D | 2.00 ± 0.09 | NA | |
| Human cervical cancer cells | HeLa | 4.41 ± 0.75 | 7.74 ± 0.30 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.-C.; Peng, B.-R.; Hsu, K.-C.; El-Shazly, M.; Shih, S.-P.; Lin, T.E.; Kuo, F.-W.; Chou, Y.-C.; Lin, H.-Y.; Lu, M.-C. 13-Acetoxysarcocrassolide Exhibits Cytotoxic Activity against Oral Cancer Cells through the Interruption of the Keap1/Nrf2/p62/SQSTM1 Pathway: The Need to Move Beyond Classical Concepts. Mar. Drugs 2020, 18, 382. https://0-doi-org.brum.beds.ac.uk/10.3390/md18080382
Liu Y-C, Peng B-R, Hsu K-C, El-Shazly M, Shih S-P, Lin TE, Kuo F-W, Chou Y-C, Lin H-Y, Lu M-C. 13-Acetoxysarcocrassolide Exhibits Cytotoxic Activity against Oral Cancer Cells through the Interruption of the Keap1/Nrf2/p62/SQSTM1 Pathway: The Need to Move Beyond Classical Concepts. Marine Drugs. 2020; 18(8):382. https://0-doi-org.brum.beds.ac.uk/10.3390/md18080382
Chicago/Turabian StyleLiu, Yi-Chang, Bo-Rong Peng, Kai-Cheng Hsu, Mohamed El-Shazly, Shou-Ping Shih, Tony Eight Lin, Fu-Wen Kuo, Yi-Cheng Chou, Hung-Yu Lin, and Mei-Chin Lu. 2020. "13-Acetoxysarcocrassolide Exhibits Cytotoxic Activity against Oral Cancer Cells through the Interruption of the Keap1/Nrf2/p62/SQSTM1 Pathway: The Need to Move Beyond Classical Concepts" Marine Drugs 18, no. 8: 382. https://0-doi-org.brum.beds.ac.uk/10.3390/md18080382