
Bioactive Bromotyrosine Derivatives from the Pacific Marine Sponge Suberea clavata (Pulitzer-Finali, 1982)

 ,
,  , ,
, ,  , ,
, ,
Abstract
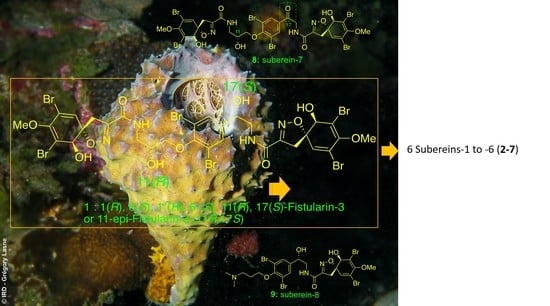
:
1. Introduction
2. Results and Discussion
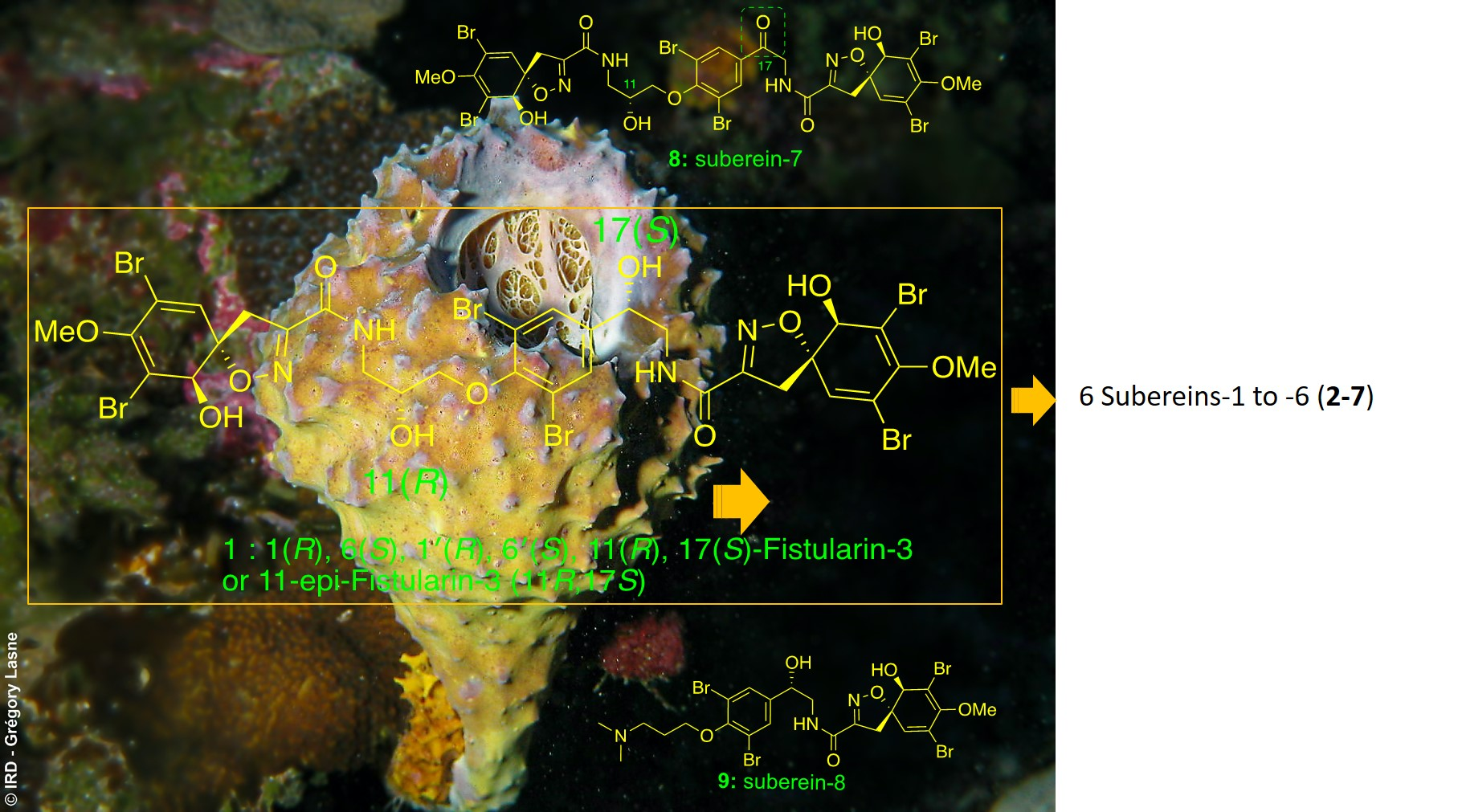
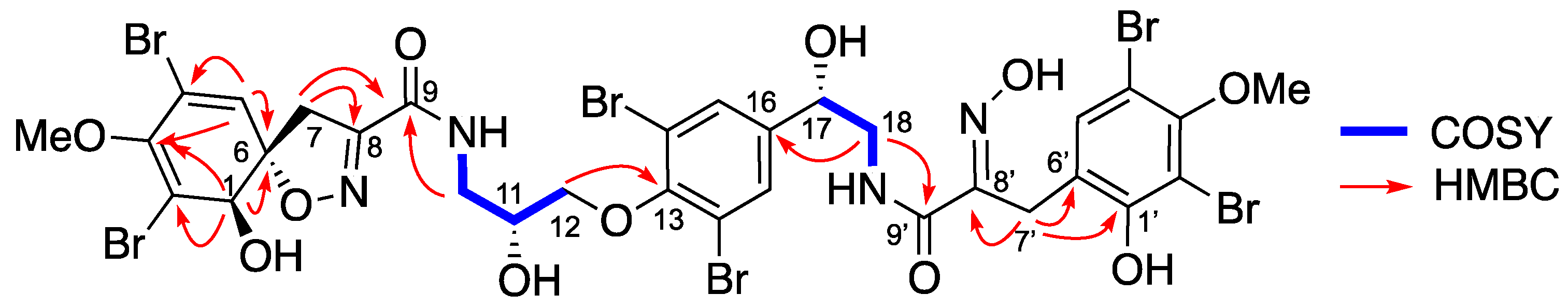
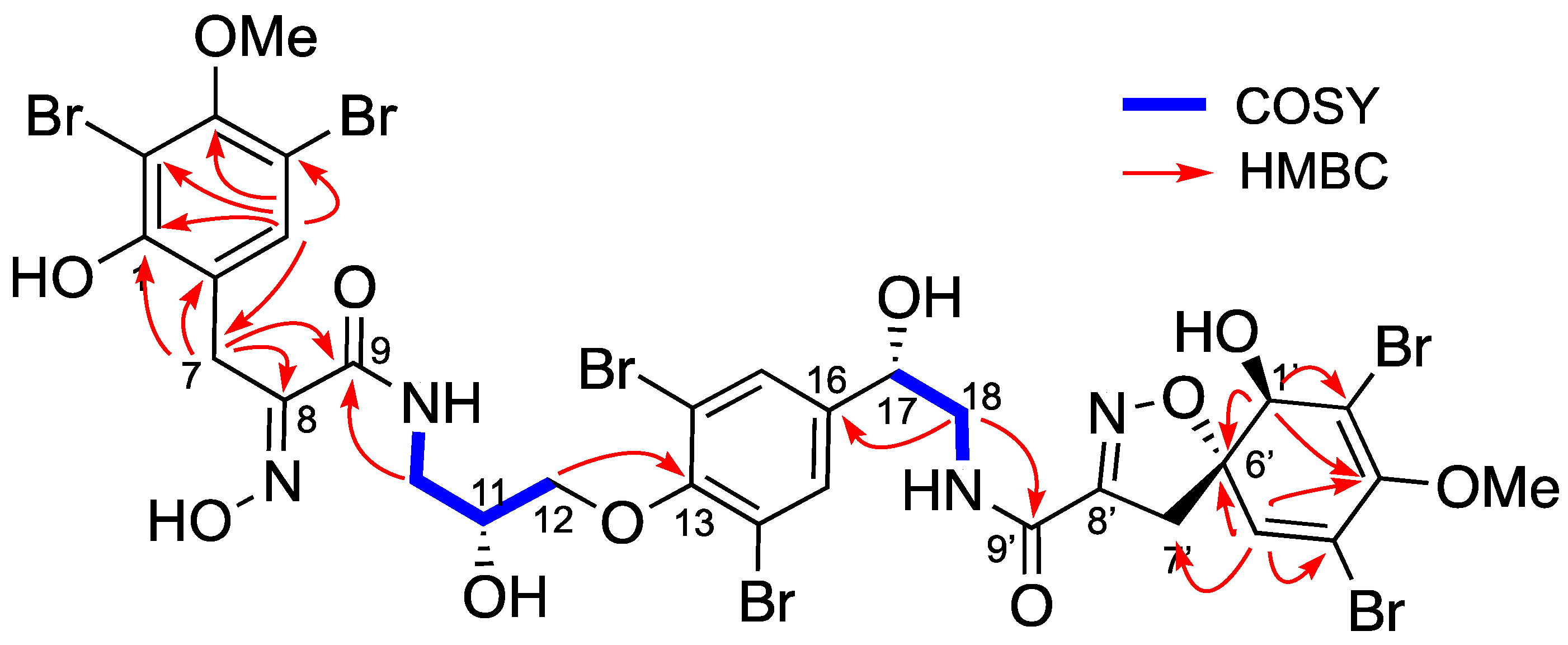
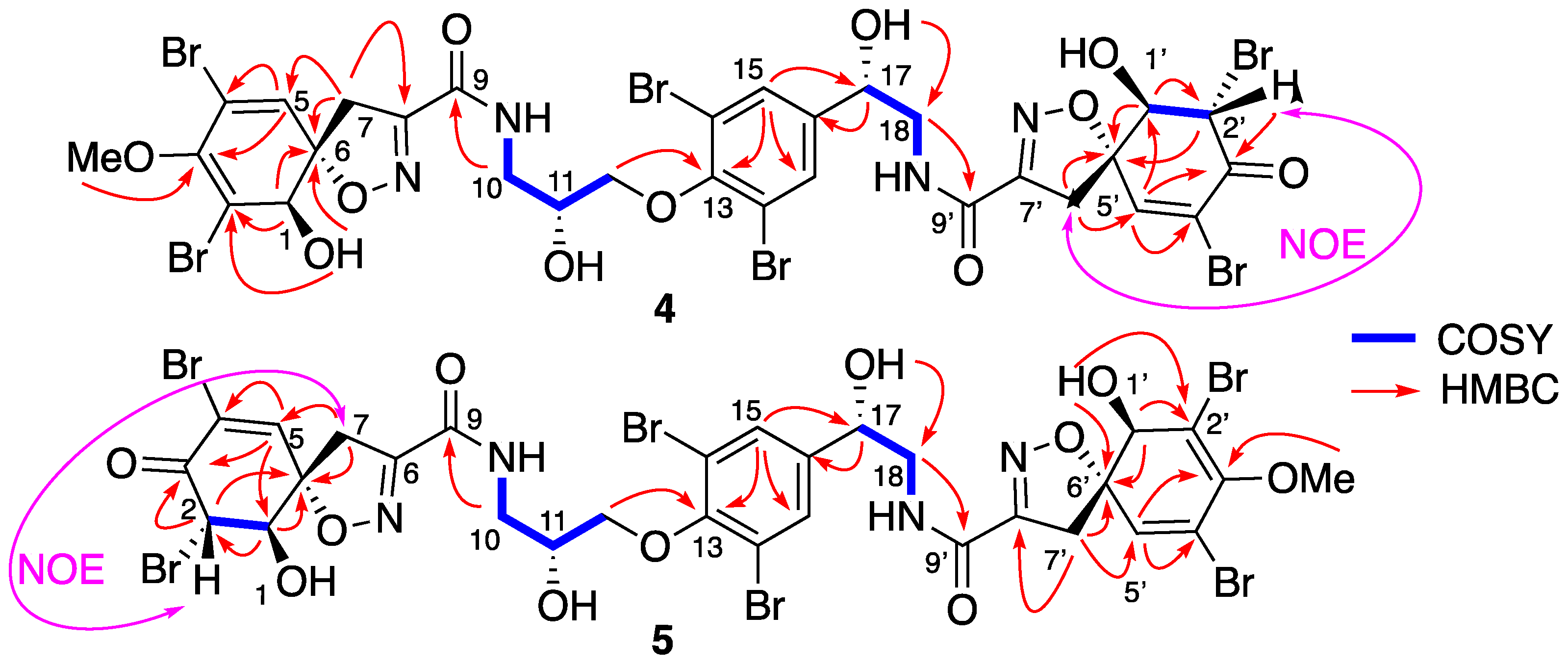
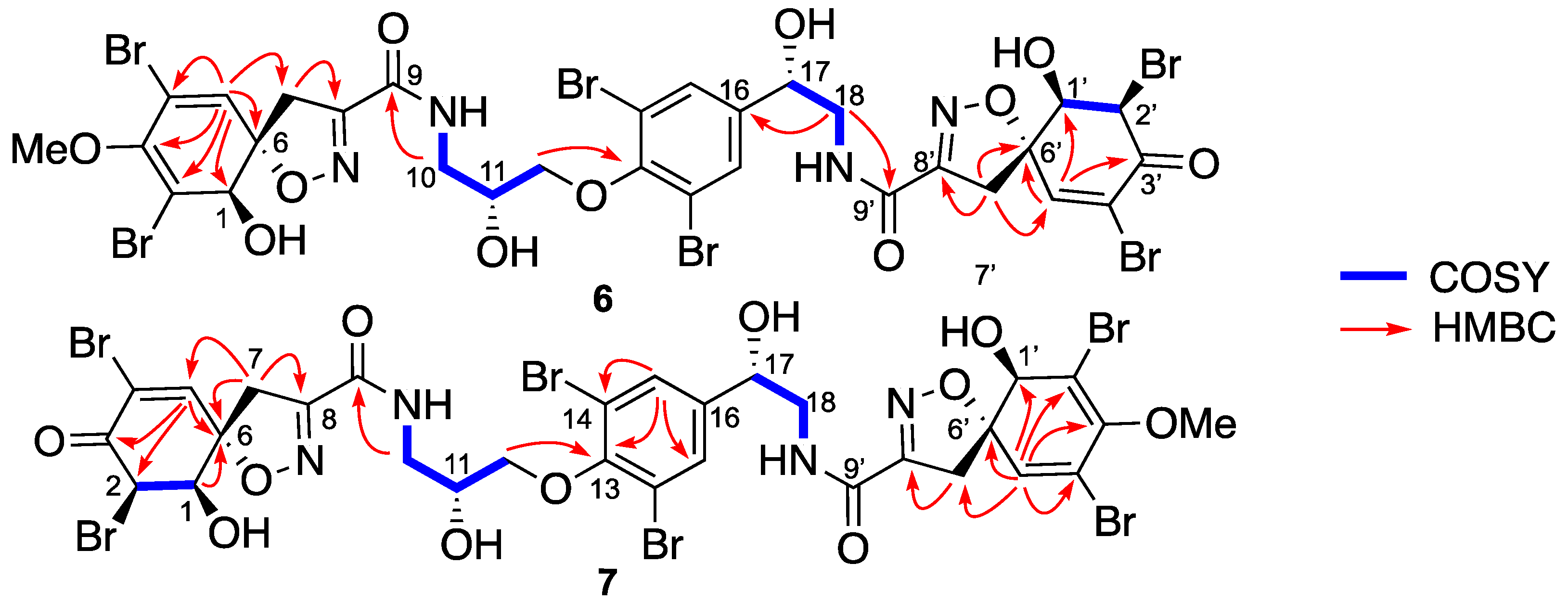
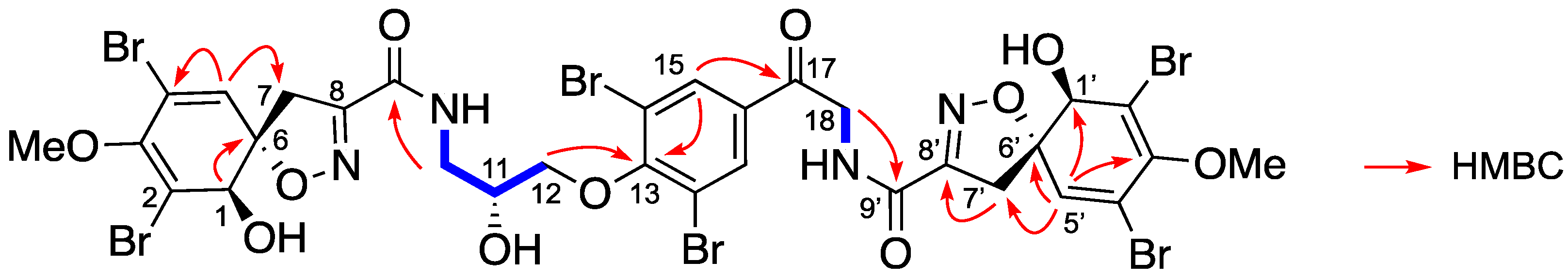
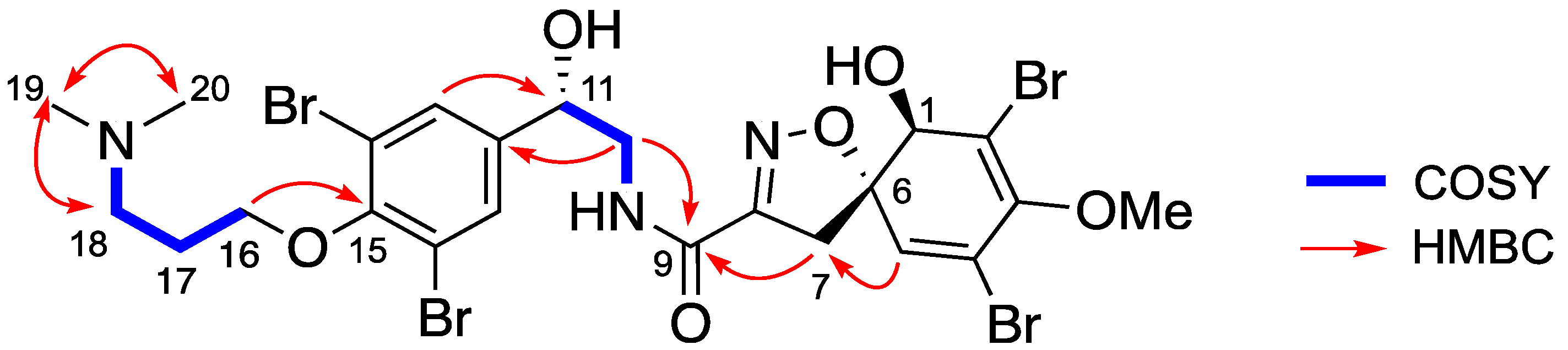
Isolation and Structure Elucidation
3. Biological Activities
3.1. Antibacterial Activity Inhibition
3.2. Acetylcholinesterase Activity Inhibition
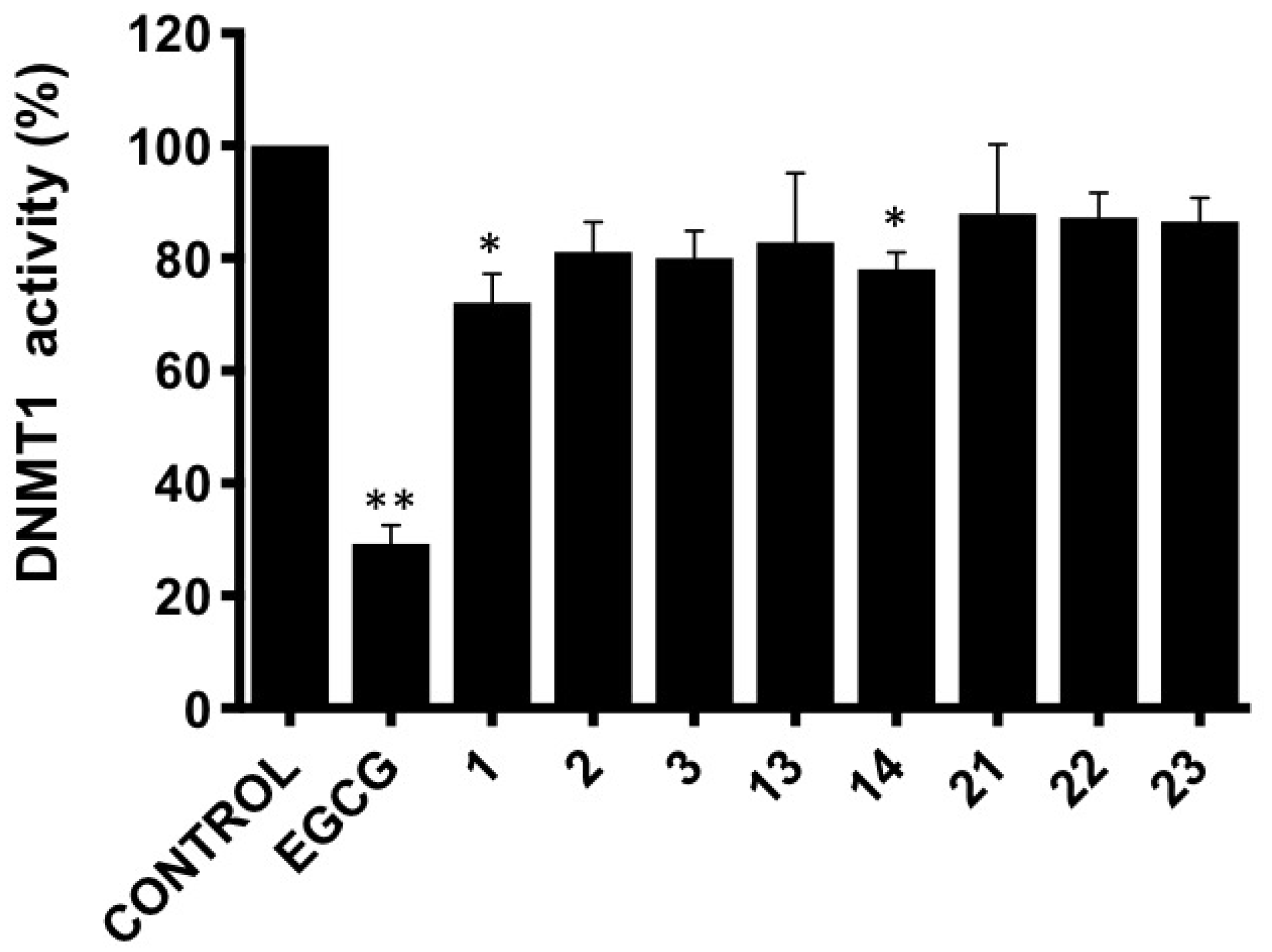
3.3. DNMT1 Activity Inhibition
4. Materials and Methods
4.1. General Experimental Procedure
4.2. Biological Material
4.3. Isolation and Spectroscopic Data
4.3.1. Isolation
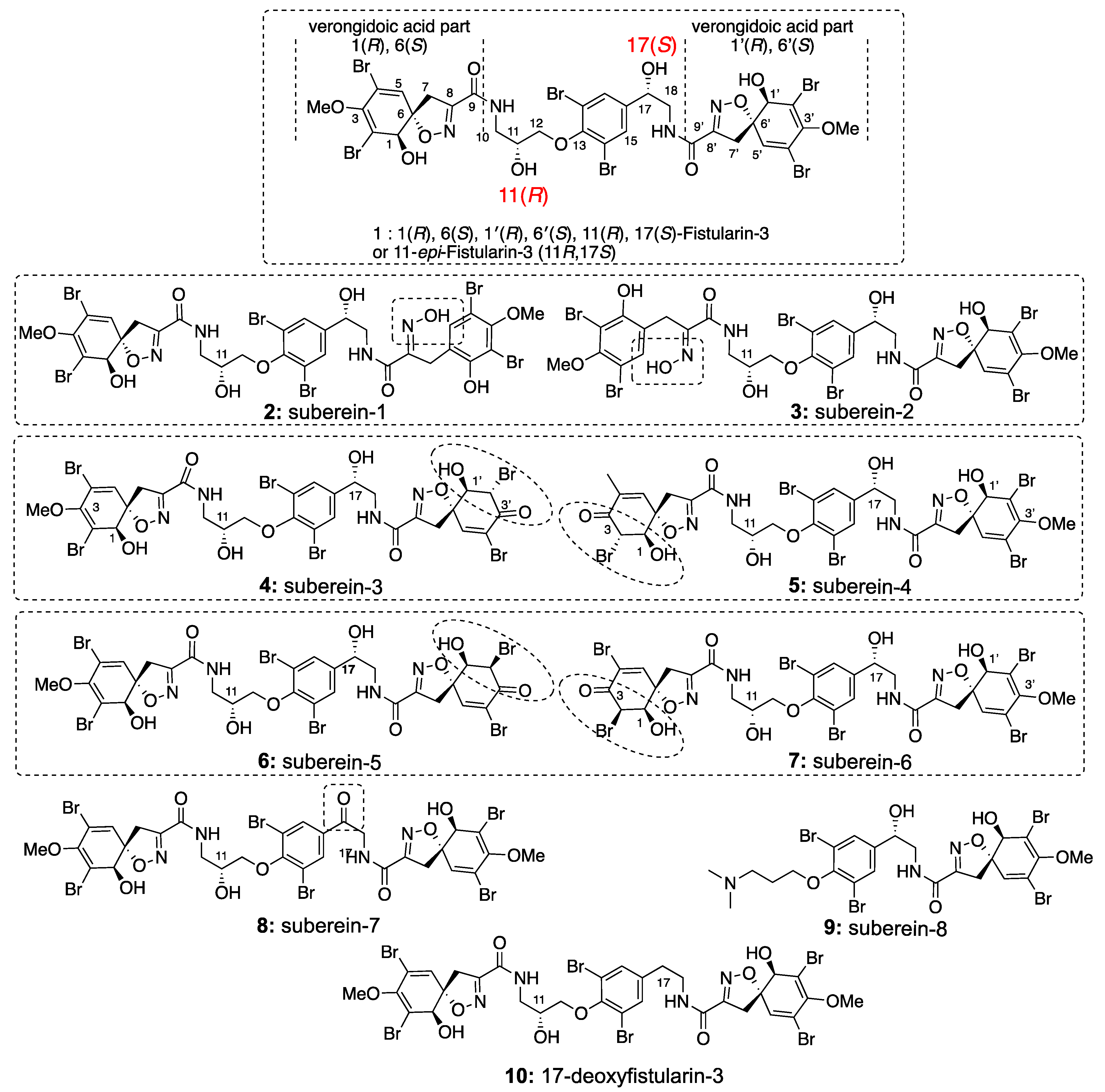
4.3.2. Spectroscopic Data and Absolute Configurations Determination
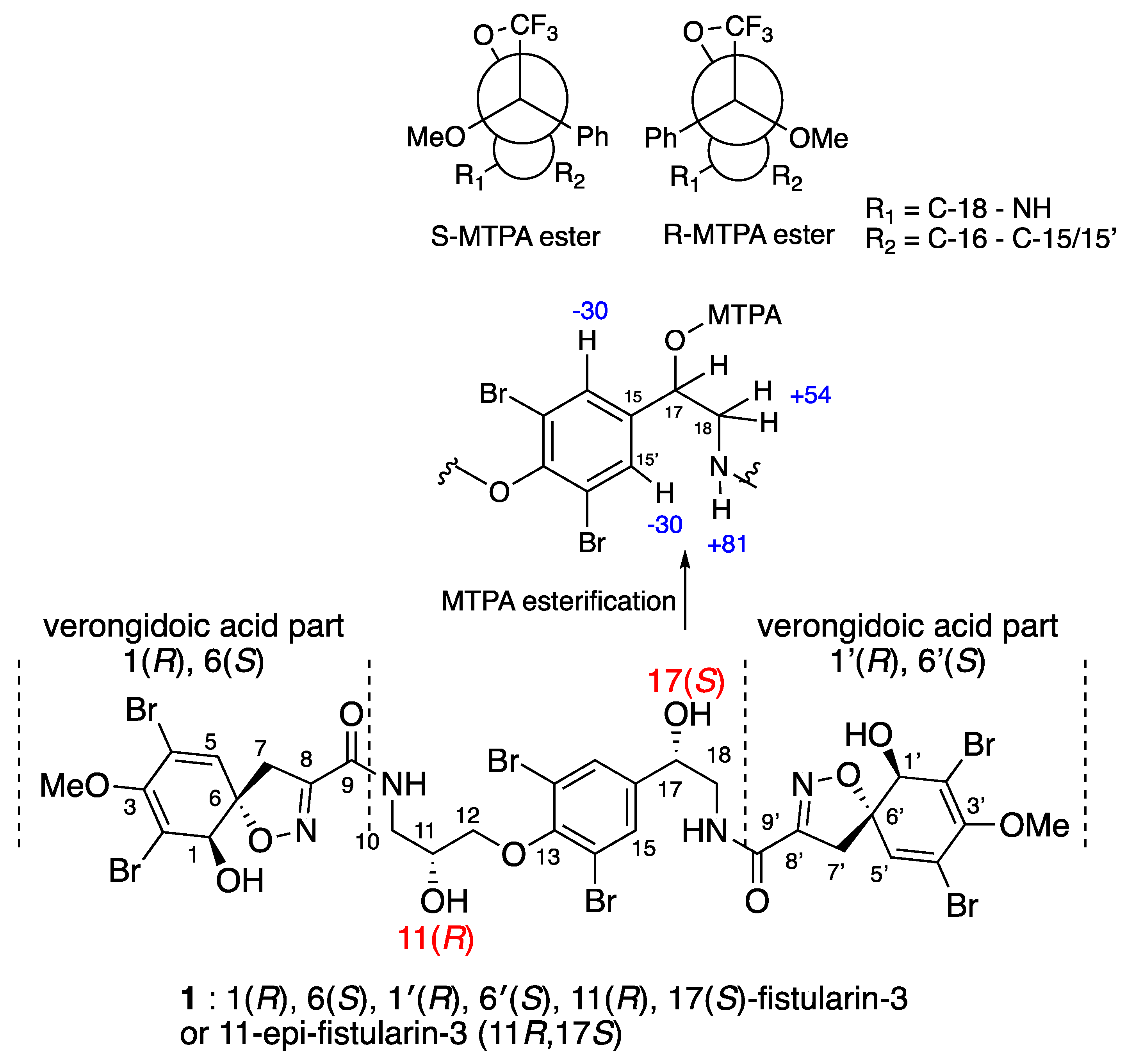
Determination of the C-17 Configuration for 11-epi-fistularin-3 (1)
Determination of C-11 Configuration for epi-fistularin-3 (1):
Determination of C-11 Configuration for Compounds 2 and 3.
Determination of C-11 Configuration for Compound 8
Determination of C-11 Configuration for 17-deoxy-epi-fistularin-3 (10)
4.4. Antibacterial Bioassay
4.4.1. Biological Material
4.4.2. Bioassay
4.5. Acetylcholinesterase Inhibition Bioassay
4.6. DNMT1 Activity Inhibition Bioassay
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kornprobst, J.M. Encyclopedia of Marine Natural Products; Wiley-Blackwell: Weinheim, Germany, 2010; ISBN 978-3-527-32703-4. [Google Scholar]
- Nuñez, C.V.; de Almelda, E.V.R.; Granato, A.C.; Marques, S.O.; Santos, K.O.; Pereira, F.R.; Macedo, M.L.; Ferreira, A.G.; Hajdu, E.; Pinheiro, U.S.; et al. Chemical variability within the marine sponge Aplysina fulva. Biochem. Syst. Ecol. 2008, 36, 283–296. [Google Scholar]
- Nicacio, K.J.; Ioca, L.P.; Froes, A.M.; Leomil, L.; Appolinario, L.R.; Thompson, C.C.; Thompson, F.L.; Ferreira, A.G.; Williams, D.E.; Andersen, R.J.; et al. Cultures of the Marine Bacterium Pseudovibrio denitrificans Ab134 Produce Bromotyrosine-Derived Alkaloids Previously Only Isolated from Marine Sponges. J. Nat. Prod. 2017, 80, 235–240. [Google Scholar] [CrossRef] [PubMed]
- König, G.M.; Wright, A.D. Agelorins A and B, and 11-epi-fistularin-3, three new antibacterial fistularin-3 derivatives from the tropical marine sponge Agelas oroides. Heterocycles 1993, 36, 1351–1358. [Google Scholar]
- Ebel, R.; Brenzinger, M.; Kunze, A.; Gross, H.J.; Proksch, P. Wound Activation of Protoxins in Marine Sponge Aplysina Aerophobaj. Chem. Ecol. 1997, 23, 1451–1462. [Google Scholar] [CrossRef]
- Thoms, C.; Wolff, M.; Padmakumar, K.; Ebel, R.; Proksch, P. Chemical Defense of Mediterranean Sponges Aplysina cavernicola and Aplysina aerophoba. Z. Für Nat. C 2004, 59, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Davis, R.A.; Buchanan, M.S.; Duffy, S.; Avery, V.M.; Camp, D.; Quinn, R.J. Antimalarial Bromotyrosine Derivatives from the Australian Marine Sponge Hyattella sp. J. Nat. Prod. 2010, 73, 985–987. [Google Scholar] [CrossRef]
- Diers, J.A.; Pennaka, H.K.; Peng, J.; Bowling, J.J.; Duke, S.O.; Hamann, M.T. Structural Activity Relationship Studies of Zebra Mussel Antifouling and Antimicrobial Agents from Verongid Sponges. J. Nat. Prod. 2004, 67, 2117–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsukamoto, S.; Kato, H.; Hirota, H.; Fusetani, N. Ceratinamides A and B: New antifouling dibromotyrosine derivatives from the marine sponge Pseudoceratina purpurea. Tetrahedron 1996, 52, 8181–8186. [Google Scholar] [CrossRef]
- Lebouvier, N.; Jullian, V.; Desvignes, I.; Maurel, S.; Parenty, A.; Dorin-Semblat, D.; Doerig, C.; Sauvain, M.; Laurent, D. Antiplasmodial Activities of Homogentisic Acid Derivative Protein Kinase Inhibitors Isolated from a Vanuatu Marine Sponge Pseudoceratina sp. Mar. Drugs 2009, 7, 640–653. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, M.F.; de Oliveira, J.H.H.L.; de Galetti, F.C.S.; Souza, A.O.; de Silva, C.L.; Hajdu, E.; Peixinho, S.; Berlinck, R.G.S. Antimycobacterial Brominated Metabolites from Two Species of Marine Sponges. Planta Med. 2006, 72, 437–441. [Google Scholar] [CrossRef]
- Gunasekera, S.P.; Cross, S.S. Fistularin 3 and 11-Ketofistularin 3. Feline Leukemia Virus Active Bromotyrosine Metabolites from the Marine Sponge Aplysina archeri. J. Nat. Prod. 1992, 55, 509–512. [Google Scholar] [CrossRef]
- Ross, S.A.; Weete, J.D.; Schinazi, R.F.; Wirtz, S.S.; Tharnish, P.; Scheuer, P.J.; Hamann, M.T.; Mololipids, A. New Series of Anti-HIV Bromotyramine-Derived Compounds from a Sponge of the Order Verongiida. J. Nat. Prod. 2000, 63, 501–503. [Google Scholar] [CrossRef] [PubMed]
- Kernan, M.R.; Cambie, R.C.; Bergquist, P.R. Chemistry of Sponges, VII. 11,19-Dideoxyfistularin 3 and 11-Hydroxyaerothionin, Bromotyrosine Derivatives from Pseudoceratina durissima. J. Nat. Prod. 1990, 53, 615–622. [Google Scholar] [CrossRef]
- Koulman, A.; Proksch, P.; Ebel, R.; Beekman, A.C.; van Uden, W.; Konings, A.W.T.; Pedersen, J.A.; Pras, N.; Woerdenbag, H.J. Cytoxicity and Mode of Action of Aeroplysinin-1 and a Related Dienone from the Sponge Aplysina Aerophoba. J. Nat. Prod. 1996, 59, 591–594. [Google Scholar] [CrossRef] [PubMed]
- Mierzwa, R.; King, A.; Conover, M.A.; Tozzi, S.; Puar, M.S.; Patel, M.; Coval, S.J.; Pomponi, S.A. Verongamine, a Novel Bromotyrosine-Derived Histamine H3-Antagonist from the Marine Sponge Verongula gigantean. J. Nat. Prod. 1994, 57, 175–177. [Google Scholar] [CrossRef]
- Kobayashi, J.; Honma, K.; Sasaki, T.; Tsuda, M. Purealidins J-R, New Bromotyrosine Alkaloids from the Okinawan Marine Sponge Psammaplysilla purea. Chem. Pharm. Bull. 1995, 43, 403–407. [Google Scholar] [CrossRef]
- Sallam, A.A.; Ramasahayam, S.; Meyer, S.A.; Sayed, K.A.E. Design, synthesis, and biological evaluation of dibromotyrosine analogues inspired by marine natural products as inhibitors of human prostate cancer proliferation, invasion, and migration. Bioorganic Med. Chem. 2010, 18, 7446–7457. [Google Scholar] [CrossRef] [PubMed]
- El-Demerdash, A.; Moriou, C.; Toullec, J.; Besson, M.; Soulet, S.; Schmitt, N.; Petek, S.; Lecchini, D.; Debitus, C.; Al-Mourabit, A. Bioactive Bromotyrosine-Derived Alkaloids from the Polynesian Sponge Suberea ianthelliformis. Mar. Drugs 2018, 16, 146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinn, R.J.; Buchanan, M.S.; Carroll, A.R.; Wessling, D.; Jobling, M.; Avery, V.M.; Davis, R.A.; Feng, Y.; Xue, Y.; Öster, L.; et al. A sponge with the same name Suberea clavata was chemically studied. Med. Chem. 2008, 51, 3583–3587. [Google Scholar]
- Buchanan, M.S.; Carroll, A.R.; Wessling, D.; Jobling, M.; Avery, V.M.; Davis, R.A.; Feng, Y.; Hooper, J.N.A.; Quinn, R.J.J. Clavatadines C−E, Guanidine Alkaloids from the Australian Sponge Suberea clavata. Nat. Prod. 2009, 72, 973–975. [Google Scholar] [CrossRef] [PubMed]
- Mancini, I.; Guella, G.; Laboute, P.; Debitus, C.; Pietra, F. Hemifistularin 3: A degraded peptide or biogenetic precursor? Isolation from a sponge of the order verongiida from the coral sea or generation from base treatment of 11-oxofistularin 3. J. Chem. Soc. Perkin Trans. 1993, 1, 3121–3125. [Google Scholar] [CrossRef]
- Compagnone, R.S.; Avila, R.; Suárez, A.I.; Abrams, O.V.; Rangel, H.R.; Arvelo, F.; Piña, I.C.; Merentes, E. 11-Deoxyfistularin-3, a New Cytotoxic Metabolite from the Caribbean Sponge Aplysina fistularis insularis. J. Nat. Prod. 1999, 62, 1443–1444. [Google Scholar] [CrossRef] [PubMed]
- Gunasekera, M.; Gunasekera, S.P. Dihydroxyaerothionin and Aerophobin 1. Two Brominated Tyrosine Metabolites from the Deep Water Marine Sponge Verongula rigida. J. Nat. Prod. 1989, 52, 753–756. [Google Scholar] [CrossRef]
- Kossuga, M.H.; MacMillan, J.B.; Rogers, E.W.; Molinski, T.F.; Nascimento, G.G.F.; Rocha, R.M.; Berlinck, R.G.S. (2S,3R)-2-Aminododecan-3-ol, a New Antifungal Agent from the Ascidian Clavelina oblonga. J. Nat. Prod. 2004, 67, 1879–1881. [Google Scholar] [CrossRef]
- Shaker, K.H.; Zinecker, H.; Ghani, M.A.; Imhoff, J.F.; Schneider, B. Bioactive Metabolites from the Sponge Suberea sp. Chem. Biodivers. 2010, 7, 2880–2887. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.D.; Pham, N.B.; Fechner, G.; Hooper, J.N.A.; Quinn, R.J. Bromotyrosine Alkaloids from the Australian Marine Sponge Pseudoceratina verrucosa. J. Nat. Prod. 2013, 76, 516–523. [Google Scholar] [CrossRef]
- Florean, C.; Kim, K.R.; Schnekenburger, M.; Kim, H.-J.; Moriou, C.; Debitus, C.; Dicato, M.; Al-Mourabit, A.; Han, B.W.; Diederich, M. Synergistic AML Cell Death Induction by Marine Cytotoxin (+)-1(R), 6(S), 1′(R), 6′(S), 11(R), 17(S)-Fistularin-3 and Bcl-2 Inhibitor Venetoclax. Mar. Drugs 2018, 16, 518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogers, E.W.; de Oliveira, M.F.; Berlinck, R.G.; Konig, G.M.; Molinski, T.F. Stereochemical heterogeneity in Verongid sponge metabolites. Absolute stereochemistry of (+)-fistularin-3 and (+)-11-epi-fistularin-3 by microscale LCMS-Marfey’s analysis. J. Nat. Prod. 2005, 68, 891–896. [Google Scholar] [CrossRef]
- Peng, J.; Li, J.; Hamann, M.T. The Marine Bromotyrosine Derivatives. Alkaloids Chem. Biol. 2005, 61, 59–262. [Google Scholar]
- Me Millan, J.A.; Paul, I.C.; Goo, Y.M.; Rinehart, K.L.; Krueger, W.C.; Pschigoda, L.M. An x-raystudy of aerothionin from Aplysina fistularis (PALLAS). Tetrahedron Lett. 1981, 22, 39–42. [Google Scholar] [CrossRef]
- Wang, Q.; Tang, X.L.; Luo, X.C.; de Voog, N.J.; Li, P.L.; Li, G.Q. Aplysinopsin-type and Bromotyrosine-derivedAlkaloids from the South China Sea Sponge Fascaplysinopsis reticulate. Sci. Rep. 2019, 9, 2248. [Google Scholar] [CrossRef] [Green Version]
- Hoye, T.R.; Jeffrey, J.; Shao, F. Mosher ester analysis for the determination of absolute configuration of stereogenic (chiral) carbinol carbons. Nat. Protoc. 2007, 2, 2451–2458. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Debbab, A.; Wray, V.; Lin, W.; Schulz, B.; Trepos, R.; Pile, C.; Hellio, C.; Proksch, P.; Aly, A.H. Marine bacterial inhibitors from the sponge-derived fungus Aspergillus sp. Tetrahedron Lett. 2014, 55, 2789–2792. [Google Scholar] [CrossRef] [Green Version]
- Von Baum, H.; Marre, R. Antimicrobial resistance of Escherichia coli and therapeutic implications. Int. J. Med Microbiol. 2005, 295, 503–511. [Google Scholar] [CrossRef]
- Labreuche, Y.; Soudant, P.; Gonçalves, M.; Lambert, C.; Nicolas, J.-L. Effects of extracellular products from the pathogenic Vibrio aestuarianus strain 01/32 on lethality and cellular immune responses of the oyster Crassostrea gigas. Dev. Comp. Immunol. 2006, 30, 367–379. [Google Scholar] [CrossRef] [Green Version]
- Labreuche, Y.; Lambert, C.; Soudant, P.; Boulo, V.; Huvet, A.; Nicolas, J.-L. Cellular and molecular hemocyte responses of the Pacific oyster, Crassostrea gigas, following bacterial infection with Vibrio aestuarianus strain 01/32. Microbes Infect. 2006, 8, 2715–2724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labreuche, Y.; Le Roux, F.; Henry, J.; Zatylny, C.; Huvet, A.; Lambert, C.; Soudant, P.; Mazel, D.; Nicolas, J.-L. Vibrio aestuarianus zinc metalloprotease causes lethality in the Pacific oyster Crassostrea gigas and impairs the host cellular immune defenses. Fish Shellfish. 2010, 29, 753–758. [Google Scholar] [CrossRef] [Green Version]
- Cochennec-Laureau, N.; Baud, J.-P.; Pepin, J.-F.; Benabdelmouna, A.; Soletchnik, P.; Lupo, C.; Garcia, C.; Arzul, I.; Boudry, P.; Huvet, A.; et al. Les Surmortalités des Naissains D’huîtres Creuses, Crassostrea Gigas: Acquis des Recherches en 2010. RST/LER/MPL/11.07. 2011. Available online: https://archimer.ifremer.fr/doc/00033/14423/ (accessed on 25 February 2021).
- Qurashi, A.W.; Sabri, A.N. Biofilm formation in moderately halophilic bacteria is influenced by varying salinity levels. J. Basic Microbiol. 2012, 52, 566–572. [Google Scholar] [CrossRef]
- Chambers, L.D.; Hellio, C.; Stokes, K.R.; Dennington, S.P.; Goodes, L.R.; Wood, R.J.K.; Walsh, F.C. Investigation of Chondrus crispus as a potential source of new antifouling agents. Int. Biodeterior. Biodegrad. 2011, 65, 939–946. [Google Scholar] [CrossRef]
- Austin, B.; Rodgers, C.J.; Forns, J.M.; Colwell, R.R. Alcaligenes faecalis subsp. homari subsp. nov., a New Group of Bacteria Isolated from Moribund Lobsters. Int. J. Syst. Bacteriol. 1981, 31, 72–76. [Google Scholar] [CrossRef]
- Drake, L.A.; Meyer, A.E.; Forsberg, R.L.; Baier, R.E.; Doblin, M.A.; Heinemann, S.; Johnson, W.P.; Koch, M.; Rublee, P.A.; Dobbs, F.C. Potential Invasion of Microorganisms and Pathogens via ‘Interior Hull Fouling’: Biofilms Inside Ballast Water Tanks. Biol. Invasions 2005, 7, 969–982. [Google Scholar] [CrossRef]
- Huchard, E.; Martinez, M.; Alout, H.; Douzery, E.J.P.; Lutfalla, G.; Berthomieu, A.; Berticat, C.; Raymond, M.; Weill, M. Acetylcholinesterase genes within the Diptera: Takeover and loss in true flies. Proc. R. Soc. B Biol. Sci. 2006, 273, 2595–2604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilg, T.; Cramer, J.; Lutz, J.; Noack, S.; Schmitt, H.; Williams, H.; Newton, T. The characterization of Lucilia cuprina acetylcholinesterase as a drug target, and the identification of novel inhibitors by high throughput screening. Insect Biochem. Mol. Biol. 2011, 41, 470–483. [Google Scholar] [CrossRef] [PubMed]
- Rickwood, C.J.; Galloway, T.S. Acetylcholinesterase inhibition as a biomarker of adverse effect: A study of Mytilus edulis exposed to the priority pollutant chlorfenvinphos. Aquat. Toxicol. 2004, 67, 45–56. [Google Scholar] [CrossRef]
- Faimali, M.; Falugi, C.; Gallus, L.; Piazza, V.; Tagliafierro, G. Involvement of Acetyl Choline in Settlement of Balanus Amphitrite. Biofouling 2003, 19, 213–220. [Google Scholar] [CrossRef]
- Piazza, V.; Dragić, I.; Sepčić, K.; Faimali, M.; Garaventa, F.; Turk, T.; Berne, S. Antifouling Activity of Synthetic Alkylpyridinium Polymers Using the Barnacle Model. Mar. Drugs 2014, 12, 1959–1976. [Google Scholar] [CrossRef] [Green Version]
- Florean, C.; Schnekenburger, M.; Lee, J.; Kim, K.R.; Mazumder, A.; Song, S.; Kim, J.-M.; Grandjenette, C.; Kim, J.-G.; Yoon, A.-Y.; et al. Discovery and characterization of Isofistularin-3, a marine brominated alkaloid, as a new DNA demethylating agent inducing cell cycle arrest and sensitization to TRAIL in cancer cells. Oncotarget 2016, 7, 24027–24049. [Google Scholar] [CrossRef]
- Payri, C. BSMS-1 cruise. RV Alis. 2004. [Google Scholar] [CrossRef]
- Plouguerne, E.; Ioannou, E.; Georgantea, P.; Vagias, C.; Roussis, V.; Hellio, C.; Kraffe, E.; Stiger-Pouvreau, V. Anti-microfouling Activity of Lipidic Metabolites from the Invasive Brown Alga Sargassum muticum (Yendo) Fensholt. Mar. Biotechnol. 2010, 12, 52–61. [Google Scholar] [CrossRef]
- Maréchal, J.-P.; Culioli, G.; Hellio, C.; Thomas-Guyon, H.; Callow, M.E.; Clare, A.S.; Ortalo-Magné, A. Seasonal variation in antifouling activity of crude extracts of the brown alga Bifurcaria bifurcata (Cystoseiraceae) against cyprids of Balanus amphitrite and the marine bacteria Cobetia marina and Pseudoalteromonas haloplanktis. J. Exp. Mar. Biol. Ecol. 2004, 313, 47–62. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Andres, V.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | δS-Ester (ppm) | δR-Ester (ppm) | ΔδSR (ppm) | ΔδSR (Hz) |
|---|---|---|---|---|
| 15, 15′ | 7.63 | 7.73 | −0.10 | −30 |
| 17 | 6.13 | 6.21 | −0.08 | −24 |
| 18 | 3.98 | 3.80 | +0.18 | +54 |
| NH’ | 7.95 | 7.68 | +0.27 | +81 |
| Suberein-1 (2) (500 MHz) | Suberein-2 (3) (600 MHz) | |||
|---|---|---|---|---|
| Position | δC, Type | δH Mult, (J in Hz) | δC, Type | δH Mult, (J in Hz) |
| 1 | 75.2, CH | 4.20, s | 155.1, C | - |
| 2 | 113.9, C | - | 108.9, C | - |
| 3 | 148.8, C | - | 154.7, C | - |
| 4 | 122.1, C | - | 106.1, C | - |
| 5 | 132.1, CH | 6.53, s | 134.7, CH | 7.57, s |
| 6 | 91.9, C | - | 122.2, C | - |
| 7 | 40.1, CH2 | 3.85, d (18.4) 3.19, d (18.4) | 25.6, CH2 | 3.84, s |
| 8 | 155.2, C | - | 151.1, C | - |
| 9 | 160.4, C | - | 166.6, C | - |
| 10 | 43.5, CH2 | 3.78, m 3.52, m | 43.6, CH2 | 3.78, m 3.58, m |
| 11 | 69.8, CH | 4.24, m | 69.2, CH | 4.25, m |
| 12 | 75.9, CH2 | 4.05, dd (9.0, 5.2) 4.02, dd (9.0, 5.2) | 75.6, CH2 | 4.04, dd (9.1, 5.4) 3.99, dd (9.1, 5.4) |
| 13 | 152.7, C | - | 152.5, C | - |
| 14, 14′ | 118.4, C | - | 118.3, C | - |
| 15, 15′ | 131.5, CH | 7.65, s | 131.4, CH | 7.63, s |
| 16 | 143.1, C | - | 143.2, C | - |
| 17 | 71.0, CH | 4.92, dd (7.7, 4.5) | 71.3, CH | 4.89, dd (7.7, 4.5) |
| 18 | 47.7, CH2 | 3.64, m 3.49, m | 47.4, CH2 | 3.61, m 3.45, m |
| 1′ | 155.1, C * | - | 75.1, CH | 4.18, s |
| 2′ | 108.9, C | - | 113.9, C | - |
| 3′ | 154.8, C | - | 148.7, C | - |
| 4′ | 106.3, C | - | 122.1, C | - |
| 5′ | 134.9, CH | 7.56, s | 132.1, CH | 6.52, s |
| 6′ | 122.1, C | - | 91.8, C | - |
| 7′ | 25.7, CH2 | 3.81, s | 39.8, CH2 | 3.83, d (18.4) 3.17, d (18.4) |
| 8′ | 151.2, C | - | 155.3, C | - |
| 9′ | 166.7, C | - | 160.4, C | - |
| OMe | 60.2, CH3 | 3.73, s | 60.5, CH3 | 3.80, s |
| OMe’ | 60.6, CH3 | 3.81, s | 60.5, CH3 | 3.72, s |
| NH | - | 7.62, bt (5.7) | - | 8.10, bt (6.8) |
| NH’ | - | 8.05, bt (6.8) | - | 7.74, bt (5.7) |
| OH-1, OH-1′ | - | - | - | - |
| OH-11 | - | - | - | - |
| OH-17 | - | - | - | - |
| Suberein-3 (4) | Suberein-4 (5) | |||
|---|---|---|---|---|
| Position | δC, Type | δH Mult, (J in Hz) | δC, Type | δH Mult, (J in Hz) |
| 1 | 75.0, CH | 4.18, d (6.0) | 75.2, CH | 4.40, dd (11.4, 6.0) |
| 2 | 113.9, C | - | 57.4, CH | 5.09, d (11.4) |
| 3 | 148.8, C | - | 183.6, C | - |
| 4 | 122.2, C | - | 122.6, C | - |
| 5 | 132.3, CH | 6.53, s | 149.3, CH | 7.62, s |
| 6 | 91.8, C | - | 91.8, C | - |
| 7 | 40.0, CH2 | 3.82, d (18.4) 3.16, d (18.4) | 38.4, CH2 | 3.86, d (18.4) 3.25, d (18.4) |
| 8 | 155.2, C | - | 155.1, C | - |
| 9 | 160.3, C | - | 160.4, C | - |
| 10 | 43.7, CH2 | 3.78, m 3.52, m | 43.7, CH2 | 3.78, m 3.52, m |
| 11 | 69.9, CH | 4.25, m | 69.9, CH | 4.25, m |
| 12 | 76.0, CH2 | 4.06, m 4.04, m | 76.0, CH2 | 4.06, m 4.04, m |
| 13 | 152.7, C | - | 152.2, C | - |
| 14, 14′ | 118.4, C | - | 118.3, C | - |
| 15, 15′ | 131.5, CH | 7.66, s | 131.5, CH | 7.67, s |
| 16 | 143.3, C | - | 143.3, C | - |
| 17 | 71.4, CH | 4.91, m | 71.4, CH | 4.91, m |
| 18 | 47.6, CH2 | 3.62, m 3.48, m | 47.6, CH2 | 3.62, m 3.48, m |
| 1′ | 75.3 CH | 4.41, dd (11.4, 6.0) | 75.1, CH | 4.20, d (6.0) |
| 2′ | 57.4, CH | 5.08, d (11.4) | 113.9, C | - |
| 3′ | 183.6, C | - | 148.7, C | - |
| 4′ | 122.6, C | - | 122.2, C | - |
| 5′ | 149.4, CH | 7.64, s | 132.1, CH | 6.52, s |
| 6′ | 91.8, C | - | 91.8, C | - |
| 7′ | 38.5, CH2 | 3.88, d (18.4) 3.29, d (18.4) | 40.1, CH2 | 3.86, d (18.4) 3.20, d (18.4) |
| 8′ | 155.1, C | - | 155.2, C | - |
| 9′ | 160.4, C | - | 160.3, C | - |
| OMe | 60.2, CH3 | 3.73, s | - | - |
| OMe’ | - | - | 60.2, CH3 | 3.73, s |
| NH | - | 7.63, m | - | 7.63, m |
| NH’ | - | 7.67, m | - | 7.67, m |
| OH-1 | - | 5.41, d (6.0) | - | 5.96, d (6.0) |
| OH-1′ | - | 5.97, d (6.0) | - | 5.41, d (6.0) |
| OH-11 | - | 4.44, m | - | 4.44, m |
| OH-17 | - | 5.01, m | - | 5.01, m |
| Suberein-5 (6) | Suberein-6 (7) | |||
|---|---|---|---|---|
| Position | δC, Type | δH Mult, (J in Hz) | δC, Type | δH Mult, (J in Hz) |
| 1 | 75.2, CH | 4.18, br s | 73.1, CH | 4.57, br s |
| 2 | 113.9, C | - | 54.8, CH | 5.27, br s |
| 3 | 148.8, C | - | 183.6, C | - |
| 4 | 122.2, C | - | 122.1, C | - |
| 5 | 132.4, CH | 6.53, s | 146.4, CH | 7.47, s |
| 6 | 91.8, C | - | 90.8, C | - |
| 7 | 40.1, CH2 | 3.82, d (18.4) 3.16, d (18.4) | 41.5, CH2 | 3.88, d (18.4) 3.35, d (18.4) |
| 8 | 155.2, C | - | 155.1, C | - |
| 9 | 160.4, C | - | 160.5, C | - |
| 10 | 43.6, CH2 | 3.78, m 3.52, m | 43.6, CH2 | 3.78, m 3.52, m |
| 11 | 69.9, CH | 4.25, m | 69.9, CH | 4.25, m |
| 12 | 76.0, CH2 | 4.06, m 4.04, m | 76.0, CH2 | 4.06, m 4.04, m |
| 13 | 152.7, C | - | 152.7, C | - |
| 14, 14′ | 118.4, C | - | 118.4, C | - |
| 15, 15′ | 131.4, CH | 7.67, s | 131.3, CH | 7.66, s |
| 16 | 143.3, C | - | 143.3, C | - |
| 17 | 71.4, CH | 4.91, m | 71.4, CH | 4.91, m |
| 18 | 47.6, CH2 | 3.62, m 3.48, m | 47.6, CH2 | 3.62, m 3.48, m |
| 1′ | 73.2 CH | 4.57, br s | 75.3, CH | 4.19, br s |
| 2′ | 54.8, CH | 5.27, br s | 113.9, C | - |
| 3′ | 183.6, C | - | 148.8, C | - |
| 4′ | 122.1, C | - | 122.2, C | - |
| 5′ | 146.4, CH | 7.49, s | 132.3, CH | 6.52, s |
| 6′ | 90.8, C | - | 91.8, C | - |
| 7′ | 41.5, CH2 | 3.91, d (18.4) 3.39, d (18.4) | 40.1, CH2 | 3.86, d (18.4) 3.20, d (18.4) |
| 8′ | 155.1, C | - | 155.2, C | - |
| 9′ | 160.5, C | - | 160.4, C | - |
| OMe | 60.2, CH3 | 3.73, s | - | - |
| OMe’ | - | - | 60.2, CH3 | 3.73, s |
| NH | - | 7.63, br t (5.0) | - | 7.66, br t (5.0) |
| NH’ | - | 7.66, br t (5.0) | - | 7.73, br t (5.0) |
| OH-1 | - | 5.45, br s | - | 5.95, br s |
| OH-1′ | - | 5.95, br s | - | 5.45, br s |
| OH-11 | - | 4.47, br s | - | 4.47, br s |
| OH-17 | - | 5.04, br s | - | 5.04, br s |
| Suberein-7 (8) (600 MHz, Acetone-d6) | Suberein-8 (9) (500, MHz, MeOH-d4) | ||||
|---|---|---|---|---|---|
| Position | δC, Type | δH Mult, (J in Hz) | Position | δC, Type | δH Mult, (J in Hz) |
| 1 | 75.2, CH | 4.19, s | 1 | 74.2, CH | 4.09, s |
| 2 | 113.9, C | - | 2 | 112.6, C | - |
| 3 | 148.7, C | - | 3 | 147.9, C | - |
| 4 | 122.1, C | - | 4 | 121.5, C | - |
| 5 | 132.3, CH | 6.53, s | 5 | 130.7, CH | 6.41, s |
| 6 | 91.8, C | - | 6 | 91.1, C | - |
| 7 | 40.1, CH2 | 3.86, d (18.4) 3.22, d (18.4) | 7 | 38.3, CH2 | 3.75, d (18.0) 3.06, d (18.0) |
| 8 | 154.8, C | - | 8 | 153.0, C | - |
| 9 | 160.6, C | - | 9 | 160.8, C | - |
| 10 | 43.5, CH2 | 3.78, m 3.53, m | 10 | 47.3, CH2 | 3.74, m 3.40, m |
| 11 | 69.9, CH | 4.29, m | 11 | 70.1, CH2 | 4.76, dd (7.2, 4.7) |
| 12 | 76.3, CH2 | 4.16, m | 12 | 142.5, C | - |
| 13 | 158.0, C | - | 13, 13′ | 130.2, CH | 7.62, s |
| 14, 14′ | 119.2, C | - | 14, 14′ | 117.6, C | - |
| 15, 15′ | 133.6, CH | 8.28, s | 15 | 152.1, C | - |
| 16 | 134.3, C | - | 16 | 71.8, CH2 | 4.07, t (6.5) |
| 17 | 192.2, C | - | 17 | 26.5, CH2 | 2.11, m |
| 18 | 45.9, CH2 | 4.87, d (5.2) | 18 | 56.0, CH2 | 2.87, m |
| 1′ | 75.2, CH | 4.23, s | 19 | 43.8 CH3 | 2.47, s |
| 2′ | 113.9, C | - | 20 | 43.8, CH | 2.47, s |
| 3′ | 148.7, C | - | OMe | 59.0, CH3 | 3.73, s |
| 4′ | 122.1, C | - | |||
| 5′ | 132.3, CH | 6.57, s | |||
| 6′ | 92.0, C | - | |||
| 7′ | 39.9, CH2 | 3.86, d (18.4) 3.19, d (18.4) | |||
| 8′ | 155.2, C | - | |||
| 9′ | 160.2, C | - | |||
| OMe | 60.2, CH3 | 3.73, s | |||
| OMe’ | 60.2, CH3 | 3.73, s | |||
| NH | - | 7.67, bt (6.2) | |||
| NH’ | - | 7.91, bt (5.2) | |||
| OH-1, OH-1′ | - | 5.47, br s | |||
| OH-11 | - | 4.58, br s | |||
| OH-17 | - | - | |||
| Vibrio aesturianus | Roseobacter littoralis | Halomonas aquamarina | Escherichia coli | Acetylcholinesterase | |
|---|---|---|---|---|---|
| Compounds | MIC µM | MIC µM | MIC µM | MIC µM | IC50 µM |
| epi-fistularin-3 (1) | >1 | >1 | >1 | >1 | >10 |
| suberein-1 (2) | 0.01 | 1 | >1 | >1 | >10 |
| suberein-2 (3) | 0.01 | >1 | >1 | 0.01 | >10 |
| 17-oxo-11-epi-fistularin-3 (8) | 0.01 | >1 | >1 | >1 | >10 |
| 17-deoxy-11-epi-fistularin-3 (10) | >1 | >1 | 0,01 | >1 | >10 |
| agelorin A (11) | 0.1 | 0.1 | 0.1 | nt | 0.19 ± 0.2 |
| agelorin B (12) | >1 | >1 | >1 | >1 | nt |
| 11-deoxyfistularin-3 (13) | >1 | >1 | 0,01 | >1 | >10 |
| 11,17-dideoxyfistularin (14) | >1 | >1 | 0,01 | >1 | 10 ± 0.3 |
| 11-hydroxyaerothionin (15) | >1 | >1 | 0,01 | >1 | 10 ± 0.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moriou, C.; Lacroix, D.; Petek, S.; El-Demerdash, A.; Trepos, R.; Leu, T.M.; Florean, C.; Diederich, M.; Hellio, C.; Debitus, C.; et al. Bioactive Bromotyrosine Derivatives from the Pacific Marine Sponge Suberea clavata (Pulitzer-Finali, 1982). Mar. Drugs 2021, 19, 143. https://0-doi-org.brum.beds.ac.uk/10.3390/md19030143
Moriou C, Lacroix D, Petek S, El-Demerdash A, Trepos R, Leu TM, Florean C, Diederich M, Hellio C, Debitus C, et al. Bioactive Bromotyrosine Derivatives from the Pacific Marine Sponge Suberea clavata (Pulitzer-Finali, 1982). Marine Drugs. 2021; 19(3):143. https://0-doi-org.brum.beds.ac.uk/10.3390/md19030143
Chicago/Turabian StyleMoriou, Céline, Damien Lacroix, Sylvain Petek, Amr El-Demerdash, Rozenn Trepos, Tinihauarii Mareva Leu, Cristina Florean, Marc Diederich, Claire Hellio, Cécile Debitus, and et al. 2021. "Bioactive Bromotyrosine Derivatives from the Pacific Marine Sponge Suberea clavata (Pulitzer-Finali, 1982)" Marine Drugs 19, no. 3: 143. https://0-doi-org.brum.beds.ac.uk/10.3390/md19030143