
Approaches to Configuration Determinations of Flexible Marine Natural Products: Advances and Prospects

 ,
,
Abstract
: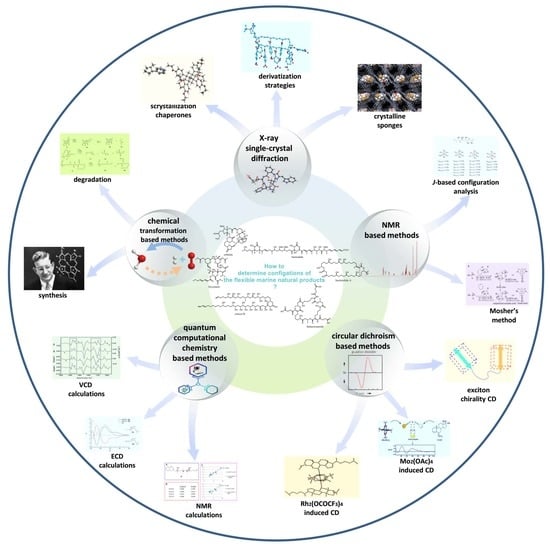
1. Introduction
2. X-ray Single-Crystal Diffraction (XRSCD)
3. NMR-Based Methods
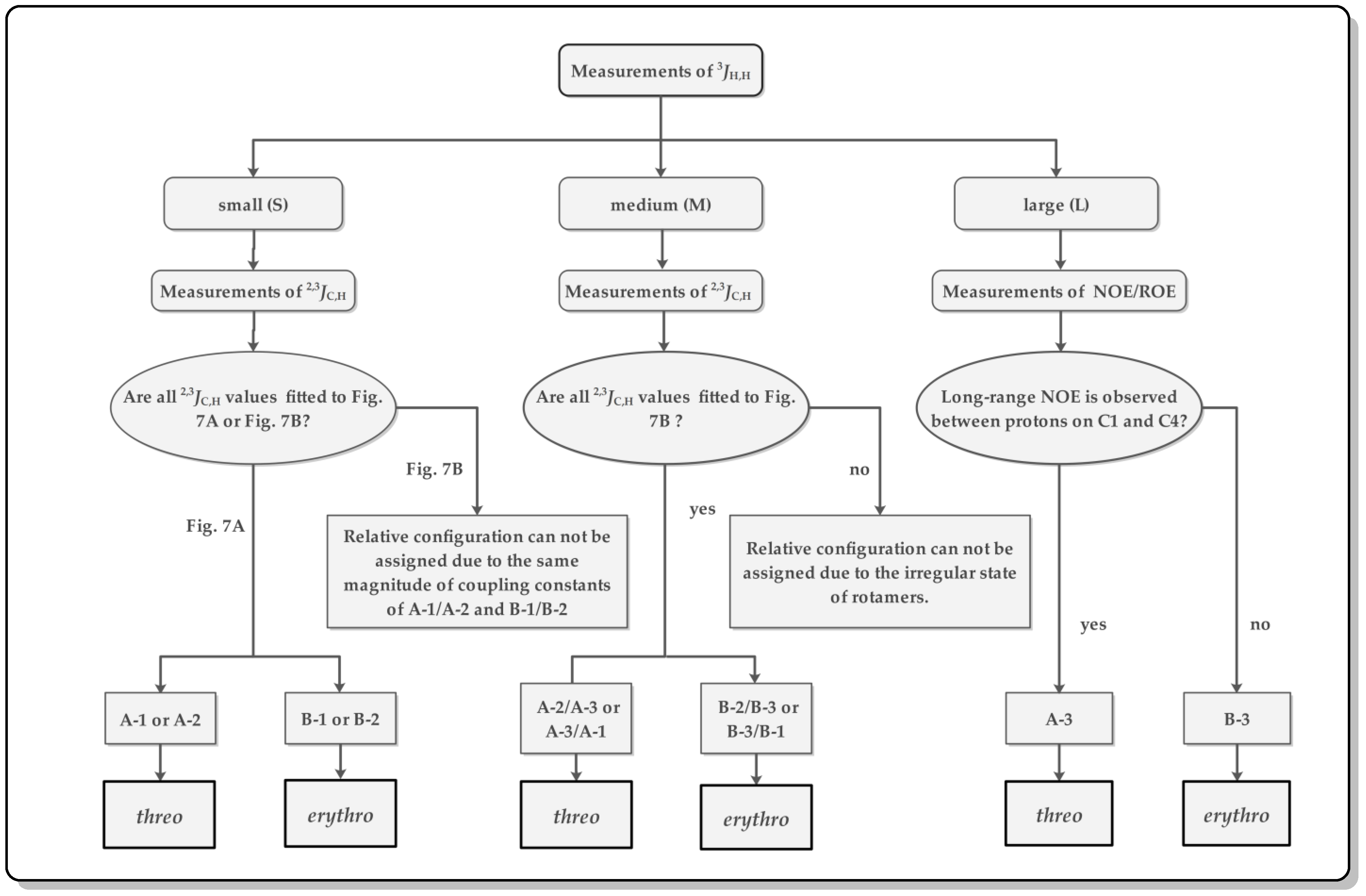
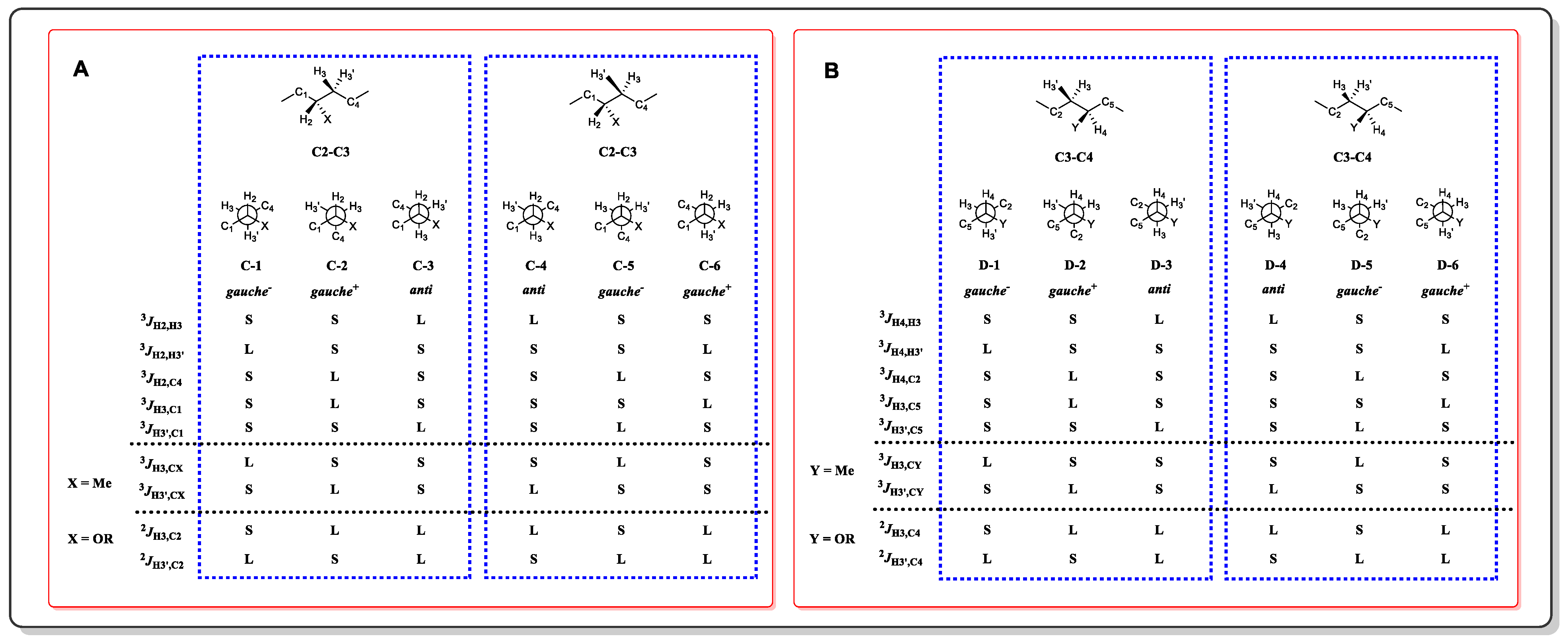
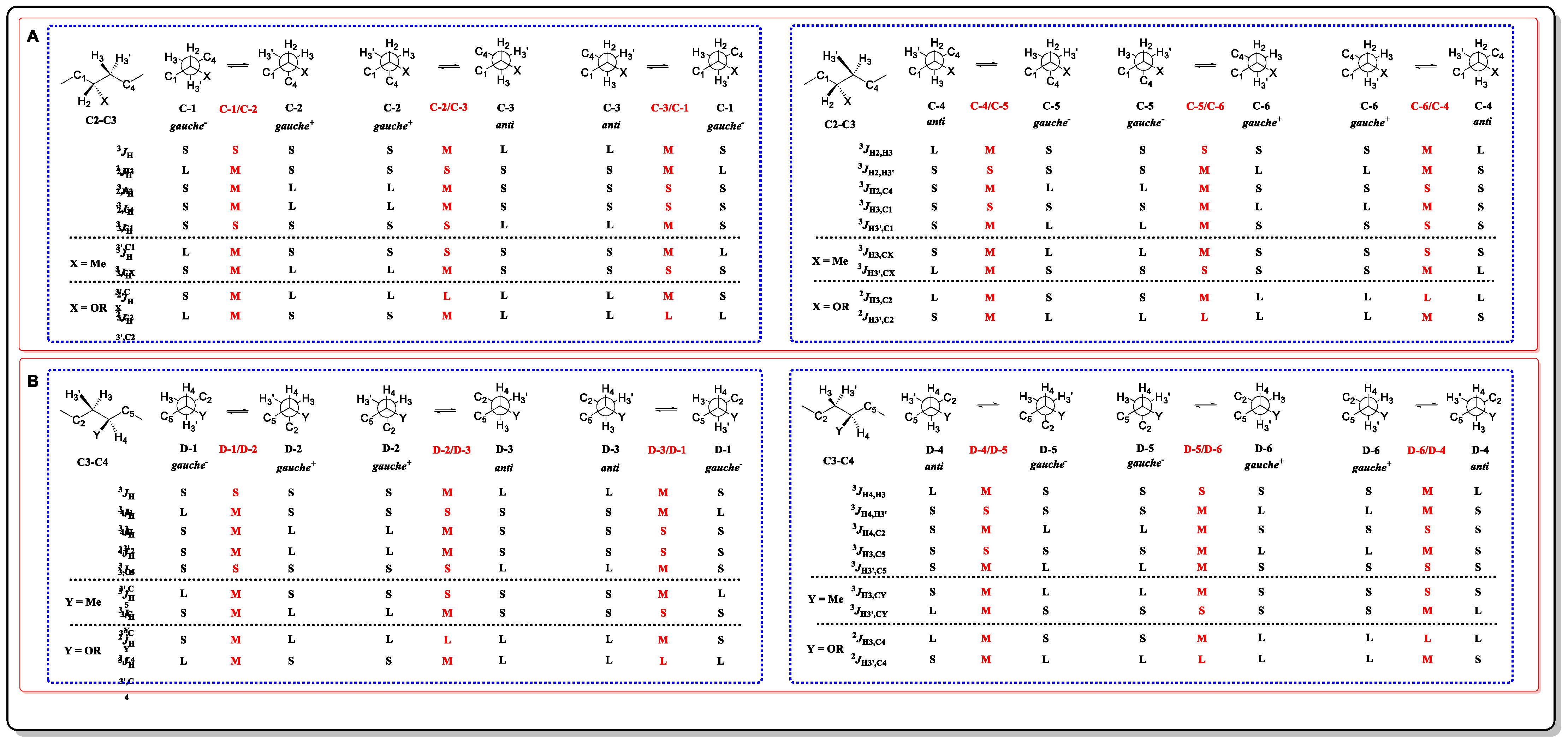
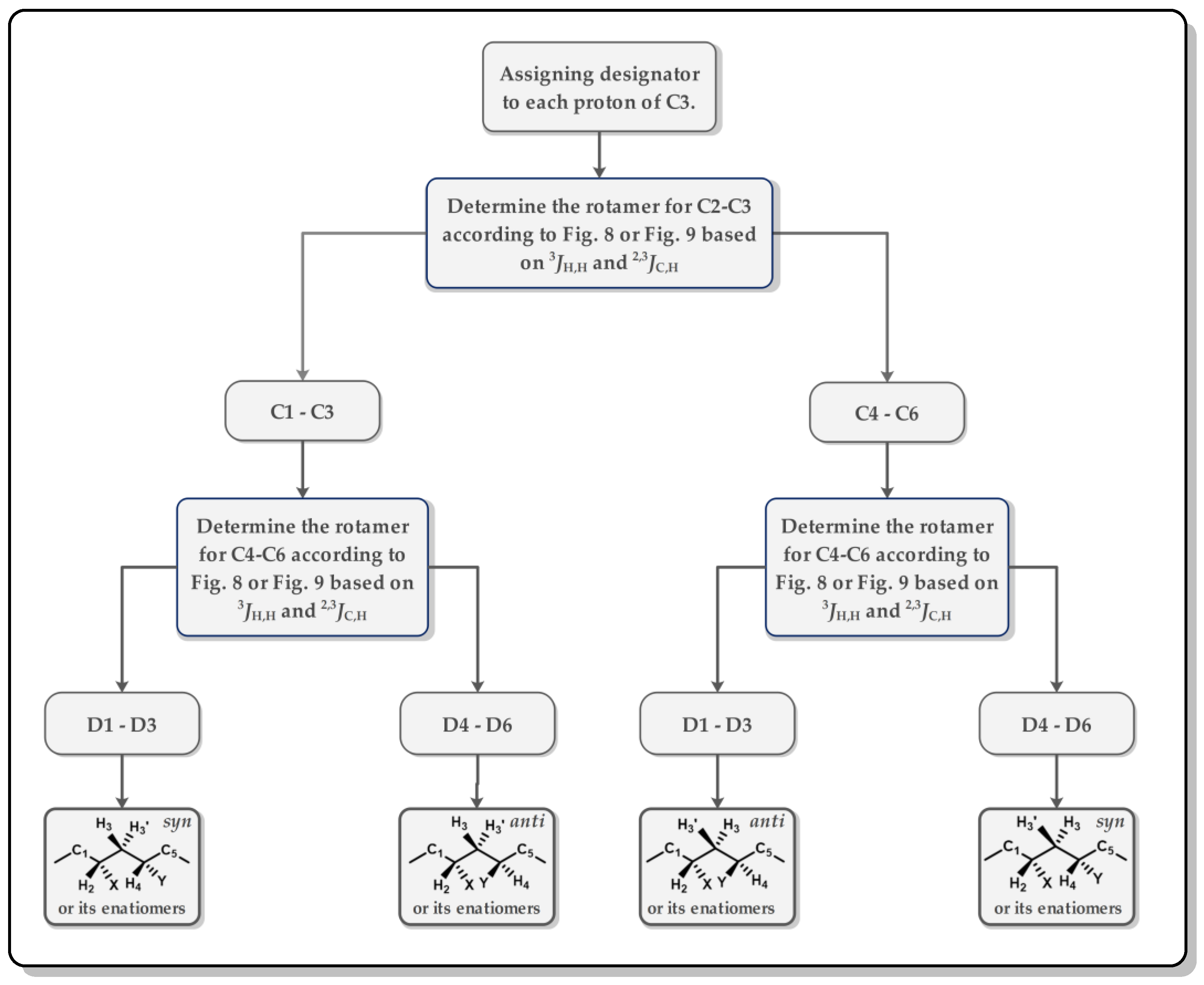
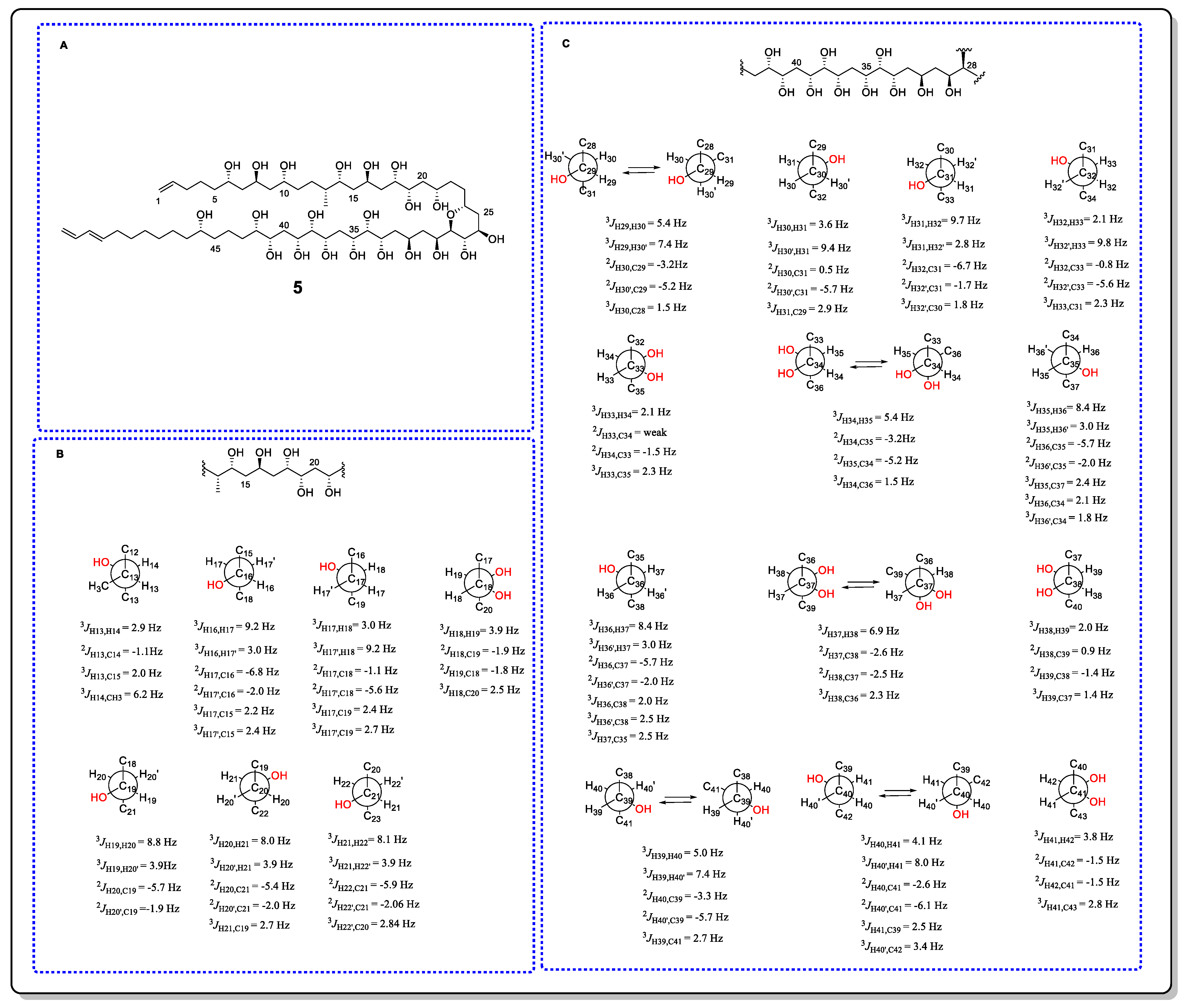
3.1. J-Based Configuration Analysis (JBCA)
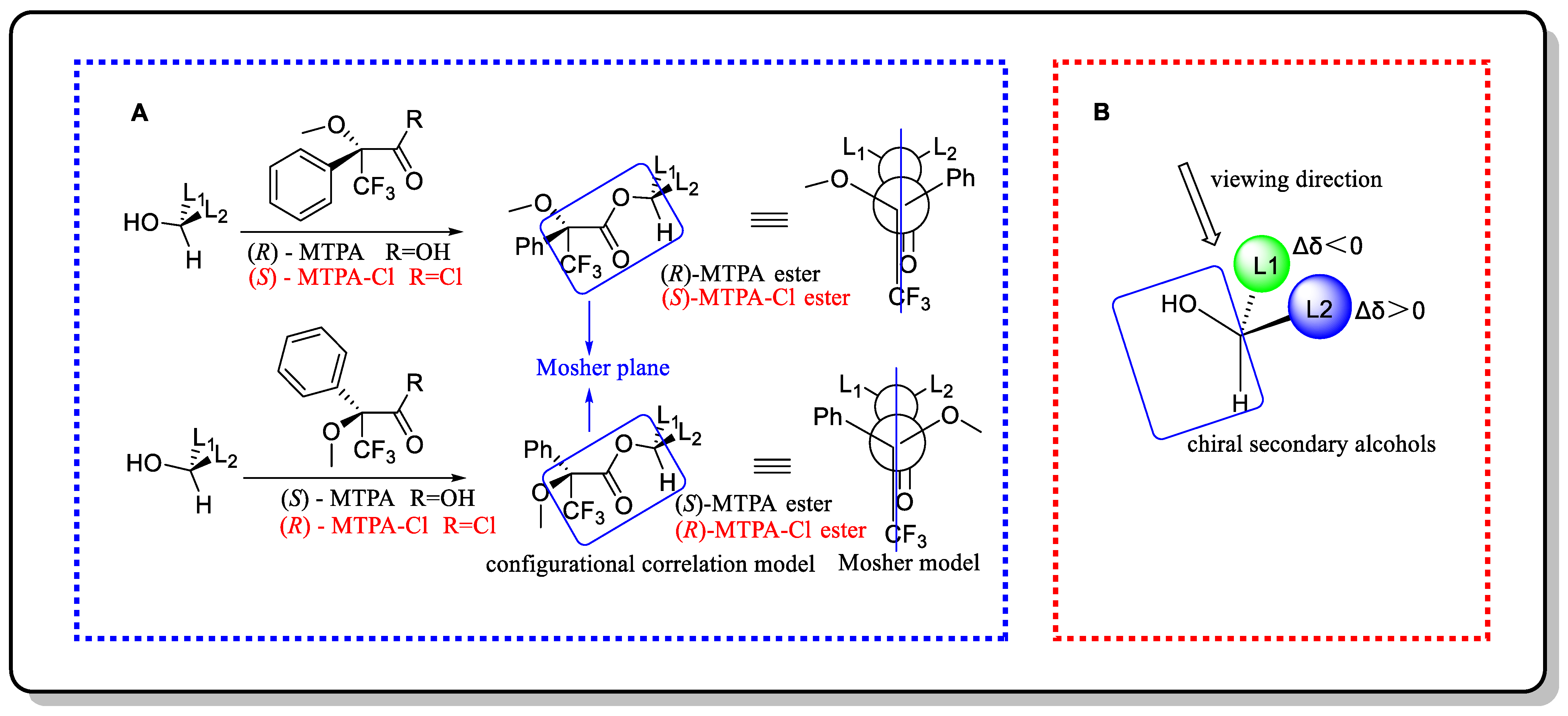
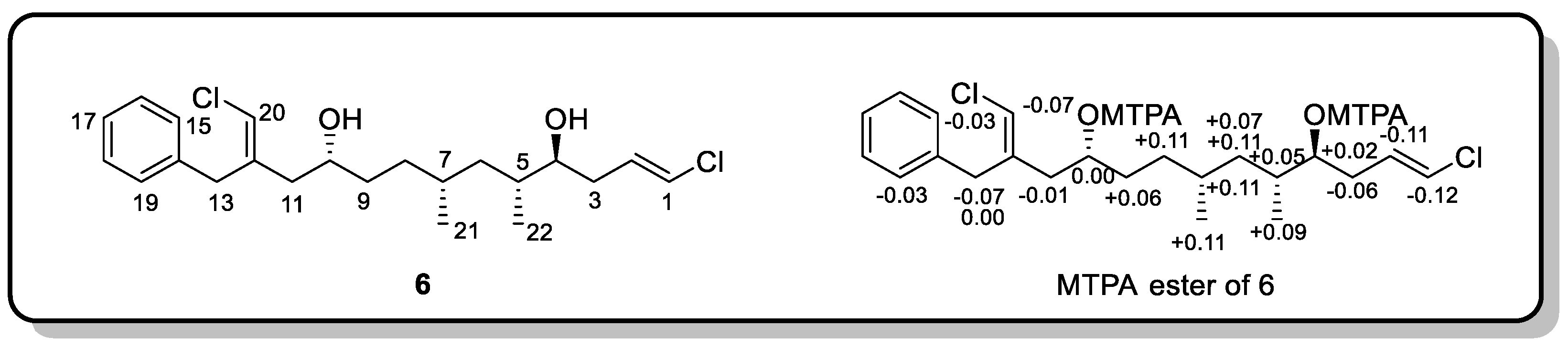
3.2. Mosher’s Method
4. Circular Dichroism (CD)-Based Methods
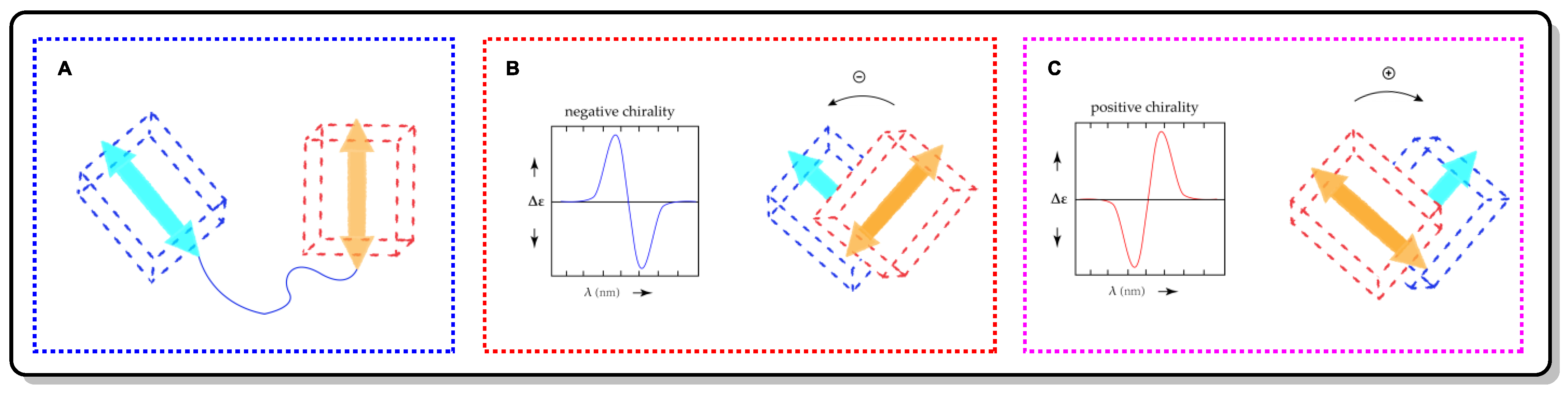
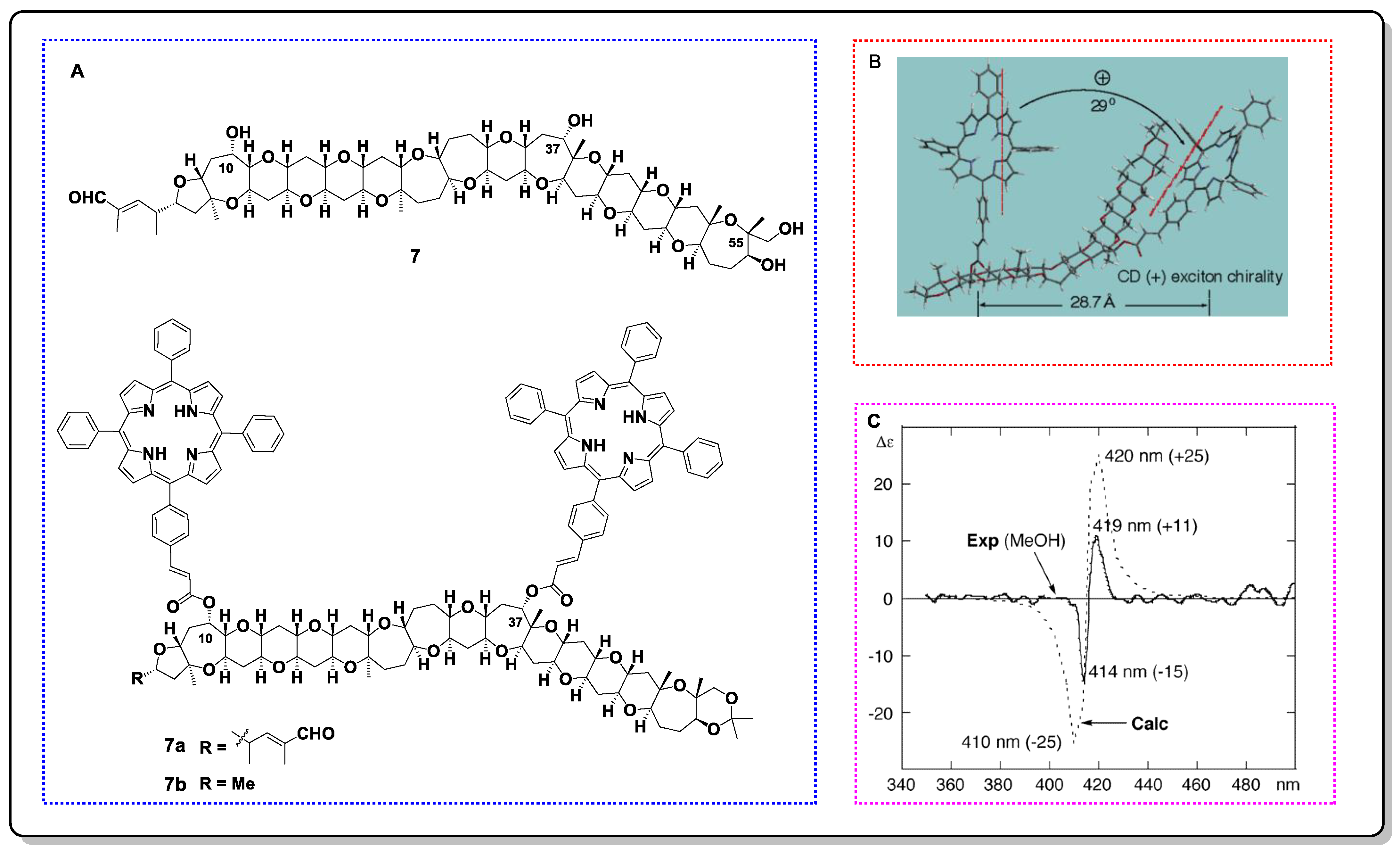
4.1. Exciton Chirality CD (ECCD)
4.2. Complexation-Induced CD (ICD)
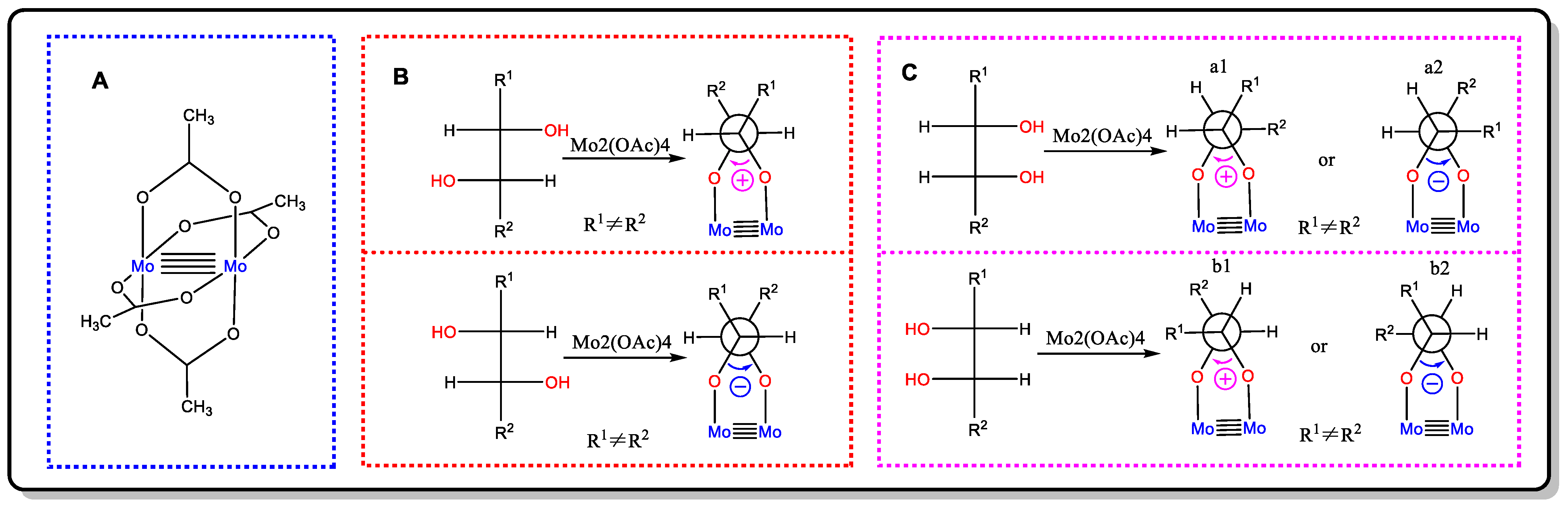
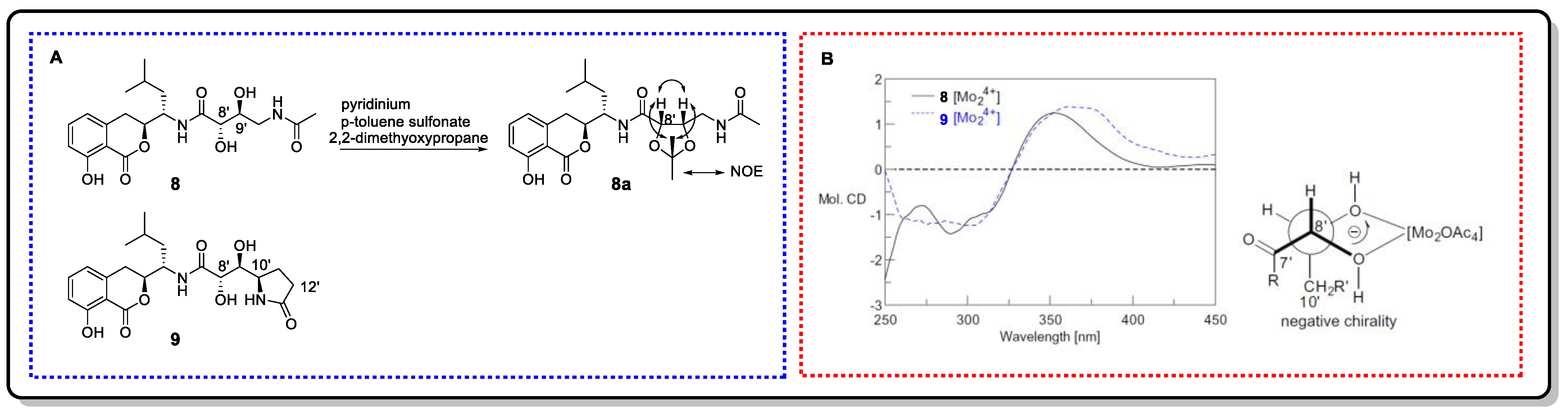
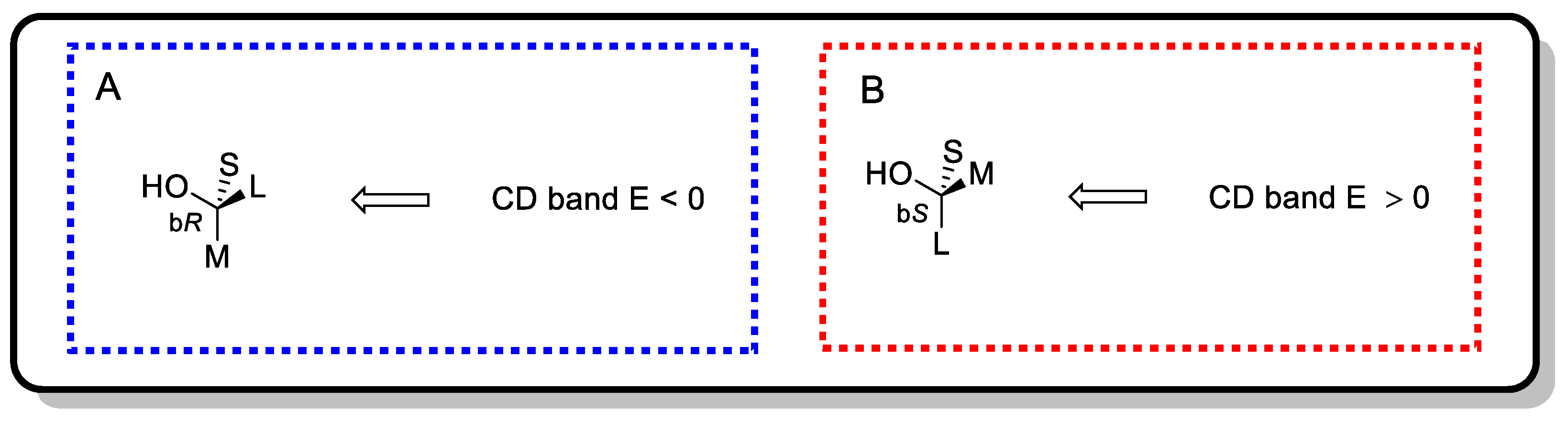
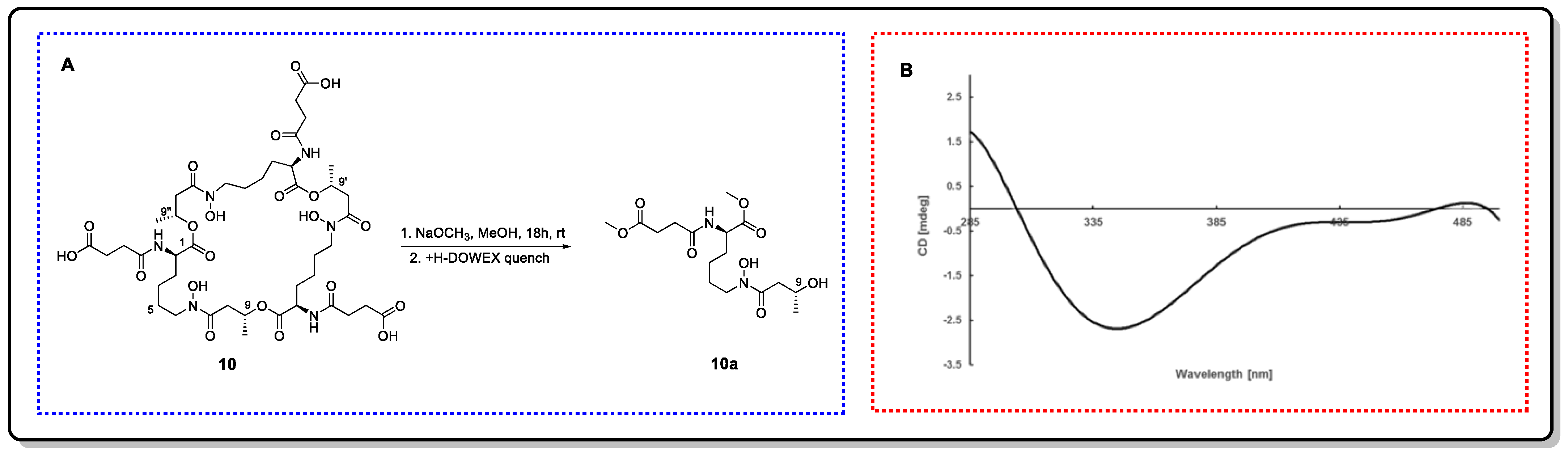
4.2.1. Snatzke’s Method (Mo2(OAc)4-Induced CD)
4.2.2. Rh2(OCOCF3)4-Induced CD
5. Quantum Computational Chemistry-Based Methods
5.1. NMR Calculations for Marine Natural Products
5.2. ECD Calculations for Marine Natural Products
5.3. VCD Calculations for Marine Natural Products
6. Chemical Transformation-Based Methods
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2021, 38, 362–413. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.Y.; Li, H.J.; Li, Q.Y.; Wu, Y.C. Application of marine natural products in drug research. Bioorganic Med. Chem. 2021, 35, 116058. [Google Scholar] [CrossRef] [PubMed]
- Cappello, E.; Nieri, P. From Life in the Sea to the Clinic: The marine drugs approved and under clinical trial. Life 2021, 11, 1390. [Google Scholar] [CrossRef]
- Ghareeb, M.A.; Tammam, M.A.; El-Demerdash, A.; Atanasov, A.G. Insights about clinically approved and preclinically investigated marine natural products. Curr. Res. Biotechnol. 2020, 2, 88–102. [Google Scholar] [CrossRef]
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.G.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2014, 31, 160–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molinski, T.F.; Morinaka, B.I. Integrated approaches to the configurational assignment of marine natural products. Tetrahedron 2012, 68, 9307–9343. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Chung-Davidson, Y.W.; Bussy, U.; Li, W.M. Recent advances and applications of experimental technologies in marine natural product research. Mar. Drugs 2015, 13, 2694–2713. [Google Scholar] [CrossRef] [Green Version]
- Grauso, L.; Teta, R.; Esposito, G.; Menna, M.; Mangoni, A. Computational prediction of chiroptical properties in structure elucidation of natural products. Nat. Prod. Rep. 2019, 36, 1005–1030. [Google Scholar] [CrossRef]
- Superchi, S.; Scafato, P.; Gorecki, M.; Pescitelli, G. Absolute configuration determination by quantum mechanical calculation of chiroptical spectra: Basics and applications to fungal metabolites. Curr. Med. Chem. 2018, 25, 287–320. [Google Scholar] [CrossRef]
- Valentín-Pérez, A.; Rosa, P.; Hillard, E.A.; Giorgi, M. Chirality determination in crystals. Chirality 2022, 34, 163–181. [Google Scholar] [CrossRef]
- Flack, H.D.; Bernardinelli, G. The use of X-ray crystallography to determine absolute configuration. Chirality 2008, 20, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.L.; Watkin, D.J. X-ray crystallography and chirality: Understanding the limitations. Tertahedron Asymmetry 2009, 20, 712–717. [Google Scholar] [CrossRef]
- Pescitelli, G.; Kurtán, T.; Flörke, U.; Krohn, K. Absolute structural elucidation of natural products—A focus on quantum-mechanical calculations of solid-state CD spectra. Chirality 2009, 21, E181–E201. [Google Scholar] [CrossRef] [PubMed]
- Shao, C.L.; Linington, R.G.; Balunas, M.J.; Centeno, A.; Boudreau, P.; Zhang, C.; Engene, N.; Spadafora, C.; Mutka, T.S.; Kyle, D.E. Bastimolide A, a potent antimalarial polyhydroxy macrolide from the marine cyanobacterium Okeania hirsuta. J. Org. Chem. 2015, 80, 7849–7855. [Google Scholar] [CrossRef] [PubMed]
- Burns, A.S.; Dooley, C.; Carlson, P.R.; Ziller, J.W.; Rychnovsky, S.D. Relative and absolute structure assignments of alkenes using crystalline osmate derivatives for X-ray analysis. Org. Lett. 2019, 21, 10125–10129. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.L.; Entzminger, K.C.; Hyun, J.; Kalyoncu, S.; Heaner, D.P.; Morales, I.A.; Sheppard, A.; Gumbart, J.C.; Maynard, J.A.; Lieberman, R.L. Structural and biophysical characterization of an epitope-specific engineered Fab fragment and complexation with membrane proteins: Implications for co-crystallization. Acta Crystallogr. Sect. D 2015, 71, 896–906. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, P.M.; Desiraju, G.R. Co-crystal formation and the determination of absolute configuration. CrystEngComm 2008, 10, 1747–1749. [Google Scholar] [CrossRef]
- Krupp, F.; Frey, W.; Richert, C. Absolute configuration of small molecules by co-crystallization. Angew. Chem. Int. Ed. 2020, 59, 15875–15879. [Google Scholar] [CrossRef]
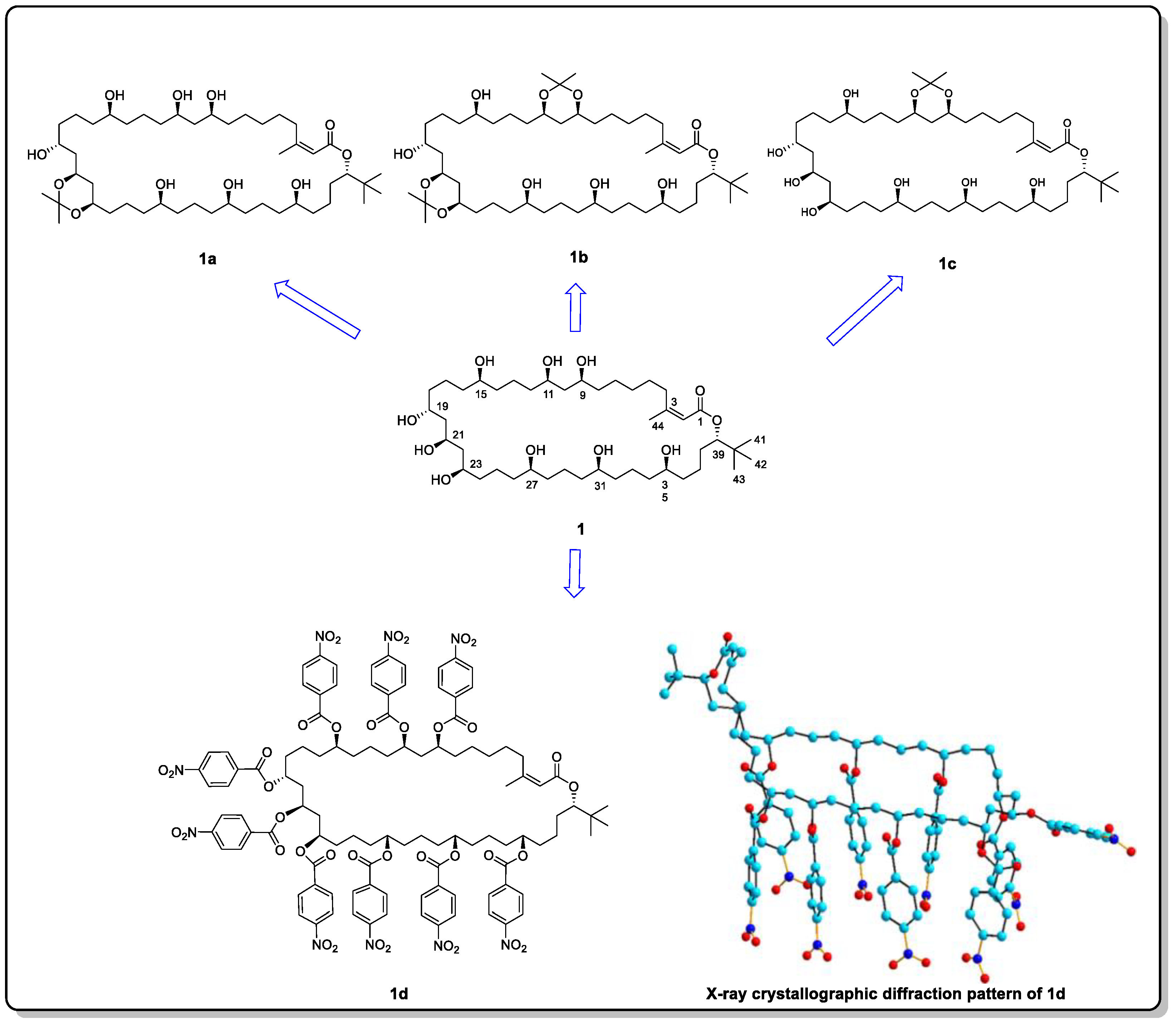
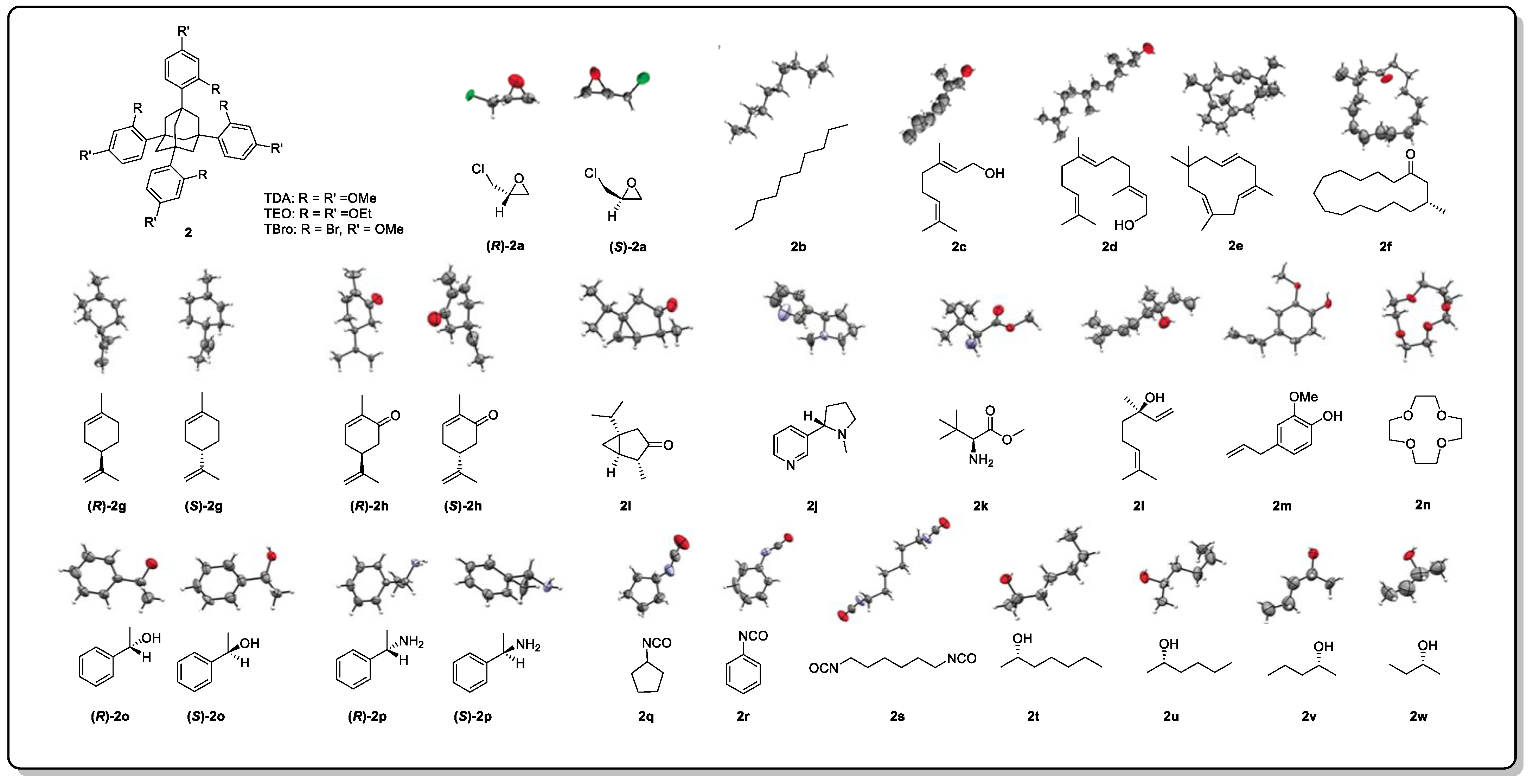
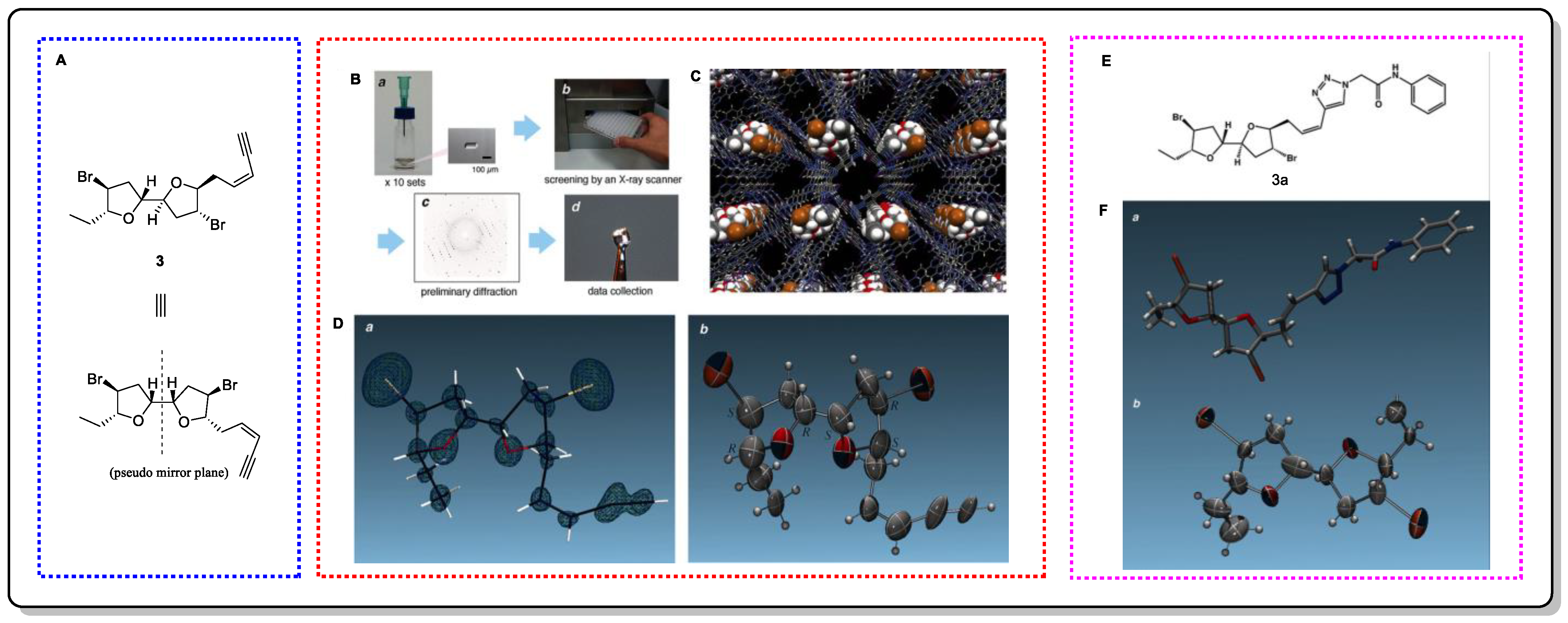
- Inokuma, Y.; Yoshioka, S.; Ariyoshi, J.; Arai, T.; Hitora, Y.; Takada, K.; Matsunaga, S.; Rissanen, K.; Fujita, M. X-ray analysis on the nanogram to microgram scale using porous complexes. Nature 2013, 495, 461–466, Erratum in Nature 2013, 501, 262. [Google Scholar] [CrossRef]
- Du, Q.; Peng, J.; Wu, P.; He, H. Review: Metal-organic framework based crystalline sponge method for structure analysis. Trend. Anal. Chem. 2018, 102, 290–310. [Google Scholar] [CrossRef]
- Zigon, N.; Duplan, V.; Wada, N.; Fujita, M. Crystalline sponge method: X-ray structure analysis of small molecules by post-orientation within porous crystals—Principle and proof-of-concept studies. Angew. Chem. Int. Ed. 2021, 60, 25204–25222. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.G.; Reiss, J.A. Elatenyne—A pyrano[3,2-β]pyranyl vinyl acetylene from the red alga. Aust. J. Chem. 1986, 39, 1401–1409. [Google Scholar] [CrossRef]
- Urban, S.; Brkljača, R.; Hoshino, M.; Lee, S.; Fujita, M. Determination of the absolute configuration of the pseudo-symmetric natural product elatenyne by the crystalline sponge method. Angew. Chem. Int. Ed. 2016, 128, 2728–2732. [Google Scholar] [CrossRef]
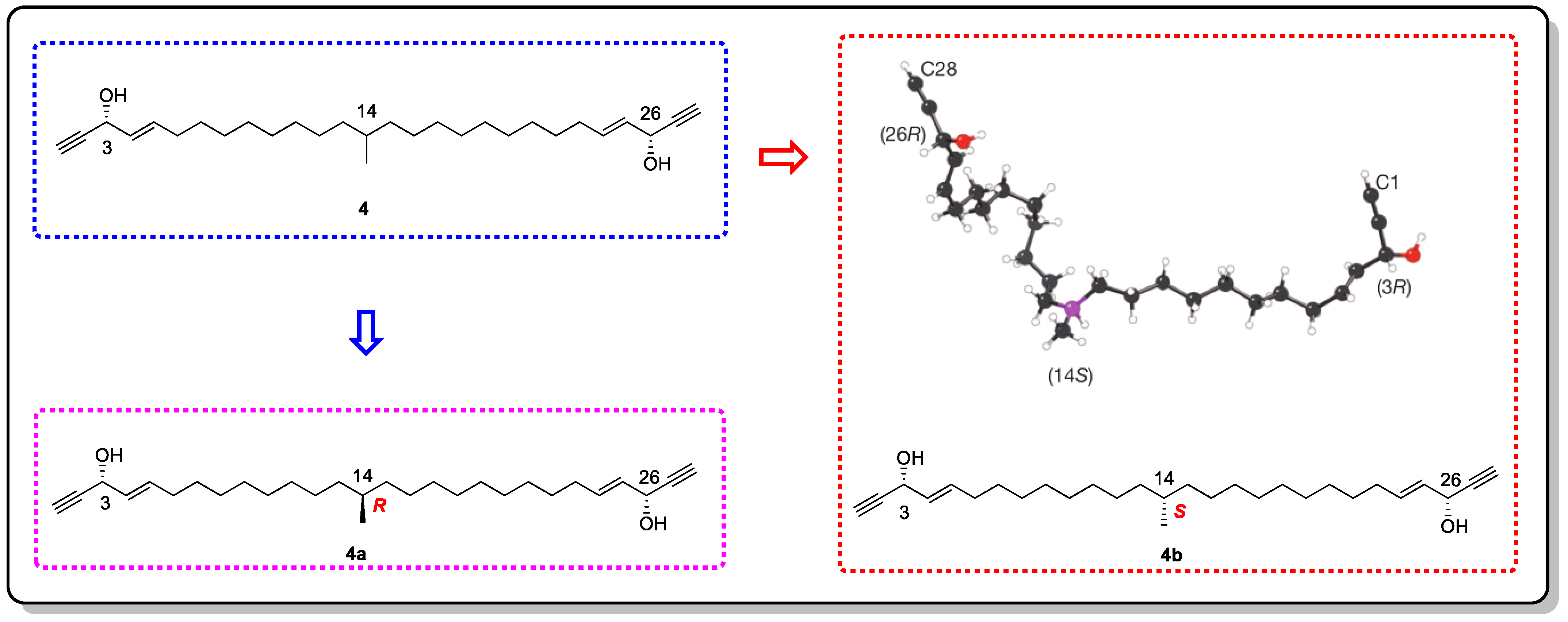
- Hitora, Y.; Takada, K.; Okada, S.; Matsunaga, S. Miyakosynes A–F, cytotoxic methyl branched acetylenes from a marine sponge Petrosia sp. Tetrahedron 2011, 67, 4530–4534. [Google Scholar] [CrossRef]
- Hitora, Y.; Takada, K.; Matsunaga, S. On the assignment of the absolute configuration at the isolated methyl branch in miyakosyne A, cytotoxic linear acetylene, from the deep-sea marine sponge Petrosia sp. Tetrahedron 2013, 69, 11070–11073. [Google Scholar] [CrossRef]
- Mori, K.; Akasaka, K.; Matsunaga, S. Chemoenzymatic synthesis and HPLC analysis of the stereoisomers of miyakosyne A [(4E,24E)-14-methyloctacosa-4,24-diene-1,27- diyne-3,26-diol], a cytotoxic metabolite of a marine sponge Petrosia sp., to determine the absolute configuration of its major component as 3R,14R,26R. Tetrahedron 2014, 70, 392–401. [Google Scholar]
- Matsumori, N.; Kaneno, D.; Murata, M.; Nakamura, H.; Tachibana, K. Stereochemical determination of acyclic structures based on carbon−Proton spin-coupling constants. A method of configuration analysis for natural products. J. Org. Chem. 1999, 64, 866–876. [Google Scholar] [CrossRef]
- Cimino, P.; Bifulco, G.; Evidente, A.; Abouzeid, M.; Riccio, R.; Gomez-Paloma, L. Extension of the J-Based configuration analysis to multiple conformer equilibria: An application to sapinofuranone A. Org. Lett. 2002, 4, 2779–2782. [Google Scholar] [CrossRef]
- Boot, C.M.; Gassner, N.C.; Compton, J.E.; Tenney, K.; Tamble, C.M.; Lokey, R.S.; Holman, T.R.; Crews, P. Pinpointing pseurotins from a marine-derived Aspergillus as tools for chemical genetics using a synthetic lethality yeast screen. J. Nat. Prod. 2007, 70, 1672–1675. [Google Scholar] [CrossRef]
- Menche, D.; Arikan, F.; Perlova, O.; Horstmann, N.; Ahlbrecht, W.; Wenzel, S.C.; Jansen, R.; Irschik, H.; Müller, R. Stereochemical determination and complex biosynthetic assembly of etnangien, a highly potent RNA polymerase inhibitor from the myxobacterium Sorangium cellulosum. J. Am. Chem. Soc. 2008, 130, 14234–14243. [Google Scholar] [CrossRef]
- Matsumori, N.; Murata, M.; Tachibana, K. Conformational analysis of natural products using long-range carbon-proton coupling constants: Three-dimensional structure of okadaic acid in solution. Tetrahedron 1995, 51, 12229–12238. [Google Scholar] [CrossRef]
- Bifulco, G.; Dambruoso, P.; Gomez-Paloma, L.; Riccio, R. Determination of relative configuration in organic compounds by NMR spectroscopy and computational methods. Chem. Rev. 2007, 107, 3744–3779. [Google Scholar] [CrossRef] [PubMed]
- Kurz, M.; Schmieder, P.; Kessler, H. HETLOC, an efficient method for determining heteronuclear long-range couplings with heteronuclei in natural abundance. Angew. Chem. Int. Ed. 1991, 30, 1329–1331. [Google Scholar] [CrossRef]
- Zhu, G.; Live, D.; Bax, A. Analysis of sugar puckers and glycosidic torsion angles in a DNA G-tetrad structure by heteronuclear three-bond J couplings. J. Am. Chem. Soc. 1994, 116, 8370–8371. [Google Scholar] [CrossRef]
- Zhu, G.; Bax, A. Measurement of long-range 1H-13C coupling constants from quantitative 2D heteronuclear multiple-quantum correlation spectra. J. Magn. Reson. Ser. A 1993, 104, 353–357. [Google Scholar] [CrossRef]
- Bassarello, C.; Bifulco, G.; Zampella, A.; D’Auria, M.V.; Riccio, R.; Gomez-Paloma, L. Stereochemical studies on sphinxolide: Advances in the J-based NMR determination of the relative configuration of flexible systems. Eur. J. Org. Chem. 2001, 2001, 39–44. [Google Scholar] [CrossRef]
- Hwang, B.S.; Yoon, E.Y.; Jeong, E.J.; Park, J.; Kim, E.H.; Rho, J.R. Determination of the absolute configuration of polyhydroxy compound ostreol B isolated from the dinoflagellate Ostreopsis cf. ovata. J. Org. Chem. 2018, 83, 194–202. [Google Scholar] [CrossRef]
- Dale, J.A.; Mosher, H.S. Nuclear magnetic resonance enantiomer regents. Configurational correlations via nuclear magnetic resonance chemical shifts of diastereomeric mandelate, O-methylmandelate, and .alpha.-methoxy-.alpha.- trifluoromethylphenylacetate (MTPA) esters. J. Am. Chem. Soc. 1973, 95, 512–519. [Google Scholar] [CrossRef]
- Sullivan, G.R.; Dale, J.A.; Mosher, H.S. Correlation of configuration and fluorine-19 chemical shifts of .alpha.-methoxy-.alpha.-trifluoromethylphenyl acetate derivatives. J. Org. Chem. 1973, 38, 2143–2147. [Google Scholar] [CrossRef]
- Latypov, S.K.; Seco, J.M.; Quinoa, E.; Riguera, R. Conformational structure and dynamics of arylmethoxyacetates: DNMR spectroscopy and aromatic shielding effect. J. Org. Chem. 1995, 60, 504–515. [Google Scholar] [CrossRef]
- Ohtani, I.; Kusumi, T.; Kashman, Y.; Kakisawa, H. A new aspect of the high-field NMR application of Mosher’s method. The absolute configuration of marine triterpene sipholenol A. J. Org. Chem. 1991, 56, 1296–1298. [Google Scholar] [CrossRef]
- Ohtani, I.; Kusumi, T.; Ishitsuka, M.O.; Kakisawa, H. Absolute configurations of marine diterpenes possessing a xenicane skeleton. An application of an advanced mosher’s method. Tetrahedron Lett. 1989, 30, 3147–3150. [Google Scholar] [CrossRef]
- Hoye, T.R.; Jeffrey, C.S.; Shao, F. Mosher ester analysis for the determination of absolute configuration of stereogenic (chiral) carbinol carbons. Nat. Protoc. 2007, 2, 2451–2458. [Google Scholar] [CrossRef]
- Ohtani, I.; Kusumi, T.; Kashman, Y.; Kakisawa, H. High-field FT NMR application of Mosher’s method. The absolute configurations of marine terpenoids. J. Am. Chem. Soc. 1991, 113, 4092–4096. [Google Scholar] [CrossRef]
- Sakamoto, T.; Kondo, Y.; Sato, S.; Yamanaka, H. Total synthesis of a marine alkaloid, rigidin. Tetrahedron Lett. 1994, 35, 2919–2920. [Google Scholar] [CrossRef]
- Teng, R.W.; Shen, P.; Wang, D.Z.; Yang, C.Z. Methods to determine the absolute stereochemistry of organic compounds by NMR spectroscopy. Chin. J. Magn. Reson. 2002, 19, 203–223. [Google Scholar]
- Fukushi, Y.; Yajima, C.; Mizutani, J. A new method for establishment of absolute configurations of secondary alcohols by NMR spectroscopy. Tetrahedron Lett. 1994, 35, 599–602. [Google Scholar] [CrossRef]
- Harada, K.; Shimizu, Y.; Fujii, K. A chiral anisotropic reagent for determination of the absolute configuration of a primary amino compound. Tetrahedron Lett. 1998, 39, 6245–6248. [Google Scholar] [CrossRef]
- Nagai, Y.; Kusumi, T. New chiral anisotropic reagents for determining the absolute configuration of carboxylic acids. Tetrahedron Lett. 1995, 36, 1853–1856. [Google Scholar] [CrossRef]
- Seco, J.M.; Quiñoá, E.; Riguera, R. The assignment of absolute configuration by NMR. Chem. Rev. 2004, 104, 17–117. [Google Scholar] [CrossRef]
- Latypov, S.K.; Seco, J.M.; Quiñoá, E.; Riguera, R. MTPA vs MPA in the determination of the absolute configuration of chiral alcohols by 1H NMR. J. Org. Chem. 1996, 61, 8569–8577. [Google Scholar] [CrossRef]
- Bertin, M.J.; Saurí, J.; Liu, Y.Z.; Via, C.W.; Roduit, A.F.; Williamson, R.T. Trichophycins B–F, chlorovinylidene-containing polyketides isolated from a cyanobacterial bloom. J. Org. Chem. 2018, 83, 13256–13266. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Sun, M. Electronic circular dichroism and Raman optical activity: Principle and applications. Appl. Spectrosc. Rev. 2021, 56, 553–587. [Google Scholar] [CrossRef]
- Mándi, A.; Kurtán, T. Applications of OR/ECD/VCD to the structure elucidation of natural products. Nat. Prod. Rep. 2019, 36, 889–918. [Google Scholar] [CrossRef] [PubMed]
- Harada, N.; Nakanishi, K. Exciton chirality method and its application to configurational and conformational studies of natural products. Acc. Chem. Res. 1972, 5, 257–263. [Google Scholar] [CrossRef]
- Pescitelli, G. ECD exciton chirality method today: A modern tool for determining absolute configurations. Chirality 2022, 34, 333–363. [Google Scholar] [CrossRef] [PubMed]
- Matile, S.; Berova, N.; Nakanishi, K.; Fleischhauer, J.; Woody, R.W. Structural studies by exciton coupled circular dichroism over a large distance: Porphyrin derivatives of steroids, dimeric steroids, and brevetoxin B. J. Am. Chem. Soc. 1996, 118, 5198–5206. [Google Scholar] [CrossRef]
- Harada, N.; Berova, N. Spectroscopic analysis: Exciton circular dichroism for chiral analysis. Compr. Chirality 2012, 8, 449–477. [Google Scholar]
- Satake, M.; Tanaka, Y.; Ishikura, Y.; Oshima, Y.; Naoki, H.; Yasumoto, T. Gymnocin-B with the largest contiguous polyether rings from the red tide dinoflagellate, Karenia (formerly Gymnodinium) mikimotoi. Tetrahedron Lett. 2005, 46, 3537–3540. [Google Scholar] [CrossRef]
- Tanaka, K.; Itagaki, Y.; Satake, M.; Naoki, H.; Yasumoto, T.; Nakanishi, K.; Berova, N. Three challenges toward the assignment of absolute configuration of gymnocin-B. J. Am. Chem. Soc. 2005, 127, 9561–9570. [Google Scholar] [CrossRef] [Green Version]
- Snatzke, G.; Wagner, U.; Wolff, H.P. Circular dichroism—LXXV1: Cottonogenic derivatives of chiral bidentate ligands with the complex [Mo2 (O2CCH3)4]. Tetrahedron 1981, 37, 349–361. [Google Scholar] [CrossRef]
- Górecki, M.; Jabłońska, E.; Kruszewska, A.; Suszczyńska, A.; Urbańczyk-Lipkowska, Z.; Gerards, M.; Morzycki, J.W.; Szczepek, W.J.; Frelek, J. Practical method for the absolute configuration assignment of tert/tert 1,2-diols using their complexes with Mo2(OAc)4. J. Org. Chem. 2007, 72, 2906–2916. [Google Scholar] [CrossRef] [PubMed]
- Frelek, J.; Ruśkowska, P.; Suszczyńska, A.; Szewczyk, K.; Osuch, A.; Jarosz, S.; Jagodziński, J. Configurational assignment of sugar erythro-1,2-diols from their electronic circular dichroism spectra with dimolybdenum tetraacetate. Tetrahedron Asymmetry 2008, 19, 1709–1713. [Google Scholar] [CrossRef]
- Bari, D.; Pescitelli, G.; Pratelli, C.; Pini, D.; Salvadori, P. Determination of absolute configuration of acyclic 1,2-diols with Mo2(OAc)4. 1′ Snatzke’s Method Revisited. J. Org. Chem. 2001, 66, 4819–4825. [Google Scholar] [CrossRef]
- Bai, J.; Liu, D.; Yu, S.; Proksch, P.; Lin, W. Amicoumacins from the marine-derived bacterium Bacillus sp. with the inhibition of NO production. Tetrahedron Lett. 2014, 55, 6286–6291. [Google Scholar] [CrossRef]
- Gerards, M.; Snatzke, G. Circular dichroism, XCIII determination of the absolute configuration of alcohols, olefins, epoxides, and ethers from the CD of their “in situ” complexes with [Rh2(O2CCF3)4]. Tetrahedron Asymmetry 1990, 1, 221–236. [Google Scholar] [CrossRef]
- Frelek, J.; Szczepek, W.J. [Rh2(OCOCF3)4] as an auxiliary chromophore in chiroptical studies on steroidal alcohols. Tetrahedron Asymmetry 1999, 10, 1507–1520. [Google Scholar] [CrossRef]
- Frelek, J.; Jagodziński, J.; Meyer-Figge, H.; Sheldrick, W.S.; Wieteska, E.; Szczepek, W.J. Chiroptical properties of binuclear rhodium complexes of lanostane alcohols. Chirality 2001, 13, 313–321. [Google Scholar] [CrossRef]
- Zhang, F.; Barns, K.; Hoffmann, F.M.; Braun, D.R.; Andes, D.R.; Bugni, T.S. Thalassosamide, a siderophore discovered from the marine-derived bacterium Thalassospira profundimaris. J. Nat. Prod. 2017, 80, 2551–2555. [Google Scholar] [CrossRef]
- Santoro, E.; Vergura, S.; Scafato, P.; Belviso, S.; Masi, M.; Evidente, A.; Superchi, S. Absolute configuration assignment to chiral natural products by biphenyl chiroptical probes: The case of the phytotoxins colletochlorin A and agropyrenol. J. Nat. Prod. 2020, 83, 1061–1068. [Google Scholar] [CrossRef]
- Marsico, G.; Calice, U.; Scafato, P.; Belviso, S.; Evidente, A.; Superchi, S. Computational approaches and use of chiroptical probes in the absolute configuration assignment to natural products by ECD spectroscopy: A 1,2,3-trihydroxy-p-menthane as a case study. Biomolecules 2022, 12, 421. [Google Scholar] [CrossRef] [PubMed]
- Wolf, C.; Bentley, K.W. Chirality sensing using stereodynamic probes with distinct electronic circular dichroism output. Chem. Soc. Rev. 2013, 42, 5408–5424. [Google Scholar] [CrossRef] [PubMed]
- Berova, N.; Pescitelli, G.; Petrovic, A.G.; Proni, G. Probing molecular chirality by CD-sensitive dimeric metalloporphyrin hosts. Chem. Commun. 2009, 40, 5958–5980. [Google Scholar] [CrossRef] [PubMed]
- Barone, G.; Gomez-Paloma, L.; Duca, D.; Silvestri, A.; Riccio, R.; Bifulco, G. Structure validation of natural products by quantum-mechanical GIAO calculations of 13C NMR chemical shifts. Chemistry 2002, 8, 3233–3239. [Google Scholar] [CrossRef]
- Meiler, J.; Sanli, E.; Junker, J.; Meusinger, R.; Lindel, T.; Will, M.; Maier, W.; Köck, M. Validation of structural proposals by substructure analysis and 13C NMR chemical shift prediction. J. Chem. Inf. Comput. Sci. 2002, 42, 241–248. [Google Scholar] [CrossRef]
- Bagno, A. Complete prediction of the 1H NMR spectrum of organic molecules by DFT calculations of chemical shifts and spin–Spin coupling constants. Chem. Eur. J. 2001, 7, 1652–1661. [Google Scholar] [CrossRef]
- Forsyth, D.A.; Sebag, A.B. Computed 13C NMR chemical shifts via empirically scaled GIAO shieldings and molecular mechanics geometries. Conformation and configuration from 13C shifts. J. Am. Chem. Soc. 1997, 119, 9483–9494. [Google Scholar] [CrossRef]
- Lodewyk, M.W.; Siebert, M.R.; Tantillo, D.J. Computational prediction of 1H and 13C chemical shifts: A useful tool for natural product, mechanistic, and synthetic organic chemistry. Chem. Rev. 2012, 112, 1839–1862. [Google Scholar] [CrossRef]
- Costa, F.L.P.; Albuquerque, A.C.F.; Fiorot, R.G.; Lião, L.M.; Martorano, L.H.; Mota, G.V.S.; Valverde, A.L.; Carneiro, J.W.M.; Junior, F.M.S. Structural characterisation of natural products by means of quantum chemical calculations of NMR parameters: New insights. Org. Chem. Front. 2021, 8, 2019–2058. [Google Scholar] [CrossRef]
- Kim, C.S.; Oh, J.; Lee, T.H. Structure elucidation of small organic molecules by contemporary computational chemistry methods. Arch. Pharm. Res. 2020, 43, 1114–1127. [Google Scholar] [CrossRef]
- Marcarino, M.O.; Cicetti, S.; Zanardi, M.M.; Sarotti, A.M. A critical review on the use of DP4+ in the structural elucidation of natural products: The good, the bad and the ugly. A practical guide. Nat. Prod. Rep. 2022, 39, 58–76. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.G.; Goodman, J.M. Assigning stereochemistry to single diastereoisomers by GIAO NMR calculation: The DP4 probability. J. Am. Chem. Soc. 2010, 132, 12946–12959. [Google Scholar] [CrossRef] [PubMed]
- Ermanis, K.; Parkes, K.E.; Agback, T.; Goodman, J.M. Expanding DP4: Application to drug compounds and automation. Org. Biomol. Chem. 2016, 14, 3943–3949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howarth, A.; Ermanis, K.; Goodman, J.M. DP4-AI automated NMR data analysis: Straight from spectrometer to structure. Chem. Sci. 2020, 11, 4351–4359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimblat, N.; Zanardi, M.M.; Sarotti, A.M. Beyond DP4: An improved probability for the stereochemical assignment of isomeric compounds using quantum chemical calculations of NMR shifts. J. Org. Chem. 2015, 80, 12526–12534. [Google Scholar] [CrossRef] [PubMed]
- Grimblat, N.; Gavín, J.A.; Daranas, A.H.; Sarotti, A.M. Combining the power of J coupling and DP4 analysis on stereochemical assignments: The J-DP4 methods. Org. Lett. 2019, 21, 4003–4007. [Google Scholar] [CrossRef]
- Zanardi, M.M.; Marcarino, M.O.; Sarotti, A.M. Redefining the impact of boltzmann analysis in the stereochemical assignment of polar and flexible molecules by NMR calculations. Org. Lett. 2020, 22, 52–56. [Google Scholar] [CrossRef]
- Hehre, W.; Klunzinger, P.; Deppmeier, B.; Driessen, A.; Uchida, N.; Hashimoto, M.; Fukushi, E.; Takata, Y. Efficient protocol for accurately calculating 13C chemical shifts of conformationally flexible natural products: Scope, Assessment, and Limitations. J. Nat. Prod. 2019, 82, 2299–2306. [Google Scholar] [CrossRef]
- Cornilescu, G.; Alvarenga, R.F.R.; Wyche, T.P.; Bugni, T.S.; Gil, R.R.; Cornilescu, C.C.; Westler, W.M.; Markley, J.L.; Schwieters, C.D. Progressive stereo locking (PSL): A residual dipolar coupling based force field method for determining the relative configuration of natural products and other small molecules. ACS Chem. Biol. 2017, 12, 2157–2163. [Google Scholar] [CrossRef] [Green Version]
- Tzvetkova, P.; Sternberg, U.; Gloge, T.; Navarro-Vázquez, A.; Luy, B. Configuration determination by residual dipolar couplings: Accessing the full conformational space by molecular dynamics with tensorial constraints. Chem. Sci. 2019, 10, 8774–8791. [Google Scholar] [CrossRef]
- Immel, S.; Köck, M.; Reggelin, M. NMR-based configurational assignments of natural products: Gibbs sampling and bayesian inference using floating chirality distance geometry calculations. Mar. Drugs 2022, 20, 14. [Google Scholar] [CrossRef] [PubMed]
- Hallwass, F.; Schmidt, M.; Sun, H.; Mazur, A.; Kummerlöwe, G.; Luy, B.; Navarro-Vázquez, A.; Griesinger, C.; Reinscheid, U.M. Residual chemical shift anisotropy (RCSA): A tool for the analysis of the configuration of small molecules. Angew. Chem. Int. Ed. 2011, 50, 9487–9490. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Saurí, J.; Mevers, E.; Peczuh, M.W.; Hiemstra, H.; Clardy, J.; Martin, G.E.; Williamson, R.T. Unequivocal determination of complexmolecular structures using anisotropic NMR measurements. Science 2017, 356, eaam5349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.W.; Liu, H.; Qiu, F.; Wang, X.J.; Lei, X.X. Residual dipolar couplings in structure determination of natural products. Nat. Prod. Bioprospect. 2018, 8, 279–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Navarro-Vázquez, A.; Gil, R.R.; Griesinger, C.; Martin, G.E.; Williamson, R.T. Application of anisotropic NMR parameters to the confirmation of molecular structure. Nat. Protoc. 2019, 14, 217–247. [Google Scholar] [CrossRef] [PubMed]
- Li, X.L.; Chi, L.P.; Navarro-Vázquez, A.; Hwang, S.; Schmieder, P.; Li, X.M.; Li, X.; Yang, S.Q.; Lei, X.X.; Wang, B.G.; et al. Stereochemical elucidation of natural products from residual chemical shift anisotropies in a liquid crystalline phase. J. Am. Chem. Soc. 2020, 142, 2301–2309. [Google Scholar] [CrossRef] [Green Version]
- Pescitelli, G.; Bruhn, T. Good computational practice in the assignment of absolute configurations by TDDFT calculations of ECD spectra. Chirality 2016, 28, 466–474. [Google Scholar] [CrossRef] [Green Version]
- Huang, R.; Liang, L.; Guo, Y. Time-dependent density functional theory electronic circular dichroism (TDDFT ECD) calculation as a promising tool to determine the absolute configuration of natural products. J. Int. Pharm. Res. 2015, 42, 686–698. [Google Scholar]
- McCann, D.M.; Stephens, P.J. Determination of absolute configuration using density functional theory calculations of optical rotation and electronic circular dichroism: Chiral alkenes. J. Org. Chem. 2006, 71, 6074–6098. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, Q.; Li, S.; Cui, H.; Sun, Z.; Chen, D.; Lu, Y.; Liu, H.; Zhang, W. Polypropionate derivatives with mycobacterium tuberculosis protein tyrosine phosphatase B inhibitory activities from the deep-sea-derived fungus Aspergillus fischeri FS452. J. Nat. Prod. 2019, 82, 3440–3449. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Pan, J.J. The determination of the absolute configurations of chiral molecules using vibrational circular dichroism (VCD) spectroscopy. Chirality 2008, 20, 643–663. [Google Scholar] [CrossRef] [PubMed]
- Felippe, L.G.; Batista, J.M.; Baldoqui, D.C.; Nascimento, I.R.; Kato, M.J.; He, Y.; Nafie, L.A.; Furlan, M. VCD to determine absolute configuration of natural product molecules: Secolignans from Peperomia blanda. Org. Biomol. Chem. 2012, 10, 4208–4214. [Google Scholar] [CrossRef] [PubMed]
- Gussem, E.D.; Herrebout, W.; Specklin, S.; Meyer, C.; Cossy, J.; Bultinck, P. Strength by joining methods: Combining synthesis with NMR, IR, and Vibrational Circular Dichroism spectroscopy for the determination of the relative configuration in hemicalide. Chem. Eur. J. 2014, 20, 17385–17394. [Google Scholar] [CrossRef] [PubMed]
- MacGregor, C.I.; Han, B.Y.; Goodman, J.M.; Paterson, I. Toward the stereochemical assignment and synthesis of hemicalide: DP4f GIAO-NMR analysis and synthesis of a reassigned C16–C28 subunit. Chem. Commun. 2016, 52, 4632–4635. [Google Scholar] [CrossRef] [Green Version]
- Han, B.Y.; Lam, N.Y.S.; MacGregor, C.I.; Goodman, J.M.; Paterson, I. A synthesis-enabled relative stereochemical assignment of the C1–C28 region of hemicalide. Chem. Commun. 2018, 54, 3247–3250. [Google Scholar] [CrossRef] [Green Version]
- Satake, M.; Murata, M.; Yasumoto, T.; Fujita, T.; Naoki, H. Amphidinol, a polyhydroxy-polyene antifungal agent with an unprecedented structure, from a marine dinoflagellate, Amphidinium klebsii. J. Am. Chem. Soc. 1991, 113, 9859–9861. [Google Scholar] [CrossRef]
- Murata, M.; Matsuoka, S.; Matsumori, N.; Paul, G.K.; Tachibana, K. Absolute configuration of amphidinol 3, the first complete structure determination from amphidinol homologues: Application of a new configuration analysis based on carbon-hydrogen spin-coupling constants. J. Am. Chem. Soc. 1999, 121, 870–871. [Google Scholar] [CrossRef]
- Wakamiya, Y.; Ebine, M.; Murayama, M.; Omizu, H.; Matsumori, N.; Murata, M.; Oishi, T. Synthesis and stereochemical revision of the C31–C67 fragment of amphidinol 3. Angew. Chem. Int. Ed. 2018, 57, 6060–6064. [Google Scholar] [CrossRef]
- Makarieva, T.N.; Ivanchina, N.V.; Stonik, V.A. Application of oxidative and reductive transformations in the structure determination of marine natural products. J. Nat. Prod. 2020, 83, 1314–1333. [Google Scholar] [CrossRef]
- Seibert, S.F.; Eguereva, E.; Krick, A.; Kehraus, S.; Voloshina, E.; Raabe, G.; Fleischhauer, J.; Leistner, E.; Wiese, M.; Prinz, H. Polyketides from the marine-derived fungus Ascochyta salicorniae and their potential to inhibit protein phosphatases. Org. Biomol. Chem. 2006, 4, 2233–2240. [Google Scholar]
- Kobayashi, J.; Sato, M.; Ishibashi, M. Amphidinolide J: A cytotoxic macrolide from the marine dinoflagellate Amphidinium sp. Determination of the absolute stereochemistry. J. Org. Chem. 1993, 58, 2645–2646. [Google Scholar] [CrossRef]
- Imperatore, C.; Luciano, P.; Aiello, A.; Vitalone, R.; Irace, C.; Santamaria, R.; Li, J.; Guo, Y.W.; Menna, M. Structure and configuration of phosphoeleganin, a protein tyrosine phosphatase 1B inhibitor from the mediterranean ascidian Sidnyum elegans. J. Nat. Prod. 2016, 79, 1144–1148. [Google Scholar] [CrossRef] [PubMed]
- Sarotti, A.M. Successful combination of computationally inexpensive GIAO 13C NMR calculations and artificial neural network pattern recognition: A new strategy for simple and rapid detection of structural misassignments. Org. Biomol. Chem. 2013, 11, 4847–4859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanardi, M.M.; Sarotti, A.M. GIAO C–H COSY simulations merged with artificial neural networks pattern recognition analysis. pushing the structural validation a step forward. J. Org. Chem. 2015, 80, 9371–9378. [Google Scholar] [CrossRef] [PubMed]
- Gruene, T.; Holstein, J.J. Establishing electron diffraction in chemical crystallography. Nat. Rev. Chem. 2021, 5, 660–668. [Google Scholar] [CrossRef]
- Jones, C.G.; Martynowycz, M.W. The CryoEM Method MicroED as a powerful tool for small molecule structure determination. ACS. Cent. Sci. 2018, 4, 1587–1592. [Google Scholar] [CrossRef]

























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
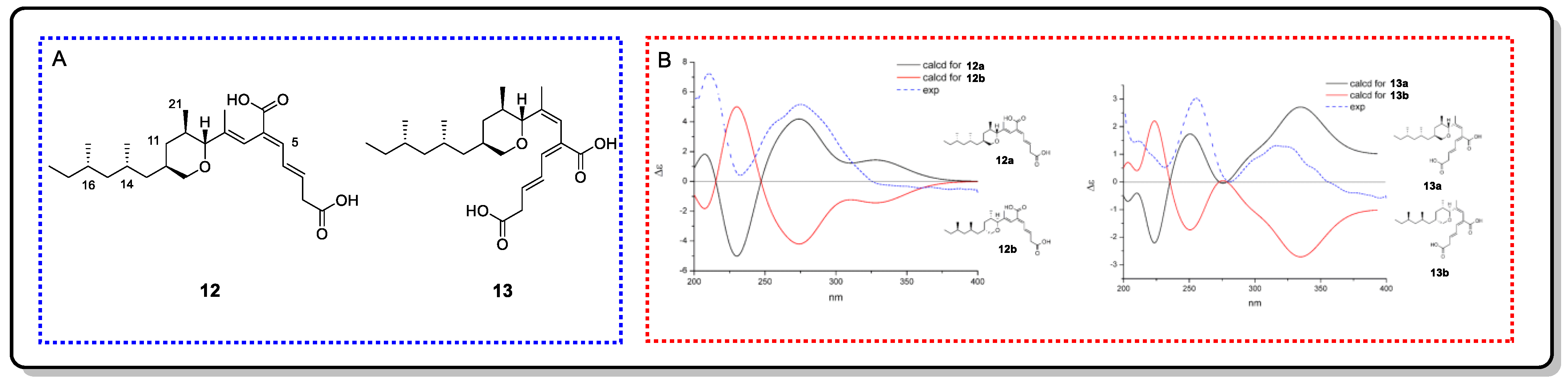{kind=link}
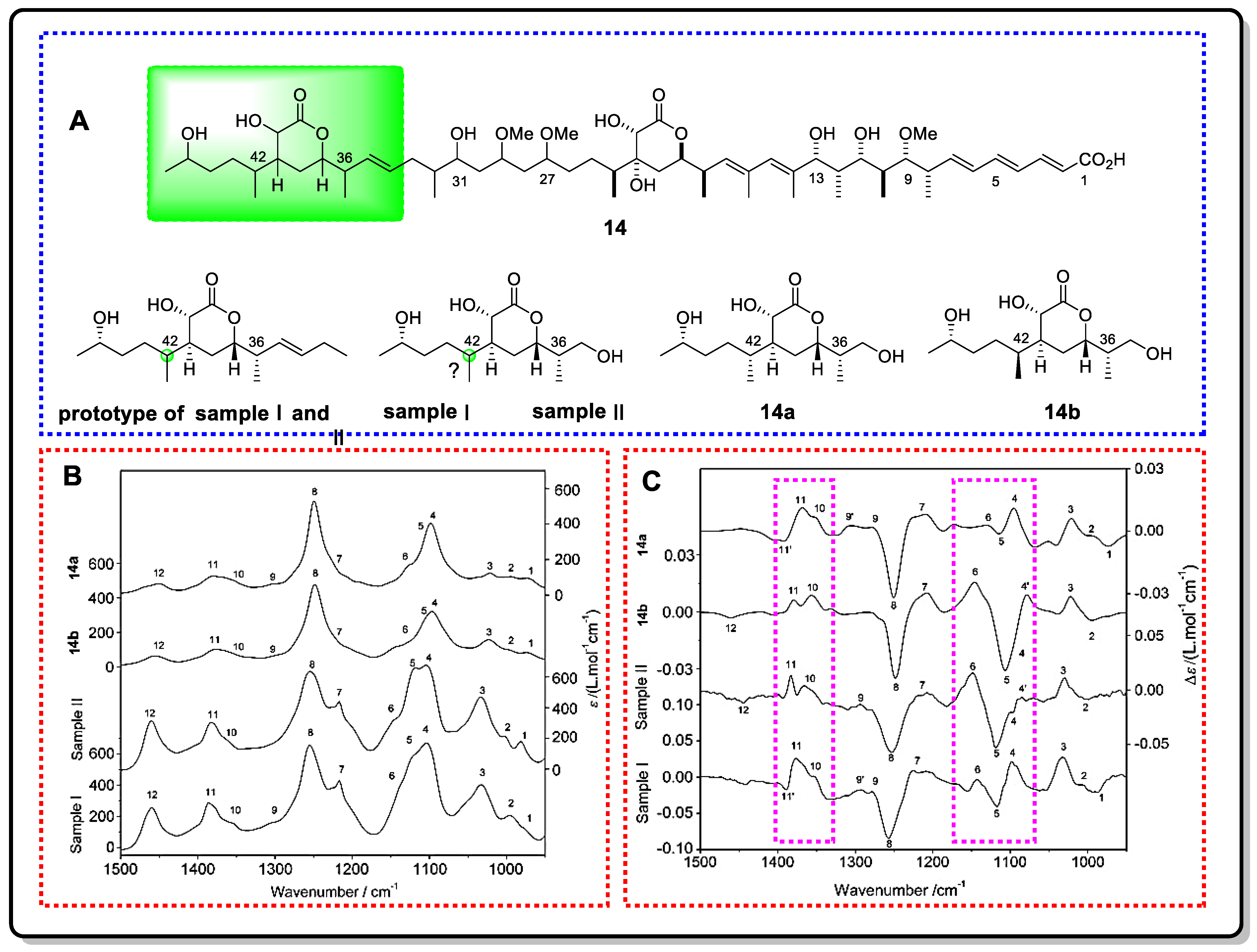{kind=link}
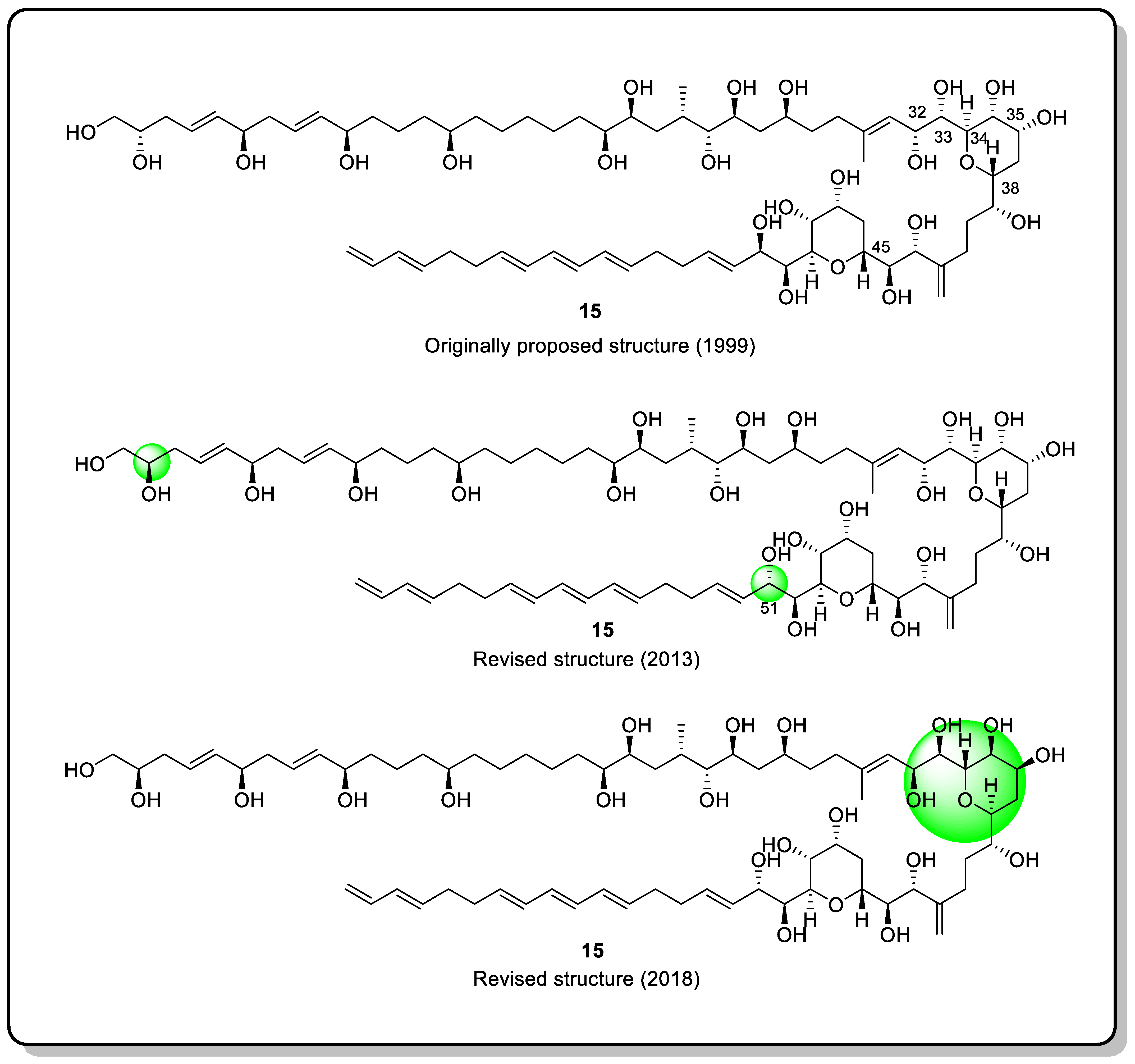{kind=link}
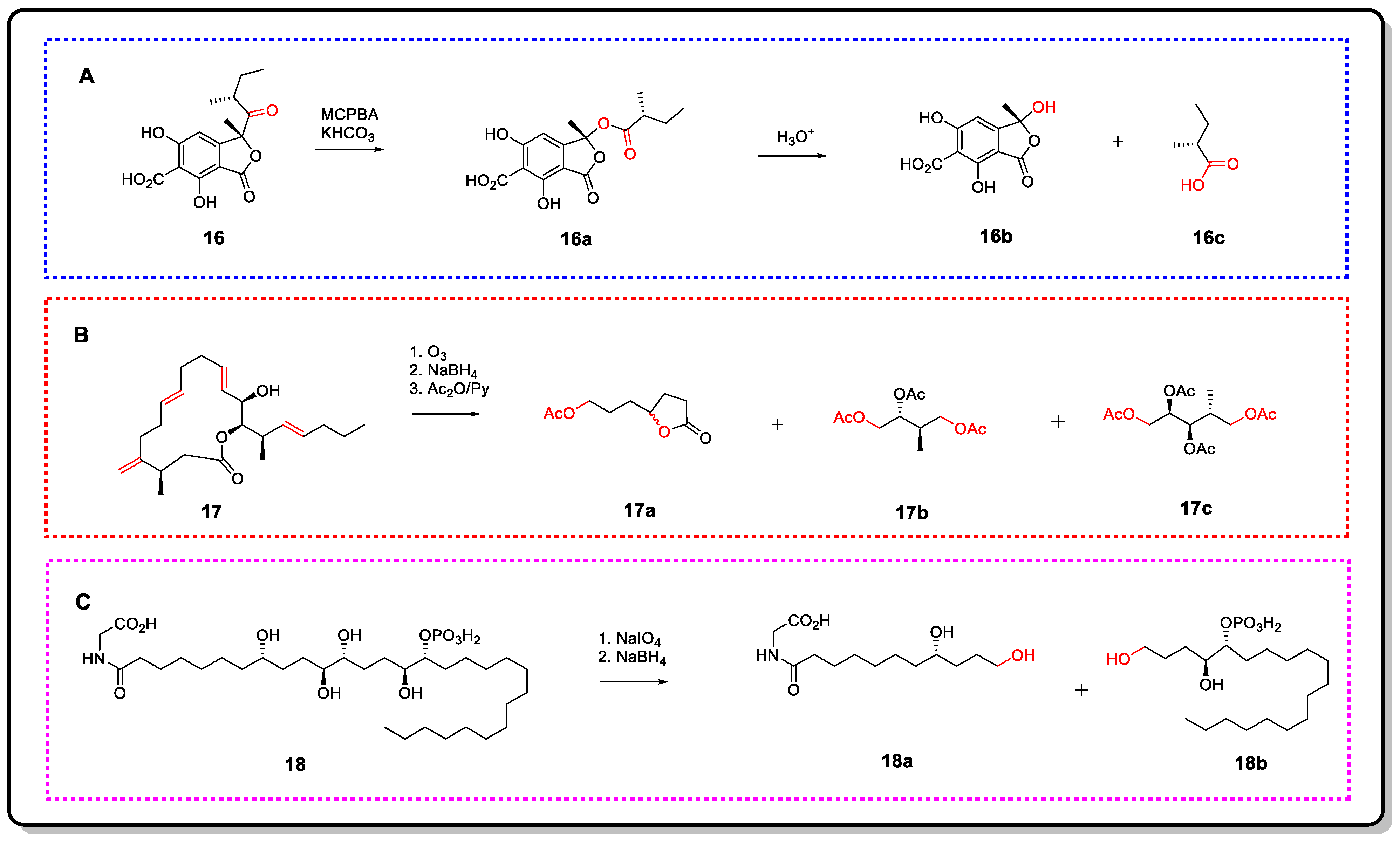{kind=link}
| 3JH,H | 3JH,C | |2JH,C| | ||||||
|---|---|---|---|---|---|---|---|---|
| None | Mono | Di | None | Mono | Di | Mono | Di | |
| S | 2–4 | 1–4 | 0–3 | 1–3 | 1–3 | 1–3 | 0–2 | 0–2 |
| M | 4–9 | 4–8 | 3–7 | 3–6 | 3–6 | 3–5 | 2–5 | 2–4 |
| L | 9–12 | 8–11 | 7–10 | 6–8 | 6–8 | 5–7 | 5–7 | 4–6 |
| Coupling | B3LYP/6-311+g(d,p)//B3LYP/6-31g(d,p) | B3LYP/6-311+g(d,p)//MO62X/6-31g(d,p) | Exp (Hz) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| C7SC5S | C7SC5R | C7RC5S | C7RC5R | C7SC5S | C7SC5R | C7RC5S | C7RC5R | ||
| H6a–C8 | 2.4 | 2.4 | 7.1 | 6.1 | 1.8 | 1.8 | 8.2 | 6.0 | 5.8 |
| H6b–C21 | 3.1 | 3 | 3.8 | 4.3 | 2.7 | 2.5 | 3.1 | 4.7 | 4.0 |
| H6b–C22 | 2.7 | 6.1 | 2.6 | 6.3 | 2.5 | 6.7 | 1.4 | 5.9 | 6.2 |
| H6a–H5 | 8.4 | 9.1 | 11.2 | 9.2 | 10.0 | 10.1 | 11.8 | 8.5 | 9.4 |
| H6b–C8 | 5.1 | 5.1 | 2.2 | 3.1 | 3.7 | 4.6 | 2.1 | 3.2 | 3.8 |
| H6a–H7 | 7.5 | 7.6 | 5.4 | 6.6 | 5.9 | 7.1 | 4.6 | 6.6 | 5.5 |
| H6b–H7 | 8.0 | 8.4 | 10.9 | 9.1 | 10.3 | 9.8 | 12.5 | 9.4 | 9.0 |
| H5–H4 | 4.5 | 3.6 | 3.0 | 4.9 | 4.2 | 3.6 | 3.2 | 6.3 | 5.3 |
| H5–C3 | 2 | 3.6 | 1.5 | 3.3 | 1.8 | 2.9 | 1.2 | 2.7 | 4.1 |
| H4−C6 | 2.1 | 2.9 | 2.1 | 2.8 | 1.9 | 3.4 | 2.0 | 4.0 | 2.7 |
| H4−C22 | 4.6 | 4.3 | 5.3 | 3.7 | 4.8 | 3.9 | 5.5 | 2.9 | 3.5 |
| H5−C4 | 1.4 a | 2.8 a | 0.2 | 3.5 a | 0.8 a | 1.8 a | 0.3 | 3.6 a | 4.7 |
| RMSD (Hz) | 2.1 | 1.6 | 2.2 | 0.7 | 2.2 | 1.8 | 2.7 | 0.9 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huo, Z.-Q.; Zhu, F.; Zhang, X.-W.; Zhang, X.; Liang, H.-B.; Yao, J.-C.; Liu, Z.; Zhang, G.-M.; Yao, Q.-Q.; Qin, G.-F. Approaches to Configuration Determinations of Flexible Marine Natural Products: Advances and Prospects. Mar. Drugs 2022, 20, 333. https://0-doi-org.brum.beds.ac.uk/10.3390/md20050333
Huo Z-Q, Zhu F, Zhang X-W, Zhang X, Liang H-B, Yao J-C, Liu Z, Zhang G-M, Yao Q-Q, Qin G-F. Approaches to Configuration Determinations of Flexible Marine Natural Products: Advances and Prospects. Marine Drugs. 2022; 20(5):333. https://0-doi-org.brum.beds.ac.uk/10.3390/md20050333
Chicago/Turabian StyleHuo, Zong-Qing, Feng Zhu, Xing-Wang Zhang, Xiao Zhang, Hong-Bao Liang, Jing-Chun Yao, Zhong Liu, Gui-Min Zhang, Qing-Qiang Yao, and Guo-Fei Qin. 2022. "Approaches to Configuration Determinations of Flexible Marine Natural Products: Advances and Prospects" Marine Drugs 20, no. 5: 333. https://0-doi-org.brum.beds.ac.uk/10.3390/md20050333