Kinase Inhibitors from Marine Sponges

1
School of Chemistry, University of Wollongong, Wollongong, NSW 2522, Australia
2
Centre for Medicinal Chemistry, University of Wollongong, Wollongong, NSW 2522, Australia
*
Author to whom correspondence should be addressed.
Mar. Drugs 2011, 9(10), 2131-2154; https://0-doi-org.brum.beds.ac.uk/10.3390/md9102131
Submission received: 6 September 2011
/
Revised: 1 October 2011
/
Accepted: 14 October 2011
/
Published: 24 October 2011
(This article belongs to the Special Issue Bioactive Compound from Marine Sponges)
Abstract
:Protein kinases play a critical role in cell regulation and their deregulation is a contributing factor in an increasing list of diseases including cancer. Marine sponges have yielded over 70 novel compounds to date that exhibit significant inhibitory activity towards a range of protein kinases. These compounds, which belong to diverse structural classes, are reviewed herein, and ordered based upon the kinase that they inhibit. Relevant synthetic studies on the marine natural product kinase inhibitors have also been included.
1. Introduction
The search for pharmaceutically active compounds from natural sources is well established, with approximately 70% of small molecule drugs produced between 1981 and 2006 possessing an important link to a natural product source [1]. The pharmaceutical value of natural products is even more exemplified in the critical area of anticancer drugs, whereupon of the 155 small-molecules produced from the 1940s, 73% are other than “synthetic”, with 47% being natural products or natural product derived [1]. With oceans covering 70% of the surface of the earth, coupled with the large and varied biodiversity of the marine environment, the oceans remain a largely unexplored, but extremely promising source of new drug candidates. Approximately half of the novel marine natural products reported in the literature are biologically active [2]. This occurrence can be contributed to the reliance of sessile, soft-bodied marine invertebrates on chemical defense for survival, as many lack the physical defense mechanisms of movement and camouflage. As these chemicals are released into the water and are rapidly diluted, these secondary metabolites produced are often extremely potent [3].
The protein kinase family encompasses all enzymes in the human body that catalyse the chemical transfer of a phosphate group from a high energy molecule such as adenine triphosphate (ATP) to a specific substrate. The human genome encodes for approximately 518 different protein kinases, which are divided into different kinase families on the basis of their selectivity for substrates [4]. The covalent attachment of a phosphate group to a substrate requires a free hydroxyl moiety, and there are three amino acids that can provide this; serine, threonine and tyrosine. Therefore, serine/threonine kinases will recognise and attach a phosphate group to a serine or threonine amino acid, while the tyrosine-specific protein kinase family will phosphorylate a protein at a tyrosine moiety.
Kinases play a large, varied and vital role in cell regulation and particularly in signal transmission pathways, controlling cell differentiation, proliferation, metabolism, DNA damage repair, cell motility, response to external stimuli and apoptosis [5]. Deregulation of kinases has been found to be a primary cause in an increasing list of diseases, including oncological diseases, central nervous system disorders, autoimmune diseases, metabolic diseases and osteoporosis, suggesting that the number of kinases with the potential to be new pharmaceutical targets is significantly large [6]. The current focus on kinases is in the development of drugs with lower side effects than previous cancer treatments which traditionally focused on DNA replication and chromosome regulation and thus also affected many healthy cells. As various kinases have been reported to be misregulated in cancerous cells, anticancer treatments involving kinases can be specifically targeted to cancer cells [4]. The development of kinase inhibitors has been predicted to be a major driver of pharmaceutical growth with more than 130 kinase inhibitors reported to be in either Phase I or Phase II clinical trials, the majority of these being tested for their potential as cancer treatments [7]. Kinase inhibitors that successfully proceed onto the pharmaceutical market will join Imitinib (Gleevec, Novartis), a tyrosine kinase inhibitor that has dramatically improved the prognosis for sufferers of chronic myeloid leukemia after being the first small-molecule kinase inhibitor to be approved for human use [6]. Herein, we review the recent highlights and developments of over 70 kinase inhibitors that have been isolated from marine sponges.
2. Reviews
Kinase inhibitors and activators from natural sources were covered in 2011 by Marston in a review that included a small number of marine natural products [8]. In 2007, Nakao and Fusetani published a review on enzyme inhibitors isolated from marine organisms which included some protein kinase inhibitors from marine sponges [9]. In 2009, Deslandes et al. reviewed the synthesis and kinase inhibitory activities of the marine natural products granulatimide and isogranulatimide [10]. In 2009, Nguyen et al. published the synthesis and evaluation of the kinase inhibitory activity of the sponge derived compound hymenialdisine and its analogues [11]. In 1998, Carter and Kane reviewed the therapeutic potential of natural compounds that regulate the activity of protein kinase C [12]. To the best of the authors’ knowledge, this is the first comprehensive review that is focussed solely on the kinase inhibitory activities of marine sponge metabolites.
3. Protein Kinase C (PKC, EC 2.7.11.13)
The family of kinases known as protein kinase C (PKC) are serine/threonine kinases that encompass eleven isozymes and through the action of phosphorylating various intracellular proteins, mediate many physiological events such as induction of cell differentiation, regulation of apoptosis and inhibition of tumor invasion [13]. Protein kinase C is composed of two distinct regions; a carboxyl-terminal catalytic site containing an adenine triphosphate (ATP) binding site and a regulatory domain at the amino terminal that possesses a phorbol-binding domain that is unique to the PKC family [14]. The catalytic site on PKC is structurally shared amongst many different classes of kinases, and as such PKC inhibitors that block this site can also inhibit the action of other functionally diverse kinases [14]. Natural activators of PKC include diacylglycerols, phosphatidyl serine, inositol triphosphate and calcium ions. The vital role that PKCs play in signal transduction pathways has marked them as potential targets for pharmaceutical inhibition of diseases such as cancer, cardiovascular disease, renal disease, immunosuppression and autoimmune disease [15].
The efficacy of the natural product staurosporine as a PKC inhibitor has been known since last century when the alkaloid was isolated from the bacteria Streptomyces staurosporeus and shown more recently to have an IC50 value of 2.7 nM against PKC [16]. In recent years, a variety of marine organisms have also provided important PKC modulators such as 11-hydroxystaurosporine from the marine tunicate Eudistoma sp. [17] and bryostatin-1, from the marine bryozoan Bugula neritina [14,18]. Marine sponges have also proven to be a particularly rich source of PKC inhibitors.
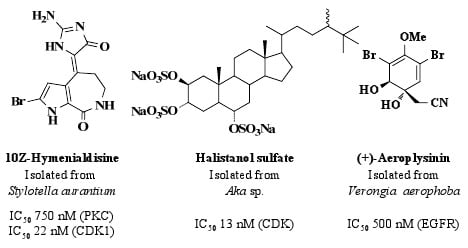
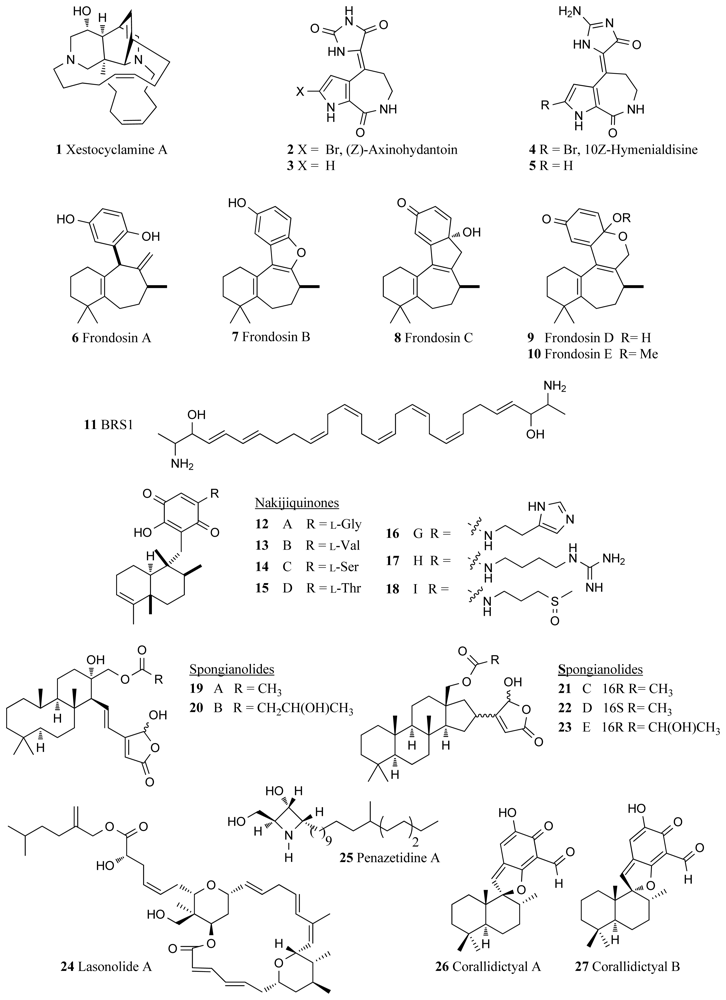
In 1994, the sponge Xestospongia sp. collected in waters off the Papua New Guinea coast, furnished xestocyclamine A (1, Figure 1) bearing a novel skeleton and found to inhibit PKC with an IC50 value of 4 μg/mL [19]. Xestocyclamine A and its pure enantiomer (−)-xestocyclamine A are considered critical PKC inhibitors for use in the development of anticancer drugs and there are many research groups focused on synthesising the stereochemically complex marine alkaloids [20,21]. (Z)-Axinohydantoin (2, Figure 1) and debromo-Z-axinohydantoin (3, Figure 1) are two PKC inhibitors with respective IC50 values of 9.0 and 22.0 μM that were isolated from the marine sponge Stylotella aurantium [22]. These novel compounds were isolated during a scale-up collection of the PKC inhibitors, hymenialdisine (4, IC50 0.8 μM, Figure 1) and debromohymenialdisine (5, IC50 1.3 μM, Figure 1) from the same sponge species [22]. Hymenialdisine is found to inhibit a range of kinases (see Section 4.1).
Five novel sesquiterpene derivatives, frondosins A–E (6–10, Figure 1), were isolated from the marine sponge Dysidea frondosa and shown to have inhibitory activity against PKC with reported IC50 values of 1.8, 4.8, 20.9, 26.0 and 30.6 μM respectively [23]. Frondosins A–E were also reported to be inhibitors of interleukin-8 in the low micromolar range [23] and more recently (−)-frondosins A (6) and D (9) have shown comparable activity against the HIV virus [24]. Various synthetic routes to frondosins A–C have been reported [25–27].
BRS1 (11, Figure 1), a polyunsaturated lipid isolated from an unidentified Australian sponge of class Calcarea was reported to be a novel inhibitor of PKC [28]. BRS1 exerts it activity by binding to the phorbol ester binding site and accounts for 0.02% of the wet weight of the sponge from which it was collected. The IC50 of BRS1 for inhibiting the binding of the phorbol ester was 9 μM, whereas 98 μM represented a 50% effective concentration for inhibiting the enzymatic activity of PKC [28].
An Okinawan marine sponge belonging to the family Spongiidae, has furnished a family of novel sesquiterpenoid quinones, including the nakijiquinones A–D (12–15, Figure 1), with reported IC50 values against PKC of 270, 200, 23 and 220 μM respectively [29,30]. A subsequent paper described the isolation of the nakijiquinones G–I (16–18) from the same sponge, which showed modest cytotoxicity in the range of 2.4 to >10 μg/mL against a range of cancer cell lines (e.g., P388 murine leukemia, L1210 murine leukemia and KB human epidermal carcinoma cells), as well as inhibitory activity against HER2 kinase [31]. The remarkable inhibitory activity of nakijiquinones A–D against a variety of kinases including epidermal growth factor receptor (EGFR), c-erbB-2 kinase and tyrosine kinase VEGFR2 has been reviewed and their biological activity and structure-activity relationships are well documented [15,32]. The synthesis of the nakijiquinones has been reported [33,34], with particular emphasis on the potential of nakijiquinone and its analogues in the prevention of angiogenesis as the nakijiquinone family is the only naturally occurring inhibitor of the Her-2/Neu receptor tyrosine kinase. Extensively implicated in tumor proliferation, the Her-2/Neu receptor tyrosine kinase is over-expressed in approximately 30% of primary breast, ovary and gastric cancers and when amplified has been linked to increases in the aggressiveness of the cancer and reduced patient survival [35].
The cytotoxic sesterpenes spongianolides A–E (19–23, Figure 1), isolated from a marine sponge belonging to the genus Spongia, were found to have inhibitory activity against PKC with IC50 values ranging between 20–30 μM [36]. The cheilanthane cyclic terpenoid contained within the structure has since been synthesized via a biomimetic approach [37] (see also Section 7).
Another potent marine sponge derived PKC inhibitor is lasonolide A (24, Figure 1). Isolated from the Caribbean sponge Forcepia sp., lasonolide A was found to inhibit the phorbol ester-stimulated adherence of EL-4.IL-2 mouse thymoma cells within 30 min with an IC50 value of 27 nM, highlighting the potential of this compound for development as a potent PKC inhibitor [38–40].
Another inhibitor of PKC enzyme is a new azetidine compound penazetidine A (25, Figure 1) isolated from the Indo-Pacific marine sponge Penares sollasi [41]. This sponge species attracted attention after its crude extract in initial screenings exhibited inhibitory activity (IC50 0.3 μg/mL) against serine kinase PKC-βI, but it was not active against protein tyrosine kinase (PTK). Penazetidine A displayed strong activity against PKC (IC50 l μM), and also showed significant cytotoxicity against human and murine cancer cell lines (A549, HT-29, B16/F10 and P388) [41]. A mixture of two diastereomeric spirosesquiterpene aldehydes, corallidictyals A and B, were isolated from the marine sponge Aka (=Siphonodictyon) coralliphaga, which was collected at Little San Salvador Island (26, 27, Figure 1) [42]. This mixture was found to show good selectivity for the inhibition of PKC (IC50 28 μM) compared to the other serine kinase enzyme PKA (IC50 300 μM). In particular, the diastereomeric mixture was selective for inhibition of the α-PKC isoform giving a lower IC50 value compared to the other isoforms of the enzyme.
4. Cyclin Dependent Kinases (CDK, EC 2.7.11.22)
Cyclin-Dependent kinases (CDKs) are a group of serine/threonine kinases that encompass approximately 25 different cyclin families, all of which are critical in the regulation of the cell cycle [4]. The distinguishing feature of the CDKs from other kinase families is the enzymatic activation requirement of the binding of the cyclin regulatory subunit [43]. The movement of the cell through the cell cycle phases is determined by the fluctuating concentrations of different activated CDK/cyclin complexes whose cellular mechanism involves the phosphorylation of many distinct proteins at serine or threonine residues in specific sequences. While CDKs are also involved in apoptosis and transcription, their pivotal role in differentiation, transformation, proliferation and metastasis has recently seen CDKs become a major target for cancer therapies, especially now that it is recognized that hyperactive CDKs (overexpression) or hypoactive CDKs (mutation, deletion) are a leading cause of uncontrolled tumor proliferation in humans [4]. Several natural and synthetic compounds that inhibit CDKs in the sub-micromolar range have been isolated and are at various stages of clinical trials, the most advanced being flavopiridol, a semi-synthetically produced analogue of an alkaloid from the Indian tree Dyoxylum binectariferum, currently in Phase II clinical trials for soft tissue sarcomas [44]. These small molecule inhibitors arrest tumor proliferation and many are also capable of inducing apoptosis in proliferating cells [45].
4.1. Cyclin Dependent Kinase-1
Cyclin Dependent Kinase-1 (CDK-1) is a critical controller of the cell cycle in multi-cellular eukaryotic organisms and operates primarily in the mitosis (M) phase. In order for the cell to pass from the growth (G2) phase into M phase, activation of the CDK-1/cyclin B1 complex must be sustained in the nucleus from prophase into metaphase [46]. Hymenialdisine (4, Figure 1), a potent inhibitor of CDK-1, was first isolated in 1982 from the marine sponges Axinella verrucosa and Acanthella aurantiaca [47]. Hymenialdisine inhibits CDK-1/cyclin B (IC50 22 nM) through competitive inhibition at the ATP-binding site, and as this site is homologous with many kinase families, hymenialdisine also shows inhibitory activity against a variety of different kinases including CDK-2/cyclin A (IC50 70 nM), CDK-2/cyclin E (IC50 40 nM), CDK-5/p25 (IC50 28 nM), glycogen synthase kinase 3 (GSK-3) (IC50 10 nM) and creatine kinase 1 (CK1) (IC50 35 nM), while still possessing good selectivity in vitro as inhibition of alternate molecular targets occurs at much higher IC50 values [48].
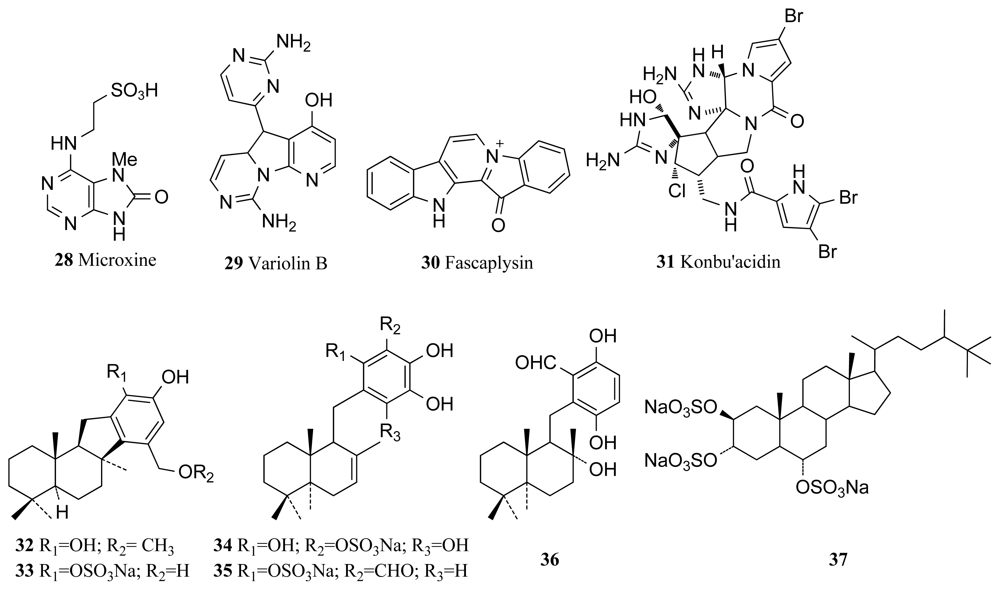
Inhibition of the CDK-1/cyclin B complex has recently been shown to induce apoptosis in cells experiencing Myc (proto-oncogene) overexpression [49], a common phenomenon in many human cancers and a mechanism by which hymenialdisine and associated analogues could potentially act as anticancer agents. Many analogues of hymenialdisine that exhibit inhibitory activity against various CDKs in the nanomolar range have been successfully synthesized as medicinal chemists recognised the potential of hymenialdisine for use against many degenerative diseases [50]. Recent patents also highlight hymenialdisine and analogues as likely future pharmaceuticals for diseases such as asthma, rheumatoid arthritis, multiple sclerosis and Alzheimer’s disease due to its ability to arrest the NF-kappa B signaling process, a critical mechanism in the above diseases [51]. Microxine (28, Figure 2), a novel purine derivative, is an inhibitor of CDK-1, isolated from the Australian marine sponge genus Microxina, with an IC50 value of 13 μM against CDK-1 [52,53]. Variolin B (29, Figure 2) was isolated from the Antarctic sponge Kirkpatrickia varialosa, and it was found to display CDK inhibitory activity exhibiting selective inhibition towards CDK-1 and CDK-2 over CDK-4 and CDK-7 [53,54]. It was hypothesized that mechanism of action of variolin B is the inhibition of cyclin-dependent kinases that interrupt the progression of the normal cell cycle. Variolin B inhibits the phosphorylation of histone H1 mediated by CDK-2/cyclin E, CDK-2/cyclin A, CDK-1/cyclin B, CDK-7/cyclin H, and CDK4/cyclin D, with IC50 values in the micromolar range [53]. Total synthesis of this compound has been performed by several research groups due to the vast biological potential of the compound with its antiviral and antitumor activity, including cytotoxicity towards the P388 murine leukemia cell line with an IC50 value of 210 ng/mL [53,55–58].
4.2. Cyclin Dependent Kinase-4
Another member of the CDK family is CDK-4, a catalytic subunit whose presence is vital for the progression of the cell cycle through the G1 phase [59]. The activity of CDK-4 is restricted to the G1-S phases and is regulated by the attachment of the regulatory subunit cyclin D and the endogenous CDK inhibitor p16(INK4a). The G1-S checkpoint is the most important regulation point in the cell cycle, exemplified by the fact that the G1-S transition is misregulated in 60–70% of cancers [60]. A major role of CDK-4 is the phosphorylation of the retinoblastoma gene product (Rb) [59]. A high incidence of mutations in Rb, along with cyclin D and p16(INK4a), has been seen in tumorigenesis in many cancers, a fact which has recently seen CDK-4 become an exciting new cancer drug target.
A major distinguishing feature of fascaplysin (30, Figure 2), a red pigment isolated from the marine sponge Fascaplysinopsis sp. is that it is a selective inhibitor of CDK-4 [61]. Poor selectivity is a common problem among kinase inhibitors due to the ATP binding site, where many inhibitors exert their actions, being conserved amongst the majority of kinase families. Fascaplysin exhibits an IC50 value of 0.35 μM against the CDK-4/cyclin D complex while IC50 values against other kinases were comparably much higher [61]. This specificity allows fascaplysin to be a useful scientific tool in investigating the direct consequences of singular CDK-4 inhibition [61] and many studies have thus been conducted establishing the potential of fascaplysin as a pharmaceutical agent. A recent study has identified fascaplysin as a natural angiogenesis inhibitor after it was found that fascaplysin selectively inhibited the proliferation of endothelial cells toward tumor cells and suppressed the vascular endothelial growth factor (VEGF), a critical player in angiogenesis [60]. Conclusions from such studies indicate that fascaplysin could in the future play a central role in preventing cancers from metastasizing and becoming malignant by preventing new vascular growth at the tumor site [60].
Konbu’acidin A (31, Figure 2) is a novel bromopyrrole alkaloid that was isolated from the Okinawan marine sponge Hymeniacidon sp. and reported to display inhibitory activity against the CDK-4/cyclin D complex [62]. Konbu’acidin A showed inhibitory activity against CDK-4 with an IC50 of 20 μg/mL but did not show any cytotoxicity against murine leukemia L1210 and epidermal carcinoma KB cell lines [62]. The marine sponge Aka sp. collected from Micronesia yielded three novel sesquiterpene quinols (32–34), two known quinols (35, 36) and halistanol sulfate (37). Four of the compounds (32, 35–37) were screened for CDK/cyclin D1 kinase inhibitory activity and compounds 35 and 37 exhibited moderate kinase inhibitory activity and inhibited complex formation with IC50 values of 9.0 and 9.5 μg/mL respectively [63].
5. Tyrosine Protein Kinase (TPK, EC 2.7.10.1)
Tyrosine protein kinase (TPK) are enzymes that catalyse the phosphorylation of tyrosine residues and can be divided into two main categories; cellular and receptor TPKs, and non-receptor TPKs. Studies into this particular class of kinase have identified them as key players in both intracellular and extracellular communication [64]. TPKs are associated with proliferative diseases such as cancer, leukemia, psoriasis and restonosis due to their role in regulating key cell functions like proliferation, differentiation, and antiapoptotic signaling [64] and it has been reported that 70% of the known oncogenes and proto-oncogenes found in cancer are associated with TPKs [65].
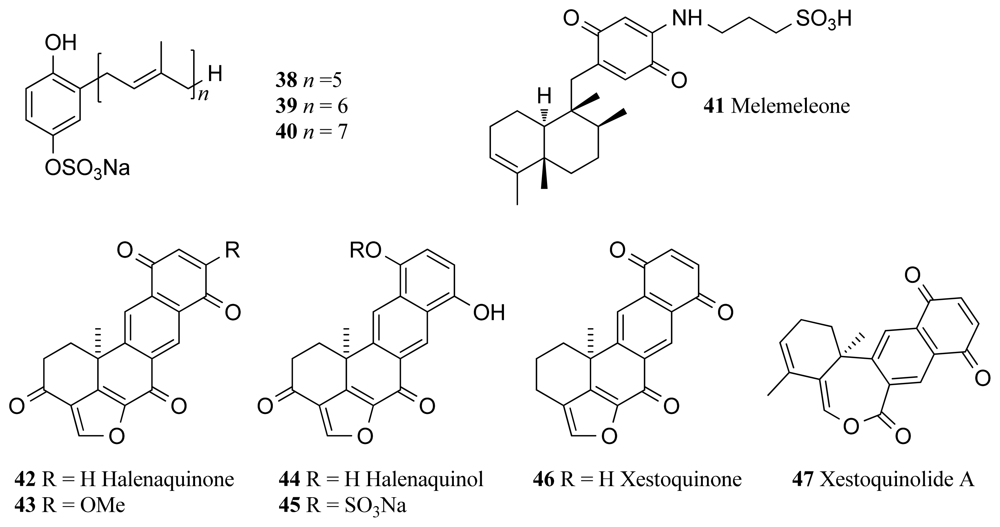
The deep-sea sponge Ircinia sp. collected off the New Caledonian coast at a depth of 425–500 m yielded three TPK inhibitors, the penta-, hexa- and hepta-prenylhydroquinone 4-sulfates (38–40, Figure 3). IC50 values for each compound against TPK were recorded as 8, 4 and 8 μg/mL respectively [66]. Penta-prenylhydroquinone sulfate (38, Figure 3) has also proven to be a potential antiviral and cytotoxic agent achieving 65% inhibition of the HIV-1 integrase enzyme at 1 μg/mL and having inhibited neuropeptide Y (NPY) receptor with an IC50 value of 50.8 μg/mL. This compound also displayed cytotoxicity against the epidermal KB carcinoma cell line [66].
Tyrosine Kinase pp60V-SRC
Tyrosine kinase pp60V-SRC is a membrane-associated protein with protein kinase activity and is also the oncogene product of the Rous Sarcoma retrovirus, which upon entry into a cell, transforms a normal cell into a rapidly proliferating cell [67]. Melemeleone (41, Figure 3) is a novel sesquiterpene quinonecompound, isolated along with another four new metabolites and two known compounds, from two sponge species of Dysidea from Solomon Island [68]. All purified compounds isolated from the sponge were tested for kinase inhibitory activity, but only melemeleone displayed activity against pp60V-SRC with an IC50 of 28 μM [68].
Several inhibitors of pp60V-SRC were isolated from the Fijian sponge Xestospongia carbonaria, namely halenaquinone (42, Figure 3), halenaquinol (44, Figure 3), halenaquinol sulfate (45, Figure 3) and xestoquinone (46, Figure 3), and reported IC50 values of 1.5, 60.0, 0.55 and 28.0 μM, respectively [69]. Of these pentacyclic polyketide compounds, halenaquinone proved to be the most pharmaceutically promising, due to its characterisation as an irreversible inhibitor. The potential of halenaquinone as an anticancer agent is evidenced by findings that it arrests the proliferation of various cell lines, including those that have been transformed by oncogenic PTKs, and halenaquinone also shows inhibitory activity against the kinase activity of the human EGFR with an IC50 value of 19 μM [69]. Halenaquinone (42) and xestoquinone (46) were also isolated from the same sponge Xestospongia sp. collected from Vanuatu and were found to inhibit several kinases. Xestoquinone inhibited Pfnek-1 kinase of Plasmodium falciparum with IC50 of 1.1 μM but displayed lower kinase inhibitory activity towards PfPK5 and no activity towards PfPK7 and PfGSK-3 [70].
Halenaquinone and halenaquinol have since been associated with antibiotic and cardiotonic activity in addition to their ability to inhibit pp60V-SRC, and have been the focus of several synthetic studies [71]. Strategies for the synthesis of the core skeletons of halenaquinone and halenaquinol have recently been described with the construction of the furan-fused tetracyclic core of the molecules. The key step involved the intramolecular [4 + 2]-cycloaddition reaction of o-quinodimethane [71]. The current highlight with halenaquinone is as its potential as an inhibitor of recombinant human Cdc25B phosphatase [72], an activator of cyclin dependent kinase Cdc2 whose presence is required for entry into the mitosis phase of the cell cycle. Displaying an IC50 value of 0.7 μM, halenaquinone stands out as a key molecule in anticancer studies revolving around this drug target [72]. Alvi et al. also isolated the two compounds 14-methoxyhalenaquinone (43, Figure 3) and xestoquinolide A (47, Figure 3) from the same sponge, for which IC50 values of 5 and 80 μM against protein tyrosine kinase (PTK) respectively were reported [73].
6. Epidermal Growth Factor Receptor (EC 2.7.10.1)
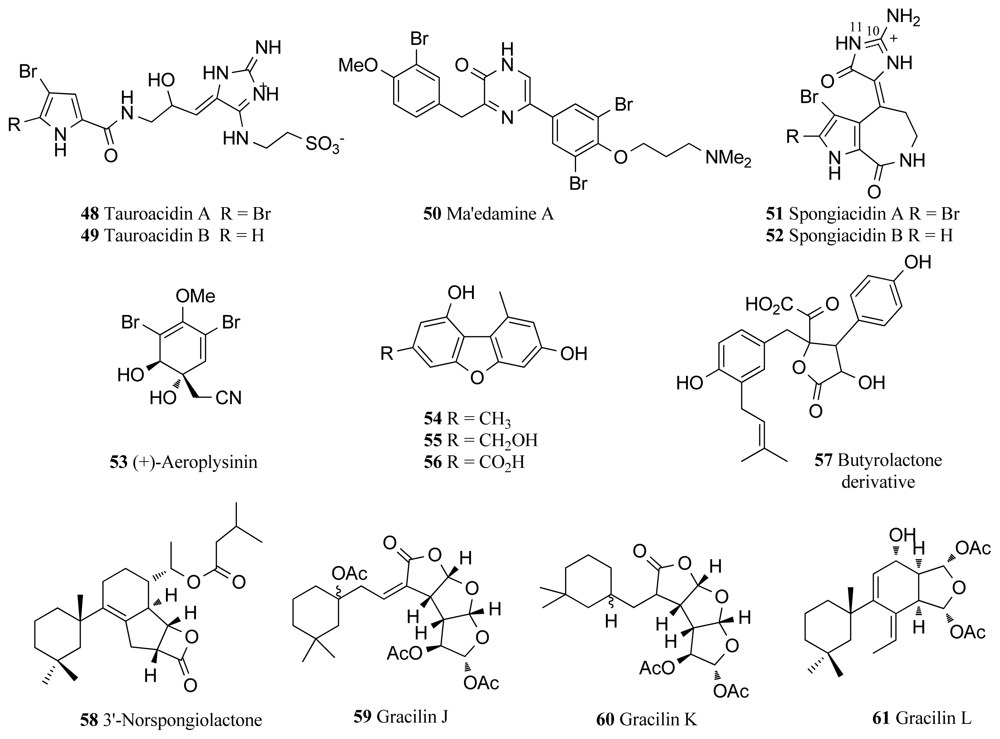
The epidermal growth factor receptor (EGFR) is a member of the type 1 growth factor receptor gene family, which also includes erbB-1, erbB-2, erbB-3 and erbB-4 [74]. This tyrosine kinase family has been heavily implicated in the mechanisms of various cancers as mutations leading to EGFR being over-expressed have often been found in cancer cases, in particular breast cancer [15]. As part of an extensive effort to identify small molecule inhibitors of this drug target, two novel bromopyrrole alkaloids were isolated from an Okinawan marine sponge Hymeniacidon sp. and named tauroacidins A and B (48, 49, Figure 4) [75]. These two compounds showed inhibitory activity against both EGFR and c-erbB-2 kinase with an IC50 value of 20 μg/mL for each respective kinase [75]. The tauroacidins A and B may be biogenetically related to other bromopyrrole alkaloids from marine sponges through the taurine residue attached to the aminoimidazole ring [75]. Okinawan marine sponges have proven to be a particularly rich source of kinase inhibitors with a bromotyrosine alkaloid, ma’edamine A (50, Figure 4), also being isolated from the Okinawan marine sponge Suberea sp. and showing inhibitory activity against c-erbB-2 kinase (IC50 6.7 μg/mL) [76]. Ma’edamine A contains a unique 2(1H)pyrazinone moiety located between the two bromotyrosine units, and also displays cytotoxicity against murine leukemia L1210 cells (IC50 4.3 μg/mL) and epidermal KB carcinoma cells (IC50 5.2 μg/mL) [76].
Spongiacidins A and B (51, 52, Figure 4) are inhibitors of c-erbB-2 kinase isolated from the Okinawan marine sponge Hymenacidon sp. [77]. These two compounds are also bromopyrrole alkaloids of the pyrrolo[2,3-c]azepine type. The respective IC50 values for spongiacidins A and B against c-erbB-2 kinase are 8.5 and 6.0 μg/mL [77]. It was later identified that spongiacidin A is actually the (E) isomer at the exocyclic C10-C11 double bond of 3-bromohymenialdisine, a metabolite of hymenialdisine discussed earlier [78].
Isolated from the marine sponge species Verongia aerophoba, (+)-aeroplysinin-1 (53, Figure 4) was found to completely inhibit EGFR at a concentration of 0.5 μM [79]. Due to this inhibitory ability, (+)-aeroplysinin-1 was found to have a strong antitumor effect on EGFR tumor cell lines, in particular blocking the proliferation of EGFR dependent human breast cancer cell lines MCF-7 and ZR-75-1 [79]. Importantly, (+)-aeroplysinin-1 displays some selectivity for cancerous cells as the application of (+)-aeroplysinin-1 at a concentration of 0.25–0.5 μM resulted in total tumor cell death, but did not have any cytotoxic effect on normal human fibroblasts at concentrations ten times higher [79]. A recent study has identified (+)-aeroplysinin-1 as an important inhibitor of several key steps of angiogenesis, the process by which tumors become mutagenic and thus a vital target for pharmaceutical intervention in cancerous diseases [80]. In detail, (+)-aeroplysinin-1 has been shown to inhibit capillary-like tube formation, induce apoptosis, promote anti-proteolysis in endothelial cells and also arrest the development of new vascular structures [80]. As angiogenesis is a major factor in fatal cancers and (+)-aeroplysinin-1 displays in vivo efficacy as an inhibitor of this process, it remains an extremely promising drug candidate.
Three novel compounds identified as 3,9-dimethyldibenzo[b,d]furan-1,7-diol (54), 3-(hydroxymethyl)-9-methyldibenzo[b,d]furan-1,7-diol (55), 1,7-dihydroxy-9-methyldibenzo[b,d] furan-3-carboxylic acid (56) and one known compound, butyrolactone derivative (57), were isolated from marine sponge Acanthella cavernosa from Fiji and all compounds displayed moderate inhibitory properties against EGFR [81]. In recent studies, bioassay-guided fractionation of the marine sponge Spongionella sp., yielded the novel bioactive diterpenes, 3′-norspongiolactone (58, Figure 4) and gracilins J–L (59–61), along with three known gracilins and the known diterpenoid tetrahydroaplysulphurin-1 [82]. All eight compounds isolated from the sponge Spongionella sp. exhibited cytotoxicity against the K562 human chronic myelogenous leukemia cell lines with IC50 values in the range of 0.6 to 15 μM, however they also showed similar levels of cytotoxicity towards human peripheral blood mononuclear cells (PBMC) [82]. All compounds displayed inhibitory activity towards EGFR tyrosine kinase with the novel diterpenes 58–61 exhibiting 25%, 19%, 75% and 57% inhibition respectively at 100 μM [82].
7. Mitogen-Activated Protein Kinase (EC 2.7.11.24)
Mitogen- and stress-activated kinase (MSK1) and mitogen-activated protein kinase (MAPK) are two stress-associated serine/threonine specific protein kinases involved in cellular signaling, regulating various processes such as cell division and proliferation, apoptosis and gene expression [83]. There are three major subclasses of this kinase family, including extracellular signal-regulated kinases (ERKs), c-Jun N-terminal kinase (JNK)/stress-activated protein kinase (SAPKs) and p38 MAPKs [83]. It has been acknowledged that selective inhibitors of these kinases are likely to affect cellular events with high specificity and are therefore molecules of significant interest in the search for anticancer pharmaceuticals [15].
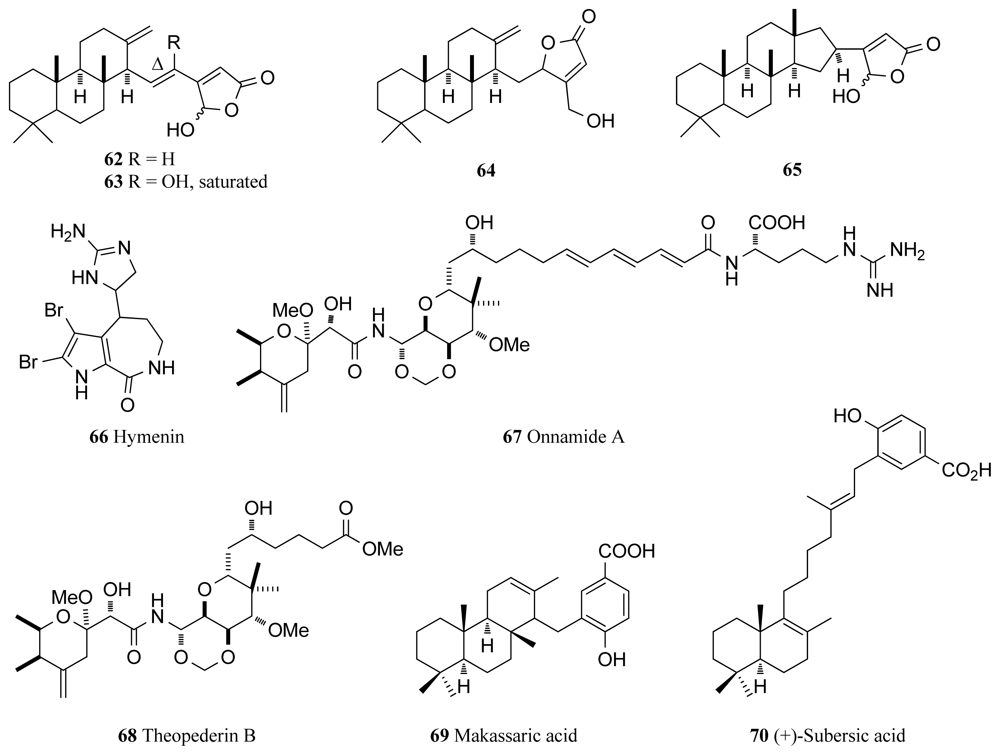
In the first description of cheilanthane sesterterpenoids from a marine sponge, three novel (62–64) and one known cheilanthane sesterterpenoids (65, Figure 5) were isolated from the marine sponge Ircinia sp., with 62, 63 and 65 obtained as inseparable 1:1 mixtures of their C-25 epimers. Intriguingly, all four compounds were reported to exhibit identical inhibitory activity against MSK1 (IC50 4 μM for each compound) and mitogen activated protein kinase activated protein kinase (MAPKAPK-2, IC50 90 μM for each compound) [84]. Extracts from two sponge species, the purple bleeding sponge Iotrochota birotulata and the West Indian bath sponge Spongia barbara were found to inhibit the MAPK/ERK cascade, a pathway that links the binding of growth factors on cell surface receptors to intracellular responses [85]. Encompassing many protein kinases, activation of this cascade leads to cell division and is thus a potential anticancer drug target [86]. The two extracts significantly inhibited the MAPK/ERK pathway to 51% and 44% of control levels respectively without affecting the survival of the cell [85].
Raf (EC 2.7.11.1)/MAP Kinase Kinase (EC 2.7.12.2)/MAPK (EC 2.7.11.24)
The Raf kinase, MAP kinase kinase (MEK) and MAPK combine to form a pathway that links extracellular signals to the phosphorylation of cellular proteins to regulate cell proliferation and differentiation [87]. The cascade is firstly activated by Ras promoting the translocation of Raf-1 to the inner cell membrane where it undergoes phosphorylation for activation. Raf-1 specifically phosphorylates and activates MEK, which will continue the process by phosphorylating MAPKs, causing them to migrate into the nucleus of the cell and influence many cellular events [87]. The oncogenic form of Ras is implicated in over 30% of all cancers, and as the Raf/MEK/MAPK cascade contains many potential sites for inhibition, this is an important and extremely promising target to be studied for pharmaceutical intervention [87].
While it has been known for some time that hymenialdisine (4, Figure 1) shows significant inhibitory activity against many cellular kinases, it has recently been reported that hymenialdisine and debromohymenialdisine (5, Figure 1) are remarkably potent inhibitors of MEK with IC50 values of 3.0 and 6.0 nM respectively [87]. These two compounds, isolated from the marine sponge Stylotella aurantium arrest the Raf/MEK/MAPK cascade by specifically binding to and inhibiting the phosphorylation of MAPK by MEK-1. It is also believed that 10E-hymenialdisine spontaneously converts to 10Z-hymenialdisine (4, Figure 1) on standing [87] and the mixture of these two compounds was shown to have the ability to inhibit the growth of LoVo and Caco-2, two human colon tumor cell lines [87]. 10Z-Hymenialdisine is now extensively used in research programs and is readily available from biochemical product suppliers as it shows good efficacy in vivo and has significant potential in a variety of different disease types as discussed earlier. Also extracted from the same sponge species was hymenin (59, Figure 5), which also showed inhibitory activity against the Raf/MEK/MAPK cascade [87]. However, with IC50 values ranging from between 128.8 and 250.0 μM for the different specific Raf, MEK and MAPK kinases, hymenin was far less potent than 10E-hymenialdisine and 10Z-hymenialdisine and was not pursued any further [87].
A methanol fraction of the sponge Batzella sp. was found to inhibit Raf kinase with an IC50 value of 2.8 μg/mL [88]. The known antimitotic compound halitoxin [89] was identified, however, it was not responsible for the observed kinase inhibitory activity.
Onnamide A (67) and theopederin B (68) are two compounds that were recently found to induce the stress-activated protein kinases, p38 kinase and JNK [90], two of the subclasses of the MAPK kinase family (Figure 5). While full understanding of the role of JNK in apoptosis has not yet been achieved, it is known that JNK and p38 kinase are predominantly activated by environmental stresses [91]. The JNK pathway is critical in the regulation of apoptosis during early brain development in mice and the p38 MAPK pathway plays a vital role in the production of inflammatory cytokines and subsequent signaling and also appears to be heavily associated with cell survival and proliferation [91]. Onnamide A and theopederin B, heterocyclic compounds that are members of the pederin family isolated from a marine sponge, activate a ribotoxic stress response and induce apoptosis [90,92,93]. As well as inducing the production of p38 and JNK, these two compounds were also found to stimulate plasminogen activator inhibitor-1 (PAI-1) gene expression in concentration ranges of 10–100 nM for onnamide A and 1–10 nM for theopederin B [90]. PAI-1 is an important current drug target as high levels of PAI-1 have consistently been found in human cancer cells and PAI-1 has also been associated with tumor growth, invasion and metastasis [94]. Thus, onnamide A and theopederin B will provide important tools in understanding more about PAI-1 expression and the induction of the ribotoxic stress response [90]. (+)-Makassaric acid (69) and (+)-subersic acid (70) are novel meroterpenoid compounds, isolated from the sponge Acanthodendrilla sp. collected in Indonesia. These compounds were found to inhibit MAPKAP kinase 2 which is involved in stress and inflammatory responses [95].
8. Glycogen Synthase Kinase-3 (GSK-3, EC 2.7.11.26)
A serine/threonine protein kinase, the main function of glycogen synthase kinase-3 (GSK-3) is the mediation of glycogen synthase but it is also involved in several key cellular events such as the response to damaged DNA and the phosphorylation of the microtubule associated mammalian protein tau. Overactivity of this phosphorylation has been identified as one of the first events in the onset of neurodegenerative diseases such as Alzheimer’s disease [96]. Over the last two decades, interest in GSK-3 has exponentially increased as its potential as a drug target in many non-curable diseases such as type-2 diabetes, stroke, Alzheimer’s disease, and bipolar disorder is recognized [96]. Current small molecule inhibitors of GSK-3 include pyridyloxadiazoles, thiadiazolidindiones, pyrazolopyrimidines and maleimides [96], but marine sponges are also proving to be a reliable source of secondary metabolites showing inhibitory activity against this drug target.
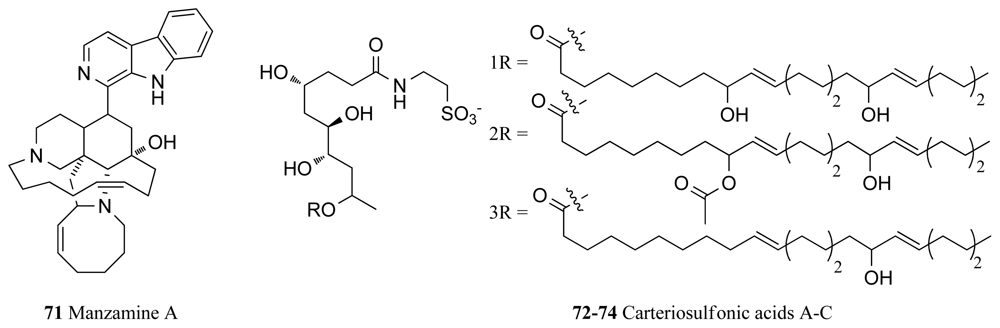
Manzamine A (71, Figure 6), a complex alkaloid isolated from an Okinawan sponge of the genus Haliclona, is one such compound showing specific non-competitive inhibition of ATP binding in GSK-3β with an IC50 value of 10.2 μM [96]. Manzamine A also inhibits CDK-5 with an IC50 value of 1.5 μM, and as this kinase coupled with GSK-3 represents the two main players in the hyperphosphorylation mechanism in Alzheimer’s disease, manzamine A is a useful drug lead for the future treatment of this disease [96]. This conclusion is supported by the fact that manzamine A has proved capable of entering cells and interfering with the tau protein as well as causing arrest in the hyperphosphorylation in human neuroblastoma cell lines [96]. Structure-activity relationships between manzamine A and the GSK-3 pharmacophore have been carried out and a variety of manzamine A analogues have also been synthesized indicating that the entire manzamine molecule is required for GSK-3 inhibitory activity [96]. Manzamine A and its synthesized derivative (−)-8-hydroxymanzamine A, have also been identified as promising new antimalarial agents producing in vivo inhibition of the growth of the malaria parasite Plasmodium berghei in rodents [97]. As the malaria parasite rapidly achieves resistance to currently administered antimalaria drugs, patents for the use of manzamine A in human antimalarial drugs have been submitted [98].
The carteriosulfonic acids A–C (72–74, Figure 6), novel compounds containing a 4,6,7,9-tetrahydroxylated decanoic acid subunit, were recently identified during a screen to identify modulators of Wnt signaling, which plays a key role in cell proliferation [99]. Phosphorylation of β-catenin by GSK-3β is involved in the negative regulation of Wnt signaling and thus it was proposed that inhibitors of GSK-3β may be associated with Wnt signaling activation. Accordingly, the compounds (72–74) were isolated from an extract of the marine sponge Carteriospongia sp., which was a Wnt signaling activator and were found to be low micromolar inhibitors of GSK-3β. Although further biological studies were foreshadowed in the above article, they had not yet appeared at the time of writing this review [99].
9. Other Kinases
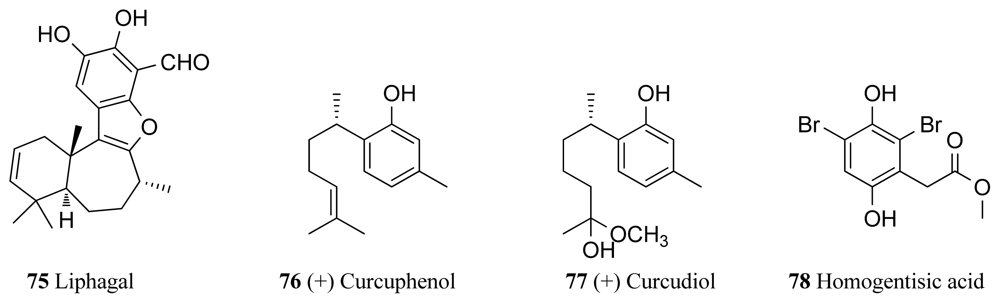
Liphagal (75, Figure 7), a meroterpenoid isolated from the marine sponge Aka coralliphaga collected in Dominica, was found to exhibit inhibitory activity against PI3K (phosphoinositide-3-kinase) with an IC50 value of 100 nM, with 10 folder higher potency against PI3Kα than towards PI3Kγ [100,101]. This compound also exhibited cytotoxicity against human colon (IC50 0.58 μM) and human breast (1.58 μM) tumor cell lines [100]. This sponge species is also known to produce the PKC inhibitors corallidictyals A and B (26 and 27) (see Section 3). Two bisabolene type sesquiterpenoids, (+)-curcuphenol (76, Figure 7) and (+)-curcudiol (77, Figure 7) were identified as bioactive compounds from the sponge Axynissa sp. from Indonesia. Curcuphenol showed Src protein kinase inhibition with an IC50 value of 7.8 μg/mL, while curcudiol inhibited focal adhesion kinase (FAK) with an IC50 value of 9.2 μg/mL [102]. Protein kinase A inhibitory activities of up to 100% (at 100 μg/mL) along with haemolytic and brine shrimp activities were also observed in a range of extracts isolated from three deep-water sponges collected from North Western Australia [103]. A novel compound, homogentisic acid (78) was isolated from the sponge Pseudoceratina collected in Vanuatu [104]. The authors previously isolated xestoquinone from a Xestospongia sp. collected from the same place and in their research for new antimalarial drugs found that this compound was an inhibitor of Pfnek-1, which is a NIMA-related protein kinase of Plasmodium falciparum. Therefore homogentisic acid was also screened against Pfnek-1 and found to display an IC50 value of 1.8 μM against this target [104] Hymenialdisine (4) has also showed Polo-Like kinase-1 inhibitory activity of 10 μM. It was isolated along with debromohymenialdisine (5) and four novel dihydrohymenialdisine derivatives from the sponge Cymbastela cantharella [105].
There are several aspects to consider regarding kinase inhibitors such as whether they are ATP-competitive or non-competitive inhibitors and whether the compounds inhibit their reported enzymatic targets in cellular assays. However, the level of mechanistic detail and characterization of the kinase inhibitory activity of the compounds described herein varies greatly. Thus, herein those articles providing a higher degree of characterization are indicated in the Table 1 by an asterisk. A further issue is one of broader kinase selectivity profiling that would be useful to see addressed in the literature, both in terms selectivity of the inhibitors towards other kinases and towards other targets.
10. Conclusions
The search for kinase inhibitors from marine sources has proven extremely successful with the advent of compounds such as bryostatin-1 into pharmaceutical development, and others such as hymenialdisine (4) and manzamine (71) looking promising. In particular, marine sponges are a rich source of highly diverse chemical compounds including lipids, terpenes and alkaloids, enhanced by a high incidence of novel carbon skeletons, such as that of xestocyclamine A (1). Marine sponge metabolites have proven to be extremely potent against a range of kinase targets heavily involved in an increasing list of disease mechanisms including cancer, Alzheimer’s disease and atherosclerosis. Several kinase inhibitors such as fascaplysin (30) possess strong selectivity not only for specific kinase subtypes, but also for cancerous cells over healthy cells and are thus promising molecules in the development of new oncological pharmaceuticals. With new technological developments bringing access to previously unexplored marine environments such as the deep sea [106], it is certain that many more sponge metabolites with novel structures and potent kinase inhibitory activities will be discovered in the future. Furthermore, as our understanding of the mechanism and regulation of various kinases continues to grow, marine sponge-derived kinase inhibitors are destined to play an expanding role in the treatment of various diseases.
Acknowledgments
We thank Mona Hamad for proof-reading of the manuscript and the University of Wollongong for financial support through the Centre for Medicinal Chemistry.
References
- Newman, DJ; Cragg, GM. Natural products as sources of new drugs over the last 25 years. J. Nat. Prod 2007, 70, 461–477. [Google Scholar]
- Faulkner, DJ. Marine pharmacology. Antonie Leeuwenhoek 2000, 77, 135–145. [Google Scholar]
- Haefner, B. Drugs from the deep: Marine natural products as drug candidates. Drug Discov. Today 2003, 8, 536–544. [Google Scholar]
- Sharma, P; Sharma, R; Tyagi, R. Inhibitors of cyclin dependent kinases: Useful targets for cancer treatment. Curr. Cancer Drug Targets 2008, 8, 53–75. [Google Scholar]
- Novak, K. Conference Report—Protein Kinase Inhibitors in Cancer Treatment: Mixing and Matching? Proceedings of the Keystone Symposium on Protein Kinases and Cancer, Lake Tahoe, CA, USA, 24–29 February 2004, Medscape General Medicine: Lake Tahoe, CA, USA, 2004. [Google Scholar]
- Goldstein, D; Gray, N; Zarrinkar, P. High-throughput kinase profiling as a platform for drug discovery. Nat. Rev. Drug Discov 2008, 7, 391–397. [Google Scholar]
- Norman, P. Overview: Kinase Therapeutics Pipelines: An Assessment of Targets and Agents in Development; Cambridge Healthtech Institute: Needham, MA, USA, 2007. [Google Scholar]
- Marston, A. Natural products as a source of protein kinase activators and inhibitors. Curr. Top. Med. Chem 2011, 11, 1333–1339. [Google Scholar]
- Nakao, Y; Fusetani, N. Enzyme inhibitors from marine invertebrates. J. Nat. Prod 2007, 70, 689–710. [Google Scholar]
- Deslandes, S; Chassaing, S; Delfourne, E. Marine pyrrolocarbazoles and analogues: Synthesis and kinase inhibition. Mar. Drugs 2009, 7, 754–786. [Google Scholar]
- Nguyen, TNT; Tepe, JJ. Preparation of hymenialdisine, analogues and their evaluation as kinase inhibitors. Curr. Med. Chem 2009, 16, 3122–3143. [Google Scholar]
- Carter, CA; Kane, CJM. Therapeutic potential of natural compounds that regulate the activity of protein kinase C. Curr. Med. Chem 2004, 11, 2883–2902. [Google Scholar]
- Newton, AC. Protein kinase C: Structure, function and regulation. J. Biol. Chem 1995, 270, 28495–28498. [Google Scholar]
- Kortmansky, J; Schwartz, GK. Bryostatin-1: A Novel PKC inhibitor in clinical development. Cancer Investig 2003, 21, 924–936. [Google Scholar]
- Fusetani, N; Nakao, Y. Enzyme inhibitors from marine invertebrates. J. Nat. Prod 2007, 70, 689–710. [Google Scholar]
- Tamaoki, T; Nomoto, H; Takahashi, I; Kato, Y; Morimoto, M; Tomita, F. Staurosporine, a potent inhibitor of phospholipid/Ca++ dependent protein kinase. Biochem. Biophys. Res. Commun 1986, 135, 397–402. [Google Scholar]
- Kinnel, RB; Scheuer, PJ. 11-Hydroxystaurosporine: A highly cytotoxic, powerful protein kinase C inhibitor from a tunicate. J. Org. Chem 1992, 57, 6327–6329. [Google Scholar]
- Pettit, GR; Herald, CL; Doubek, DL; Herald, DL; Arnold, E; Clardy, J. Isolation and structure of bryostatin 1. J. Am. Chem. Soc 1982, 104, 6846–6848. [Google Scholar]
- Rodriguez, J; Peters, BM; Kurz, L; Schatzman, RC; McCarley, D; Lou, L; Crews, P. An alkaloid protein kinase C inhibitor, xestocyclamine A, from the marine sponge Xestospongia sp. J. Am. Chem. Soc 1993, 115, 10436–10437. [Google Scholar]
- Chappell, M. Total Synthesis of Xestocyclamine A; Grant No. 1F32GM019972-01; National Institute of General Medical Sciences (NIGMS): Bethesda, MD, USA, 1999. [Google Scholar]
- Yun, H; Gagnon, A; Danishefsky, SJ. Toward the synthesis of xestocyclamine A: Investigation of double Michael reaction and direct aza Diels-Alder reaction. Tetrahedron Lett 2006, 47, 5311–5315. [Google Scholar]
- Patil, AD; Freyer, AJ; Killmer, L; Hofmann, G; Johnson, RK. Z-Axinohydantoin and debromo-Z-axinohydantoin from the sponge Stylotella aurantium: Inhibitors of protein kinase C. Nat. Prod. Res 1997, 9, 201–207. [Google Scholar]
- Freyer, AJ; Patil, AD; Killmer, L; Offen, P; Carte, B; Jurewicz, AJ; Johnson, RK. Frondosins, five new sesquiterpene hydroquinone derivatives with novel skeletons from the sponge Dysidea frondosa: Inhibitors of interleukin-8 receptors. Tetrahedron 1997, 53, 5047–5060. [Google Scholar]
- Boyd, MR; Hallock, YF; Cardellina, JH. (−)-Frondosins A and D, HIV-inhibitory sesquiterpene hydroquinone derivatives from Euryspongia sp. Nat. Prod. Res 1998, 11, 153–160. [Google Scholar]
- Trost, BM; Hu, Y; Horne, DB. Total synthesis of (+)-frondosin A. Application of the Ru-catalyzed [5 + 2] cycloaddition. J. Am. Chem. Soc 2007, 129, 11781–11790. [Google Scholar]
- Inoue, M; Frontier, AJ; Danishefsky, SJ. The total synthesis of frondosin B. Angew. Chem. Int. Ed 2000, 39, 761–764. [Google Scholar]
- Li, X; Kynea, RE; Ovaska, TV. Total syntheses of (±)-frondosin C and (±)-8-epi-frondosin C via a tandem anionic 5-exo dig cyclization—Claisen rearrangement sequence. Tetrahedron 2007, 63, 1899–1906. [Google Scholar]
- Willis, RH; de Vries, DJ. BRS1, A C30 BIS-amino, BIS-hydroxy polyunsaturated lipid from an Australian calcareous sponge that inhibits protein kinase C. Toxicon 1997, 35, 1125–1129. [Google Scholar]
- Shigemori, H; Madono, T; Sasaki, T; Mikami, Y; Kobayashi, J. Nakijiquinones A and B, new antifungal sesquiterpenoid quinones with an amino acid residue from an Okinawan marine sponge. Tetrahedron 1994, 50, 8347–8354. [Google Scholar]
- Kobayashi, J; Madono, T; Shigemori, H. Nakijiquinones C and D, new sesquiterpenoid quinones with a hydroxy amino acid residue from a marine sponge inhibiting c-erbB-2 kinase. Tetrahedron 1995, 51, 10867–10874. [Google Scholar]
- Takahashi, Y; Kubota, T; Ito, J; Mikami, Y; Fromont, J; Kobayashi, J. Nakijiquinones G–I, new sesquiterpenoid quinones from marine sponge. Bioorg. Med. Chem 2008, 16, 7561–7564. [Google Scholar]
- Kissau, L; Stahl, P; Mazitschek, R; Giannis, A; Waldmann, H. Development of natural product-derived receptor tyrosine kinase inhibitors based on conservation of protein domain fold. J. Med. Chem 2003, 46, 2917–2931. [Google Scholar]
- Stahl, P; Waldmann, H. Asymmetric synthesis of the nakijiquinones—Selective inhibitors of the Her-2/Neu protooncogene. Angew. Chem. Int. Ed 1999, 38, 3710–3713. [Google Scholar]
- Stahl, P; Kissau, L; Mazitschek, R; Huwe, A; Furet, P; Giannis, A; Waldmann, H. Total synthesis and biological evaluation of the nakijiquinones. J. Am. Chem. Soc 2001, 123, 11586–11593. [Google Scholar]
- Slamon, DJ; Clark, GM; Wong, SG; Levin, WJ; Ullrich, A; McGuire, WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/Neu oncogene. Science 1987, 235, 177–182. [Google Scholar]
- He, H; Kulanthaivel, P; Baker, BJ. New cytotoxic sesterterpenes from the marine sponge Spongia sp. Tetrahedron Lett 1994, 35, 7189–7192. [Google Scholar]
- Kulciţki, V. A biomimetic approach to some specifically functionalized cyclic terpenoids. Acta Biochim. Pol 2007, 54, 679–693. [Google Scholar]
- Longley, RE; Harmody, D. A rapid colorimetric microassay to detect agonist/antagonists of protein-kinase-C based on adherence of EL-4.IL-2 cells. J. Antibiot 1991, 44, 93–102. [Google Scholar]
- Horton, PA; Koehn, FE; Longley, RE; McConnell, OJ. Lasonolide A, a new cytotoxic macrolide from the marine sponge Forcepia sp. J. Am. Chem. Soc 1994, 116, 6015–6016. [Google Scholar]
- Isbrucker, RA; Guzman, EA; Pitts, TP; Wright, AE. Early effects of lasonolide A on pancreatic cancer cells. J. Pharmacol. Exp. Ther 2009, 331, 733–739. [Google Scholar]
- Alvi, KA; Jaspars, M; Crews, P; Strulovici, B; Oto, E. Penazetidine-A, an alkaloid inhibitor of protein kinase C. Bioorg. Med. Chem. Lett 1994, 4, 2447–2450. [Google Scholar]
- Chan, JA; Freyer, AJ; Carte, BK; Hemling, ME; Hofmann, GA; Mattern, MR; Mentzer, MA; Westley, JW. Protein kinase C inhibitors: Novel spirosesquiterpene aldehydes from a marine sponge Aka (=Siphonodictyon) coralliphagum. J. Nat. Prod 1994, 57, 1543–1548. [Google Scholar]
- Morgan, D. The Cell-Cycle Control System. In The Cell Cycle: Principles of Control; Oxford University Press: Oxford, UK, 2007; pp. 30–31. [Google Scholar]
- Morris, D; Bramwell, V; Turcotte, R; Figueredo, A; Blackstein, M; Verma, S; Matthews, S; Eisenhauer, E. A phase II study of flavopiridol in patients with previously untreated advanced soft tissue sarcoma. Sarcoma 2006, 1, 1–7. [Google Scholar]
- Muhtasib, H. Cyclin-dependent kinase inhibitors from natural sources: Recent advances and future prospects for cancer treatment. Adv. Phytomed 2006, 2, 155–167. [Google Scholar]
- Castedo, M; Perfettini, J-L; Roumier, T; Kroemer, G. Cyclin-dependent kinase-1: Linking apoptosis to cell cycle and mitotic castastrophe. Cell Death Differ 2002, 9, 1287–1293. [Google Scholar]
- Cimino, G; de Rosa, S; de Stefano, S; Mazzarella, L; Puliti, R; Sodano, G. Isolation and X-ray crystal structure of a novel bromo-compound from two marine sponges. Tetrahedron Lett 1982, 23, 767–768. [Google Scholar]
- Meijer, L; Thunnissen, AM; White, AW; Garnier, M; Nikolic, M; Tsai, LH; Walter, J; Cleverley, KE; Salinas, PC; Wu, YZ; et al. Inhibition of cyclin-dependent kinases, GSK-3[beta] and CK1 by hymenialdisine, a marine sponge constituent. Chem. Biol 2000, 7, 51–63. [Google Scholar]
- Goga, A; Yang, D; Tward, A; Morgan, D; Bishop, J. Inhibition of CDK1 as a potential therapy for tumours over-expressing MYC. Nat. Med 2007, 13, 820–827. [Google Scholar]
- Wan, Y; Hur, W; Cho, C; Liu, Y; Adrian, F; Lozach, O; Bach, S; Mayer, T; Fabbro, D; Meijer, L; et al. Synthesis and target identification of hymenialdisine analogs. Chem. Biol 2004, 11, 247–259. [Google Scholar]
- Tepe, J. Preparation of Hymenialdisine Derivatives and Use Thereof. US Patent 7,193,079, 20 March 2007. [Google Scholar]
- Killday, B; Yarwood, D; Sills, M; Murphy, P; Hooper, J; Wright, A. Microxine, a new cdc2 kinase inhibitor from the Australian marine sponge Microxima species. J. Nat. Prod 2001, 64, 525–526. [Google Scholar]
- Walker, SR; Carter, EJ; Huff, BC; Morris, JC. Variolins and related alkaloids. Chem. Rev 2009, 109, 3080–3098. [Google Scholar]
- Trimurtulu, G; Faulkner, DJ; Perry, NB; Ettouati, L; Litaudon, M; Blunt, JW; Munro, MHG; Jameson, GB. Alkaloids from the antarctic sponge Kirkpatrickia varialosa. Part 2: Variolin A and N(3′)-methyl tetrahydrovariolin B. Tetrahedron 1994, 50, 3993–4000. [Google Scholar]
- Anderson, RJ; Hill, JB; Morris, JC. Concise total syntheses of variolin B and deoxyvariolin B. J. Org. Chem 2005, 70, 6204–6212. [Google Scholar]
- Baeza, A; Mendiola, J; Burgos, C; Alvarez-Builla, J; Vaquero, JJ. Palladium-mediated C-N, C-C, and C-O functionalization of azolopyrimidines: A new total synthesis of variolin B. Tetrahedron Lett 2008, 49, 4073–4077. [Google Scholar]
- Ahaidar, A; Fernandez, D; Danelon, G; Cuevas, C; Manzanares, I; Albericio, F; Joule, JA; Alvarez, M. Total syntheses of variolin B and deoxyvariolin B. J. Org. Chem 2003, 68, 10020–10029. [Google Scholar]
- Molina, P; Fresneda, PM; Delgado, S; Bleda, JA. Synthesis of the potent antitumoral marine alkaloid variolin B. Tetrahedron Lett 2002, 43, 1005–1007. [Google Scholar]
- Sherr, CJ; Roberts, JM. CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes Dev 1999, 13, 1501–1512. [Google Scholar]
- Lin, J; Yan, X-J; Chen, H-M. Fascaplysin, a selective CDK4 inhibitor, exhibit anti-angiogenic activity in vitro and in vivo. Cancer Chemother. Pharmacol 2007, 59, 439–445. [Google Scholar]
- Soni, R; Muller, L; Furet, P; Schoepfer, J; Stephan, C; Zumstein-Mecker, S; Fretz, H; Chaudhuri, B. Inhibition of cyclin-dependent kinase 4 (Cdk4) by fascaplysin, a marine natural product. Biochem. Biophys. Res. Commun 2000, 275, 877–884. [Google Scholar]
- Kobayashi, J; Suzuki, M; Tsuda, M. Konbu’acidin A, a new bromopyrrole alkaloid with cdk4 inhibitory activity from hymeniacidon sponge. Tetrahedron 1997, 53, 15681–15684. [Google Scholar]
- Mukku, V; Edrada, RA; Schmitz, FJ; Shanks, MK; Chaudhuri, B; Fabbro, D. New sesquiterpene quinols from a micronesian sponge, Aka sp. J. Nat. Prod 2003, 66, 686–689. [Google Scholar]
- Levitzki, A; Mishani, E. Tyrphostins and other tyrosine kinase inhibitors. Annu. Rev. Biochem 2006, 75, 93–109. [Google Scholar]
- Carapancea, M. Strategies to Increase Effectiveness of Growth Factor Receptors-Targeted Therapy in Glioblastoma. Licentiate Thesis, Karolinska Institutet, Stockholm, Sweden, December 2007. [Google Scholar]
- Bifulco, G; Bruno, I; Minale, L; Riccio, R; Debitus, C; Bourdy, G; Vassas, A; Lavayre, J. Bioactive prenylhydroquinone sulfates and a novel C31 furanoterpene alcohol sulfate from the marine sponge, Ircinia sp. J. Nat. Prod 1995, 58, 1444–1449. [Google Scholar]
- Gray, G; Macara, I. The pp60V-SRC tyrosine kinase desensitizes epidermal growth factor binding to 3T3 fibroblasts by two distinct protein kinase C-independent mechanisms. J. Biol. Chem 1988, 263, 10714–10719. [Google Scholar]
- Alvi, KA; Diaz, MC; Crews, P; Slate, DL; Lee, RH; Moretti, R. Evaluation of new sesquiterpene quinones from two Dysidea sponge species as inhibitors of protein tyrosine kinase. J. Org. Chem 1992, 57, 6604–6607. [Google Scholar]
- Lee, RH; Slate, DL; Moretti, R; Alvi, KA; Crews, P. Marine sponge polyketide inhibitors of protein tyrosine kinase. Biochem. Biophys. Res. Commun 1992, 184, 765–772. [Google Scholar]
- Laurent, D; Jullian, V; Parenty, A; Knibiehler, M; Dorin, D; Schmitt, S; Lozach, O; Lebouvier, N; Frostin, M; Alby, F; et al. Antimalarial potential of xestoquinone, a protein kinase inhibitor isolated from a Vanuatu marine sponge Xestospongia sp. Bioorg. Med. Chem 2006, 14, 4477–4482. [Google Scholar]
- Toyooka, N; Nagaoka, M; Sasaki, E; Qin, H; Kakuda, H; Nemoto, H. Model studies toward the total synthesis of halenaquinol and halenaquinone. Tetrahedron 2002, 58, 6097–6101. [Google Scholar]
- Cao, S; Foster, C; Brisson, M; Lazo, JS; Kingston, DGI. Halenaquinone and xestoquinone derivatives, inhibitors of Cdc25B phosphatase from a Xestospongia sp. Bioorg. Med. Chem 2005, 13, 999–1003. [Google Scholar]
- Alvi, KA; Rodriguez, J; Diaz, MC; Moretti, R; Wilhelm, RS; Lee, RH; Slate, DL; Crews, P. Protein tyrosine kinase inhibitory properties of planar polycyclics obtained from the marine sponge Xestospongia cf. carbonaria and from total synthesis. J. Org. Chem 1993, 58, 4871–4880. [Google Scholar]
- Carpenter, G; Cohen, S. Epidermal growth factor. Annu. Rev. Biochem 1979, 48, 193–216. [Google Scholar]
- Kobayashi, J; Inaba, K; Tsuda, M. Tauroacidins A and B, new bromopyrrole alkaloids possessing a taurine residue from hymeniacidon sponge. Tetrahedron 1997, 53, 16679–16682. [Google Scholar]
- Kobayashi, J; Hirano, K; Kubota, T; Tsuda, M; Watanabe, K; Fromont, J. Ma’edamines A and B, cytotoxic bromotyrosine alkaloids with a unique 2(1H)pyrazinone ring from sponge Suberea sp. Tetrahedron 2000, 56, 8107–8110. [Google Scholar]
- Inaba, K; Sato, H; Tsuda, M; Kobayashi, J. Spongiacidins A–D, new bromopyrrole alkaloids from hymeniacidon sponge. J. Nat. Prod 1998, 61, 693–695. [Google Scholar]
- Gossauer, A. Monopyrollic Natural Compounds Including Tetramic Acid Derivatives; Springer: Berlin, Germany, 2003. [Google Scholar]
- Kreuter, M-H; Leake, RE; Rinaldi, F; Müller-Klieser, W; Maidhof, A; Müller, WEG; Schröder, HC. Inhibition of intrinsic protein tyrosine kinase activity of EGF-receptor kinase complex from human breast cancer cells by the marine sponge metabolite (+)-aeroplysinin-1. Comp. Biochem. Physiol. B Biochem. Mol. Biol 1990, 97, 151–158. [Google Scholar]
- Rodriguez-Nieto, S; Gonzalez-Iriarte, M; Carmona, R; Munoz-Chapuli, R; Medina, MA; Quesada, AR. Antiangiogenic activity of aeroplysinin-1, a brominated compound isolated from a marine sponge. FASEB J 2002, 16, 261–263. [Google Scholar]
- Rateb, ME; Houssen, WE; Legrave, NM; Clements, C; Jaspars, M; Ebel, R. Dibenzofurans from the marine sponge-derived ascomycete Super1F1-09. Bot. Mar 2010, 53, 499–506. [Google Scholar]
- Rateb, ME; Houssen, WE; Schumacher, M; Harrison, WTA; Diederich, M; Ebel, R; Jaspars, M. Bioactive diterpene derivatives from the marine sponge Spongionella sp. J. Nat. Prod 2009, 72, 1471–1476. [Google Scholar]
- Kannan-Thulasiraman, P; Katsoulidis, E; Tallman, MS; Arthur, JSC; Platanias, LC. Activation of the mitogen- and stress-activated kinase 1 by arsenic trioxide. J. Biol. Chem 2006, 281, 22446–22452. [Google Scholar]
- Buchanan, MS; Edser, A; King, G; Whitmore, J; Quinn, RJ. Cheilanthane sesterterpenes, protein kinase inhibitors, from a marine sponge of the genus Ircinia. J. Nat. Prod 2001, 64, 300–303. [Google Scholar]
- Brown, J; Kesler, C; Neary, J; Fishman, L. Effects of marine sponge extracts on mitogen-activated protein kinase (MAPK/ERK1,2) activity in SW-13 huma adrenal carcinoma cells. Toxicon 2001, 39, 1835–1839. [Google Scholar]
- Kolch, W. Meaningful relationships: The regulation of the Ras/Raf/MEK/ERK pathway by protein interactions. Biochem. J 2000, 351, 289–305. [Google Scholar]
- Tasdemir, D; Mallon, R; Greenstein, M; Feldberg, L; Kim, S; Collins, K; Wojciechowicz, D; Mangalindan, G; Concepcion, G; Harper, MK; Ireland, CM. Aldisine alkaloids from the Philippine sponge Stylissa massa are potent Inhibitors of mitogen-activated protein kinase kinase-1 (MEK-1). J. Med. Chem 2002, 45, 529–532. [Google Scholar]
- Segraves, NL; Crews, P. A Madagascar sponge Batzella sp. as a source of alkylated iminosugars. J. Nat. Prod 2005, 68, 118–121. [Google Scholar]
- Freitas, J; Malpezzi, E; Costa, L; Berlinck, R; Almeida, A; Ogawa, C; Sanchez, M; Hajdu, E. Cytotoxic and neurotoxic effects induced by halitoxin isolated from Amphimedon viridis (Porifera). Toxicon 1996, 34, 335. [Google Scholar]
- Lee, K-H; Nishimura, S; Matsunaga, S; Fusetani, N; Horinouchi, S; Yoshida, M. Inhibition of protein synthesis and activation of stress-activated protein kinases by onnamide A and theopederin B, antitumor marine natural products. Cancer Sci 2005, 96, 357–364. [Google Scholar]
- Fedorov, S; Bode, A; Stonik, V; Gorshkova, I; Schmid, P; Radchenko, O; Berdyshev, E; Dong, Z. Marine alkaloid polycarpine and its synthetic derivative dimethylpolycarpine induce apoptosis in JB6 cells through p53- and caspase 3-dependent pathways. Pharm. Res 2004, 21, 2307–2319. [Google Scholar]
- Fusetani, N; Sugawara, T; Matsunaga, S. Bioactive marine metabolites. 41. Theopederins A–E, potent antitumor metabolites from a marine sponge, Theonella sp. J. Org. Chem 1992, 57, 3828–3832. [Google Scholar]
- Sakemi, S; Ichiba, T; Kohmoto, S; Saucy, G; Higa, T. Isolation and structure elucidation of onnamide A, a new bioactive metabolite of a marine sponge, Theonella sp. J. Am. Chem. Soc 1988, 110, 4851–4853. [Google Scholar]
- Andreasen, P. PAI-1—A Potential therapeutic target in cancer. Curr. Drug Targets 2007, 8, 1030–1041. [Google Scholar]
- Williams, DE; Telliez, JB; Liu, J; Tahir, A; van Soest, R; Andersen, RJ. Meroterpenoid MAPKAP (MK2) inhibitors isolated from the Indonesian marine sponge Acanthodendrilla sp. J. Nat. Prod 2004, 67, 2127–2129. [Google Scholar]
- Hamann, M; Alonso, D; Martin-Aparicio, E; Fuertes, A; Perez-Puerto, M; Castro, A; Morales, S; Navarro, M; Monte-Millan, M; Medina, M; et al. Glycogen synthase kinase-3 (GSK-3) inhibitory activity and structure activity relationship (SAR). Studies of the manzamine alkaloids. Potential for Alzheimer’s disease. J. Nat. Prod 2007, 70, 1397–1405. [Google Scholar]
- Ang, K; Holmes, M; Higa, T; Hamann, M; Kara, U. In vivo antimalarial activity of the beta-carboline alkaloid manzamine A. Antimicrob. Agents Chemother 2000, 44, 1645–1649. [Google Scholar]
- Kara, U; Higa, T; Holmes, M; Ang, K. Antimalarial activity of β-carboline alkaloids. US Patent 6,143,756, 7 November 2000. [Google Scholar]
- McCulloch, MWB; Bugni, TS; Concepcion, GP; Coombs, GS; Harper, MK; Kaur, S; Mangalindan, GC; Mutizwa, MM; Veltri, CA; Virshup, DM; et al. Carteriosulfonic acids A–C, GSK-3 beta inhibitors from a Carteriospongia sp. J. Nat. Prod 2009, 72, 1651–1656. [Google Scholar]
- Marion, F; Williams, DE; Patrick, BO; Hollander, I; Mallon, R; Kim, SC; Roll, DM; Feldberg, L; Van Soest, R; Andersen, RJ. Liphagal, a selective inhibitor of PI3 kinase alpha isolated from the sponge Aka coralliphaga: Structure elucidation and biomimetic synthesis. Org. Lett 2006, 8, 321–324. [Google Scholar]
- Alvarez-Manzaneda, E; Chahboun, R; Alvarez, E; Cano, MJ; Haidour, A; Alvarez-Manzaneda, R. Enantioselective total synthesis of the selective PI3 kinase inhibitor liphagal. Org. Lett 2010, 12, 4450–4453. [Google Scholar]
- Hertiani, T; Edrada-Ebel, RA; Kubbutat, MHG; van Soest, RWM; Proksch, P. Protein kinase inhibitors from Indonesian sponge Axynissa sp. Maj. Farm. Indones 2008, 19, 78–85. [Google Scholar]
- Zivanovic, A; Pastro, NJ; Fromont, J; Thomson, M; Skropeta, D. Kinase Inhibitory, haemolytic and cytotoxic activity of three deep-water sponges from North Western Australia and their fatty acid composition. Nat Prod Commun 2011, in press. [Google Scholar]
- Lebouvier, N; Jullian, V; Desvignes, I; Maurel, S; Parenty, A; Dorin-Semblat, D; Doerig, C; Sauvain, M; Laurent, D. Antiplasmodial activities of homogentisic acid derivative protein kinase inhibitors isolated from a vanuatu marine sponge Pseudoceratina sp. Mar. Drugs 2009, 7, 640–653. [Google Scholar]
- Sauleau, P; Retailleau, P; Nogues, S; Carletti, I; Marcourt, L; Raux, R; Al Mourabit, A; Debitus, C. Dihydrohymenialdisines, new pyrrole-2-aminoimidazole alkaloids from the marine sponge Cymbastela cantharella. Tetrahedron Lett 2011, 52, 2676–2678. [Google Scholar]
- Skropeta, D. Deep-sea natural products. Nat. Prod. Rep 2008, 25, 1131–1166. [Google Scholar]
Figure 1.
Protein kinase C inhibitors isolated from marine sponges.

Figure 2.
Cyclin dependant kinase inhibitors isolated from marine sponges.

Figure 3.
Tyrosine protein kinase inhibitors isolated from marine sponges.

Figure 4.
Epidermal growth factor receptor inhibitors isolated from marine sponges.

Figure 5.
Mitogen-activated protein kinase inhibitors isolated from marine sponges.

Figure 6.
Glycogen synthase kinase-3 inhibitors isolated from marine sponges.

Figure 7.
Inhibitors of PI3K, Src, and focal adhesion kinase isolated from marine sponges.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Kinase | Sponge species | Natural product (or compound type) | IC50 (μM) ₤ (or % inhibition) | Ref. |
|---|---|---|---|---|
| PKC | Xestospongia sp. | Xestocyclamine A (1) | 10 | [19] |
| Stylotella aurantium | Axinohydantoins (2, 3) | 9–22 | [22] | |
| Stylotella aurantium | Hymenialdisines (4, 5) | 0.8–1.3 | [22] | |
| Dysidea frondosa | Frondosins A–E (6–10) | 2–31 | [23] | |
| Class Calcarea | BRS 1 (11) | 98 | [28] * | |
| Family Spongiidae | Nakijiquinones A–D, G–I (12–18) | 23–270 | [29,30] | |
| Spongia sp. | Spongianolides A–E (19–23) | 20–30 | [36] | |
| Forecpia sp. | Lasonolide A (24) | 0.03 | [38,40] * | |
| Penares sollesi | Penazetidine A (25) | 1 | [41] | |
| Aka coralliphaga | Corallidictyals A and B (26, 27) | 28 | [42] | |
| CDK | Axinella verrucosa | Hymenialdisine (4) | 0.02 | [48] * |
| Microxina sp. | Microxine (28) | 13 | [52] | |
| Kirkpatrickia varialosa | Variolin B (29) | 0.03 | [53] * | |
| Fascaplysinopsis sp. | Fascaplysin (30) | 0.4 | [61] * | |
| Hymeniacidon sp. | Konbu’acidin A (31) | 27 | [62] | |
| Aka sp. | Quinol derivative (34) | 0.019 | [63] | |
| Aka sp. | Halistanol sulfate (37) | 0.013 | [63] | |
| TPK | Ircinia sp. | Prenylhydroquinone 4-sulfates (38–40) | 7–15 | [66] |
| Dysidea sp. | Melemeleone B (41) | 28 | [68] | |
| Xestospongia sp. | Halenoquinone (42, 43) | 1.5–5 | [69,73] * | |
| Xestospongia sp. | Halenaquinols (44, 45) | 0.6–60 | [69] * | |
| Xestospongia sp. | Xestoquinone (46) | 28 | [69] * | |
| Xestospongia sp. | Xestoquinolide A (47) | 80 | [73] * | |
| EGFR | Hymeniacidon sp. | Tauroacidins A–B (48, 49) | 38–45 | [75] |
| Suberea sp. | Ma’edamine A (50) | 11 | [76] | |
| Hymeniacidon sp. | Spongiacidins A–B (51, 52) | 19–21 | [78] | |
| Verongia aerophoba | Aeroplysinin-1 (53) | 0.5 | [79] | |
| Acanthella cavernosa | Dibenzofurandiols (54–57) | 33–59 † | [81] | |
| Spongionella sp. | 3′-Norspongiolactone (58) | 25 † | [82] | |
| Spongionella sp. | Gracilins J–L (59–61) | 19–75 † | [82] | |
| MAPK | Ircinia sp. | Cheilanthene sesterpenoids (62–65) | 4–90 | [84] |
| Raf/MAP | Stylissa massa | Hymenialdisines (4, 5) | 0.003–0.006 | [87] * |
| Stylotella aurantium | Hymenin (66) | 129 | [87] * | |
| Theonella sp. | Theopederin B (68) | - ‡ | [90,92] * | |
| Theonella sp. | Onnamide A (67) | - ‡ | [90,93] * | |
| Acanthodendrilla sp. | (+)-Makassaric acid (69) | 20 | [95] | |
| Acanthodendrilla sp. | (+)-Subersic acid (70) | 9.6 | [95] | |
| GSK-3 | Haliclona sp. | Manzamine A (71) | 10 | [96] * |
| Unidentified sp. | Glycerol lipids (72–74) | 0.1–0.4 | [99] | |
| Others | Aka coralliphaga | Liphagal (75) | 0.1 | [100,101] |
| Axynissa sp. | (+)-Curcuphenol (76) | 36 | [102] | |
| Axynissa sp. | (+)-Curcudiol (77) | 37 | [102] | |
| Pseudoceratina sp. | Homogentisic acid (78) | 1.8 | [104] |
₤Values reported in μg/mL were converted to μM;
†% Inhibition at 100 μM;
‡Induces activation of p38 and JNK;
*An asterisk denotes articles containing detailed characterisation of the kinase inhibitory activity.
© 2011 by the authors; licensee MDPI, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Skropeta, D.; Pastro, N.; Zivanovic, A. Kinase Inhibitors from Marine Sponges. Mar. Drugs 2011, 9, 2131-2154. https://0-doi-org.brum.beds.ac.uk/10.3390/md9102131
AMA Style
Skropeta D, Pastro N, Zivanovic A. Kinase Inhibitors from Marine Sponges. Marine Drugs. 2011; 9(10):2131-2154. https://0-doi-org.brum.beds.ac.uk/10.3390/md9102131
Chicago/Turabian StyleSkropeta, Danielle, Natalie Pastro, and Ana Zivanovic. 2011. "Kinase Inhibitors from Marine Sponges" Marine Drugs 9, no. 10: 2131-2154. https://0-doi-org.brum.beds.ac.uk/10.3390/md9102131