
Assessment of Offspring DNA Methylation across the Lifecourse Associated with Prenatal Maternal Smoking Using Bayesian Mixture Modelling

Abstract
:1. Introduction
2. Experimental Section
2.1. Data
2.2. Statistical Methods
3. Results

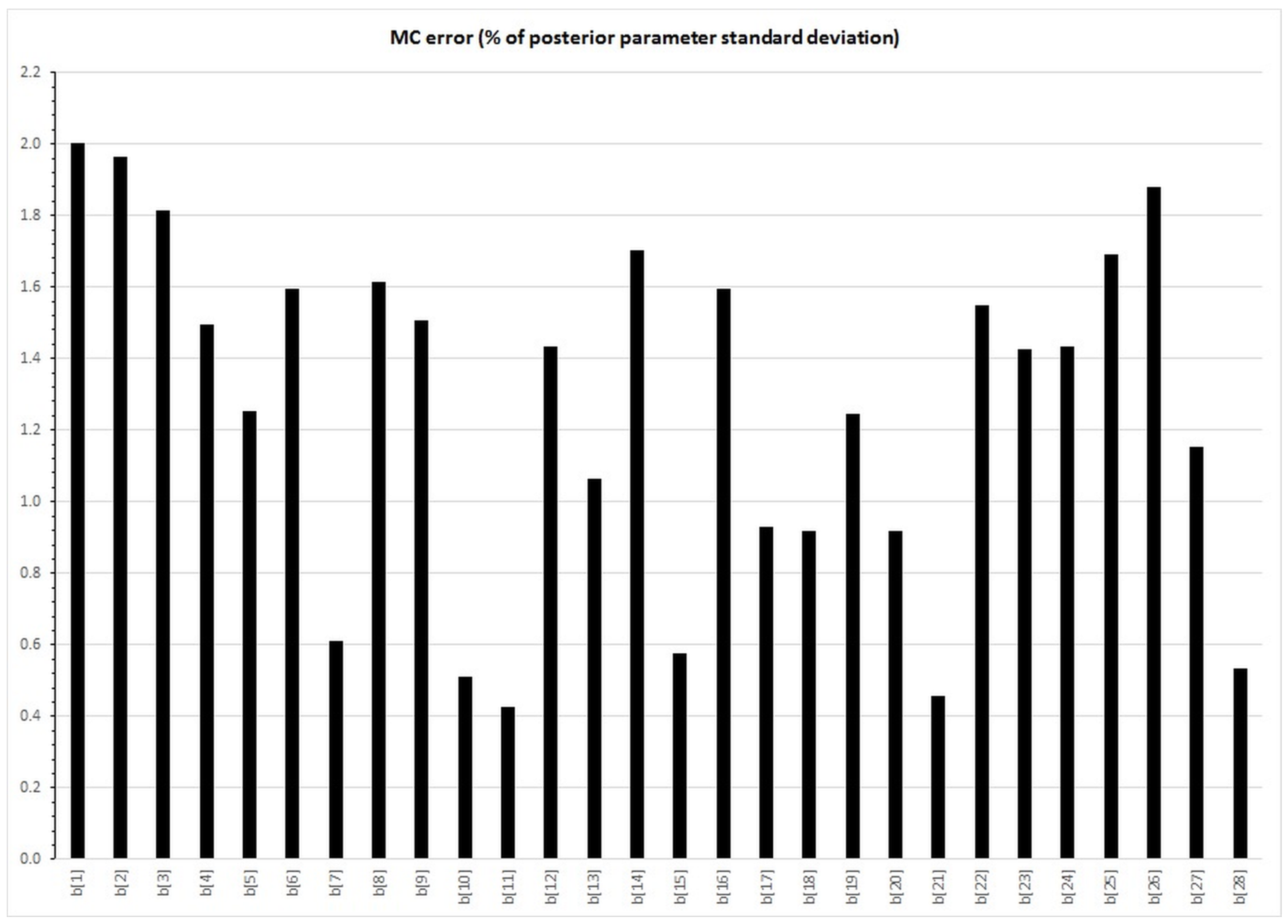{kind=link}
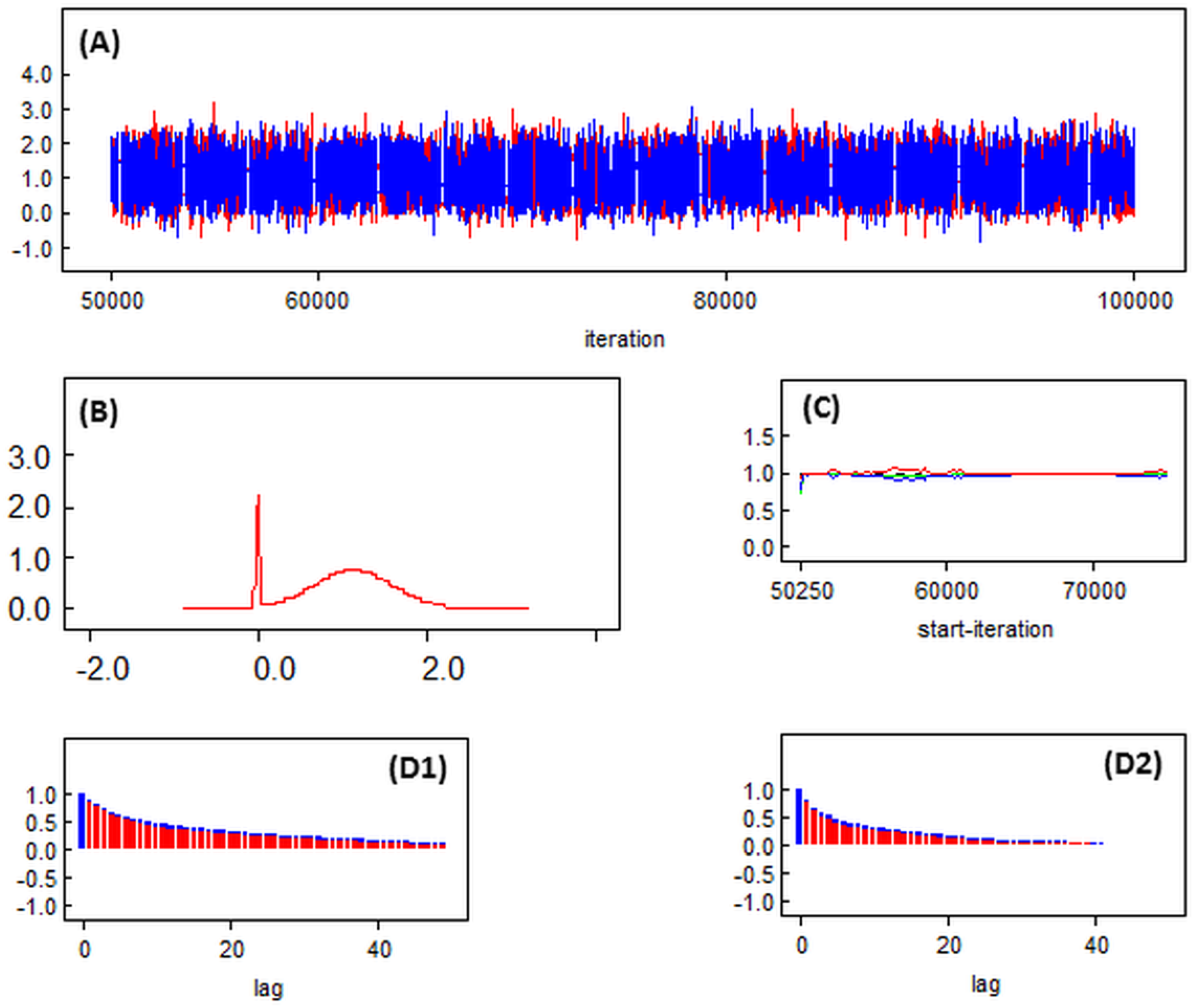{kind=link}
| Characteristic | ARIES Dataset 1 (Missing Data Imputed) | ARIES Dataset (*) | ||||
|---|---|---|---|---|---|---|
| Sustained Smoker | Non-Smokers | Sustained Smoker | Non-Smokers | |||
| N | % | N | % | % | % | |
| Sustained maternal smoking during pregnancy | 110 | 12.0 | 804 | 87.9 | 11.5 | 88.5 |
| Maternal education | ||||||
| CSE/vocational | 37 | 33.6 | 114 | 14.2 | 33.0 | 13.9 |
| O-level | 46 | 41.8 | 257 | 32.0 | 40.9 | 32.7 |
| A-level | 18 | 16.4 | 249 | 31.0 | 18.2 | 30.4 |
| Degree | 9 | 8.2 | 184 | 22.9 | 8.0 | 23.1 |
| Maternal age | ||||||
| <25 years | 32 | 29.1 | 58 | 7.2 | 30.8 | 7.6 |
| 25–30 | 40 | 36.4 | 313 | 38.9 | 37.4 | 39.2 |
| >30 years | 38 | 34.5 | 433 | 53.9 | 31.9 | 53.2 |
| Alcohol | ||||||
| Non-drinker | 43 | 39.1 | 269 | 33.5 | 40.9 | 34.2 |
| Drank before 18 weeks gestation | 5 | 4.5 | 122 | 15.2 | 5.7 | 15.6 |
| Still drinking at 18 weeks of gestation | 62 | 56.4 | 413 | 51.4 | 53.4 | 50.2 |
| Paternal smoking | 83 | 75.5 | 177 | 22.0 | 79.1 | 21.4 |
| Household social class (manual labour) | 38 | 34.5 | 72 | 9.1 | 35.4 | 8.8 |


| CpG Site | Chromosome | Gene Region | Position | Cord blood | Age 7 Years | Age ~17 Years | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Prob. of Effect (T) 1 | OR 2 | 95% Cred. Limit 3 | Prob. of Effect (T) | OR | 95% Cred. Limit | Prob. of Effect (T) | OR | 95% Cred. Limit (T) | ||||
| cg05575921 | 5 | AHRR | 373 378 | 0.68 | 0.49 | 0.13–1.02 | 0.33 | 0.85 | 0.27–1.36 | 0.34 | 0.84 | 0.28–1.31 |
| cg22132788 | 7 | MYO1G | 45 002 486 | 0.37 | 1.26 | 0.83–4.16 | 0.53 | 1.57 | 0.95–5.80 | 0.55 | 1.61 | 0.97–5.85 |
| cg12803068 | 7 | MYO1G | 45 002 919 | 0.87 | 2.70 | 0.99–7.52 | 0.98 | 4.31 | 1.30–10.98 | 0.94 | 3.33 | 1.00–8.69 |
| cg09935388 | 1 | GFI1 | 92 947 588 | 0.87 | 0.36 | 0.12–1.01 | 0.57 | 0.65 | 0.22–1.02 | 0.43 | 0.76 | 0.27–1.05 |
| cg14179389 | 1 | GFI1 | 92 947 961 | 0.51 | 0.67 | 0.20–1.06 | 0.39 | 0.79 | 0.25–1.18 | 0.36 | 0.82 | 0.26–1.23 |
| cg18146737 | 1 | GFI1 | 92 946 700 | 0.38 | 0.80 | 0.26–1.14 | 0.24 | 0.99 | 0.48–1.84 | 0.24 | 0.99 | 0.50–1.82 |
| cg05549655 | 15 | CYP1A1 | 75 019 143 | 0.26 | 1.05 | 0.54–2.49 | 0.27 | 1.04 | 0.56–2.43 | 0.25 | 1.05 | 0.59–2.45 |
| cg06338710 | 1 | GFI1 | 92 946 187 | 0.72 | 0.54 | 0.20–1.02 | 0.20 | 1.02 | 0.68–1.75 | 0.22 | 0.95 | 0.49–1.29 |
| cg12876356 | 1 | GFI1 | 92 946 825 | 0.48 | 0.72 | 0.24–1.04 | 0.20 | 1.02 | 0.69–1.73 | 0.28 | 0.89 | 0.40–1.13 |
| cg25949550 | 7 | CNTNAP2 | 145 814 306 | 0.25 | 0.98 | 0.45–1.84 | 0.25 | 0.98 | 0.45–1.89 | 0.26 | 0.97 | 0.44–1.86 |
| cg11902777 | 5 | AHRR | 3 68 843 | 0.25 | 0.99 | 0.46–1.92 | 0.26 | 0.98 | 0.46–1.95 | 0.25 | 0.99 | 0.47–1.94 |
| cg12101586 | 15 | CYP1A1 | 75 019 203 | 0.48 | 1.47 | 0.94–5.20 | 0.41 | 1.33 | 0.88–4.48 | 0.33 | 1.17 | 0.76–3.45 |
| cg18316974 | 1 | GFI1 | 92 947 035 | 0.22 | 0.98 | 0.51–1.68 | 0.25 | 0.96 | 0.42–1.72 | 0.24 | 0.97 | 0.45–1.69 |
| cg26146569 | 15 | KLF13 | 31 637 592 | 0.40 | 0.77 | 0.24–1.13 | 0.30 | 0.88 | 0.32–1.35 | 0.24 | 1.03 | 0.60–2.19 |
| cg07339236 | 20 | ATP9A | 50 312 490 | 0.26 | 0.95 | 0.41–1.73 | 0.25 | 0.97 | 0.44–1.83 | 0.26 | 0.96 | 0.42–1.77 |
| cg09662411 | 1 | GFI1 | 92 946 132 | 0.73 | 0.48 | 0.15–1.02 | 0.20 | 1.01 | 0.64–1.67 | 0.21 | 1.04 | 0.73–1.93 |
| cg18092474 | 15 | CYP1A1 | 75 019 302 | 0.28 | 1.11 | 0.79–2.74 | 0.25 | 1.06 | 0.67–2.44 | 0.54 | 1.55 | 0.97–5.11 |
| cg04180046 | 7 | MYO1G | 45 002 736 | 0.33 | 1.18 | 0.76–3.49 | 0.34 | 1.20 | 0.77–3.79 | 0.42 | 1.32 | 0.85–4.36 |
| cg25189904 | 1 | GNG12 | 68 299 493 | 0.41 | 0.76 | 0.23–1.16 | 0.34 | 0.85 | 0.28–1.32 | 0.24 | 0.99 | 0.49–1.87 |
| cg04598670 | 7 | ENSG00000225718 | 68 697 651 | 0.28 | 0.90 | 0.36–1.34 | 0.26 | 0.94 | 0.40–1.51 | 0.24 | 0.99 | 0.51–1.76 |
| cg27629977 | 2 | CTNNA2 | 80 531 633 | 0.25 | 1.03 | 0.56–2.29 | 0.26 | 1.01 | 0.50–2.11 | 0.25 | 0.99 | 0.47–1.97 |
| cg10835306 | 9 | NOTCH1 | 139 396 760 | 0.33 | 0.85 | 0.31–1.21 | 0.27 | 1.07 | 0.64–2.50 | 0.25 | 0.93 | 0.41–1.40 |
| cg00483459 | 3 | ALS2CL | 46 735 782 | 0.34 | 0.85 | 0.28–1.29 | 0.24 | 1.01 | 0.53–2.07 | 0.27 | 1.08 | 0.63–2.70 |
| cg22549041 | 15 | CYP1A1 | 75 019 251 | 0.59 | 1.69 | 0.97–5.77 | 0.39 | 1.28 | 0.87–4.06 | 0.32 | 1.16 | 0.77–3.28 |
| cg22937882 | 5 | AHRR | 4 05 774 | 0.29 | 1.12 | 0.71–3.06 | 0.26 | 0.95 | 0.40–1.65 | 0.25 | 0.99 | 0.48–1.89 |
| cg11196333 | 1 | CHI3L1 | 203 154 370 | 0.49 | 0.69 | 0.21–1.07 | 0.28 | 1.08 | 0.67–2.67 | 0.29 | 1.12 | 0.73–2.98 |
| cg00624799 | 15 | ZNF710 | 90 605 618 | 0.28 | 0.92 | 0.36–1.54 | 0.25 | 0.97 | 0.44–1.82 | 0.26 | 1.01 | 0.51–2.16 |
| cg00560284 | 12 | SPATS2 | 49 783 222 | 0.27 | 0.95 | 0.40–1.70 | 0.25 | 0.99 | 0.48–1.94 | 0.25 | 1.00 | 0.48–1.99 |
4. Discussion
5. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interests
References
- Florath, I.; Butterbach, K.; Muller, H.; Bewerunge-Hudler, M.; Brenner, H. Cross-sectional and longitudinal changes in DNA methylation with age: An epigenome-wide analysis revealing over 60 novel age-associated CpG sites. Hum. Mol. Genet. 2014, 23, 1186–1201. [Google Scholar] [CrossRef] [PubMed]
- Hannum, G.; Guinney, J.; Zhao, L.; Zhang, L.; Hughes, G.; Sadda, S.; Klotzle, B.; Bibikova, M.; Fan, J.B.; Gao, Y.; et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell 2013, 49, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Groom, A.; Elliott, H.R.; Embleton, N.D.; Relton, C.L. Epigenetics and child health: Basic principles. Arch. Dis. Child. 2011, 96, 863–869. [Google Scholar] [CrossRef] [PubMed]
- Mensaert, K.; Denil, S.; Trooskens, G.; Van Criekinge, W.; Thas, O.; de Meyer, T. Next-generation technologies and data analytical approaches for epigenomics. Environ. Mol. Mutagen. 2014, 55, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Winzer-Serhan, U.H. Long-term consequences of maternal smoking and developmental chronic nicotine exposure. Front. Biosci. 2008, 13, 636–649. [Google Scholar] [CrossRef] [PubMed]
- Bibby, E.; Stewart, A. The epidemiology of preterm birth. Neuroendocrinol Lett. 2004, 25, 43–47. [Google Scholar] [PubMed]
- Barker, D.J. Adult consequences of fetal growth restriction. Clin. Obstet. Gynecol. 2006, 49, 270–283. [Google Scholar] [CrossRef] [PubMed]
- Hakonsen, L.B.; Ernst, A.; Ramlau-Hansen, C.H. Maternal cigarette smoking during pregnancy and reproductive health in children: A review of epidemiological studies. Asian. J. Androl. 2014, 16, 39–49. [Google Scholar] [CrossRef] [PubMed]
- De Bie, H.M.; Oostrom, K.J.; Delemarre-van de Waal, H.A. Brain development, intelligence and cognitive outcome in children born small for gestational age. Hormone Res. Paediat. 2010, 73, 6–14. [Google Scholar] [CrossRef] [PubMed]
- HSCIC. Statistics on Women’s Smoking Status at Time of Delivery: England, Quarter 4—April 2013 to March 2014; Health & Social Care Information Centre: Leeds, UK, June 19 2014. [Google Scholar]
- Colman, G.J.; Joyce, T. Trends in smoking before, during, and after pregnancy in ten states. Amer. J. Prev. Med. 2003, 24, 29–35. [Google Scholar] [CrossRef]
- Egebjerg Jensen, K.; Jensen, A.; Nohr, B.; Kruger Kjaer, S. Do pregnant women still smoke? A study of smoking patterns among 261,029 primiparous women in Denmark 1997–2005. Acta. Obstet. Gynecol. Scand. 2008, 87, 760–767. [Google Scholar] [CrossRef] [PubMed]
- Breitling, L.P.; Yang, R.; Korn, B.; Burwinkel, B.; Brenner, H. Tobacco-smoking-related differential DNA methylation: 27K Discovery and replication. Amer. J. Hum. Genet. 2011, 88, 450–457. [Google Scholar] [CrossRef] [PubMed]
- Elliott, H.R.; Tillin, T.; McArdle, W.L.; Ho, K.; Duggirala, A.; Frayling, T.M.; Davey Smith, G.; Hughes, A.D.; Chaturvedi, N.; Relton, C.L. Differences in smoking associated DNA methylation patterns in South Asians and Europeans. Clin. Epigenetics 2014. [Google Scholar] [CrossRef] [PubMed]
- Shenker, N.S.; Polidoro, S.; van Veldhoven, K.; Sacerdote, C.; Ricceri, F.; Birrell, M.A.; Belvisi, M.G.; Brown, R.; Vineis, P.; Flanagan, J.M. Epigenome-wide association study in the European Prospective Investigation into Cancer and Nutrition (EPIC-Turin) identifies novel genetic loci associated with smoking. Hum. Mol. Genet. 2013, 22, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Joubert, B.R.; Haberg, S.E.; Nilsen, R.M.; Wang, X.; Vollset, S.E.; Murphy, S.K.; Huang, Z.; Hoyo, C.; Midttun, O.; Cupul-Uicab, L.A.; et al. 450K Epigenome-wide scan identifies differential DNA methylation in newborns related to maternal smoking during pregnancy. Environ. Health Perspect. 2012, 120, 1425–1431. [Google Scholar] [CrossRef] [PubMed]
- Markunas, C.A.; Xu, Z.; Harlid, S.; Wade, P.A.; Lie, R.T.; Taylor, J.A.; Wilcox, A.J. Identification of DNA methylation changes in newborns related to maternal smoking during pregnancy. Environ. Health Perspect. 2014, 122, 1147–1153. [Google Scholar] [CrossRef] [PubMed]
- Novakovic, B.; Ryan, J.; Pereira, N.; Boughton, B.; Craig, J.M.; Saffery, R. Postnatal stability, tissue, and time specific effects of AHRR methylation change in response to maternal smoking in pregnancy. Epigenetics 2014, 9, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Lumley, J.; Oliver, S.S.; Chamberlain, C.; Oakley, L. Interventions for promoting smoking cessation during pregnancy. Cochrane Database Syst. Rev. 2004. [Google Scholar] [CrossRef] [Green Version]
- Breton, C.V.; Siegmund, K.D.; Joubert, B.R.; Wang, X.; Qui, W.; Carey, V.; Nystad, W.; Haberg, S.E.; Ober, C.; Nicolae, D.; et al. Prenatal tobacco smoke exposure is associated with childhood DNA CpG methylation. PloS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.W.; Richmond, R.; Hu, P.; French, L.; Shin, J.; Bourdon, C.; Reischl, E.; Waldenberger, M.; Zeilinger, S.; Gaunt, T.; et al. Prenatal exposure to maternal cigarette smoking and DNA methylation: Epigenome-wide association in a discovery sample of adolescents and replication in an independent cohort at birth through 17 years of age. Environ. Health Perspect. 2015, 123, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Ng, J.W.; Barrett, L.M.; Wong, A.; Kuh, D.; Smith, G.D.; Relton, C.L. The role of longitudinal cohort studies in epigenetic epidemiology: Challenges and opportunities. Genome Biol. 2012, 13, 246. [Google Scholar] [CrossRef] [PubMed]
- Sharp, G.; Lawlor, D.; Richmond, R.; Fraser, A.; Simpkin, A.; Suderman, M.; Shihab, H.; Lyttleton, O.; McArdle, W.; Ring, S.; Gaunt, T.; et al. Maternal pre-pregnancy BMI and gestational weight gain, offspring DNA methylation and later offspring adiposity: Findings from the Avon Longitudinal Study of Parents and Children. Int. J. Epidemiol. 2015, in press. [Google Scholar] [CrossRef] [PubMed]
- Richmond, R.C.; Simpkin, A.J.; Woodward, G.; Gaunt, T.R.; Lyttleton, O.; McArdle, W.L.; Ring, S.M.; Smith, A.D.; Timpson, N.J.; Tilling, K.; et al. Prenatal exposure to maternal smoking and offspring DNA methylation across the lifecourse: Findings from the Avon Longitudinal Study of Parents and Children (ALSPAC). Hum. Mol. Genet. 2015, 24, 2201–2217. [Google Scholar] [CrossRef] [PubMed]
- Simpkin, A.J.; Suderman, M.; Gaunt, T.R.; Lyttleton, O.; McArdle, W.L.; Ring, S.M.; Tilling, K.; Smith, G.D.; Relton, C.L. Longitudinal analysis of DNA methylation associated with birth weight and gestational age. Hum. Mol. Genet. 2015, 24, 3752–3763. [Google Scholar] [CrossRef] [PubMed]
- Philibert, R.A.; Beach, S.R.; Brody, G.H. Demethylation of the aryl hydrocarbon receptor repressor as a biomarker for nascent smokers. Epigenetics 2012, 7, 1331–1338. [Google Scholar] [CrossRef] [PubMed]
- Wan, E.S.; Qiu, W.; Baccarelli, A.; Carey, V.J.; Bacherman, H.; Rennard, S.I.; Agusti, A.; Anderson, W.; Lomas, D.A.; Demeo, D.L. Cigarette smoking behaviors and time since quitting are associated with differential DNA methylation across the human genome. Hum. Mol. Genet. 2012, 21, 3073–3082. [Google Scholar] [CrossRef] [PubMed]
- Zeilinger, S.; Kuhnel, B.; Klopp, N.; Baurecht, H.; Kleinschmidt, A.; Gieger, C.; Weidinger, S.; Lattka, E.; Adamski, J.; Peters, A.; Strauch, K.; et al. Tobacco smoking leads to extensive genome-wide changes in DNA methylation. PloS ONE 2013. [Google Scholar] [CrossRef] [PubMed]
- Relton, C.L.; Gaunt, T.; McArdle, W.; Ho, K.; Duggirala, A.; Shihab, H.; Woodward, G.; Lyttleton, O.; Evans, D.M.; Reik, W.; et al. Data resource profile: Accessible Resource for Integrated Epigenomic Studies (ARIES). Int. J. Epidemiol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Boyd, A.; Golding, J.; Macleod, J.; Lawlor, D.A.; Fraser, A.; Henderson, J.; Molloy, L.; Ness, A.; Ring, S.; Davey Smith, G. Cohort Profile: the children of the 90s—the index offspring of the Avon Longitudinal Study of Parents and Children. Int. J. Epidemiol. 2013, 42, 111–127. [Google Scholar] [CrossRef] [PubMed]
- Fraser, A.; Macdonald-Wallis, C.; Tilling, K.; Boyd, A.; Golding, J.; Davey, S.G.; Henderson, J.; Macleod, J.; Molloy, L.; Ness, A.; et al. Cohort profile: the Avon Longitudinal Study of Parents and Children: ALSPAC mothers cohort. Int. J. Epidemiol. 2013, 42, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Wakefield, J.; de Vocht, F.; Hung, R.J. Bayesian mixture modeling of gene-environment and gene-gene interactions. Genet. Epidemiol. 2010, 34, 16–25. [Google Scholar] [CrossRef] [PubMed]
- De Vocht, F.; Cherry, N.; Wakefield, J. A Bayesian mixture modeling approach for assessing the effects of correlated exposures in case-control studies. J. Expo. Sci. Environ. Epidemiol. 2012, 22, 352–360. [Google Scholar] [CrossRef] [PubMed]
- MacLehose, R.F.; Dunson, D.B.; Herring, A.H.; Hoppin, J.A. Bayesian methods for highly correlated exposure data. Epidemiology 2007, 18, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Bibikova, M.; Barnes, B.; Tsan, C.; Ho, V.; Klotzle, B.; Le, J.M.; Delano, D.; Zhang, L.; Schroth, G.P.; Gunderson, K.L. High density DNA methylation array with single CpG site resolution. Genomics 2011, 98, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Teschendorff, A.E.; Zhuang, J.; Widschwendter, M. Independent surrogate variable analysis to deconvolve confounding factors in large-scale microarray profiling studies. Bioinformatics 2011, 27, 1496–1505. [Google Scholar] [CrossRef] [PubMed]
- Chene, G.; Thompson, S.G. Methods for summarizing the risk associations of quantitative variables in epidemiologic studies in a consistent form. Amer. J. Epidemiol. 1996, 144, 610–621. [Google Scholar] [CrossRef]
- Kupers, L.K.; Xu, X.; Jankipersadsing, S.A.; Vaez, A.; la Bastide-van Gemert, S.; Scholtens, S.; Nolte, I.M.; Richmond, R.C.; Relton, C.L.; Felix, J.F.; et al. DNA methylation mediates the effect of maternal smoking during pregnancy on birthweight of the offspring. Int. J. Epidemiol. 2015, 44, 1224–1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moorthy, B.; Chu, C.; Carlin, D.J. Polycyclic aromatic hydrocarbons: From metabolism to lung cancer. Toxicol. Sci. 2015, 145, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Stroud, L.R.; Papandonatos, G.D.; Rodriguez, D.; McCallum, M.; Salisbury, A.L.; Phipps, M.G.; Lester, B.; Huestis, M.A.; Niaura, R.; Padbury, J.F.; et al. Maternal smoking during pregnancy and infant stress response: Test of a prenatal programming hypothesis. Psychoneuroendocrinology 2014, 48, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Rubin, D.B. Multiple Imputation WCL Paper; John Wiley & Sons: New York, NY, USA, 2004. [Google Scholar]
- George, E.I.; McCulloch, R.E. Approaches for variable selection. Stat. Sin. 1997, 7, 339–374. [Google Scholar]
- Zhao, Z.; Han, L. CpG islands: Algorithms and applications in methylation studies. Biochem. Biophys. Res. Commun. 2009, 382, 643–645. [Google Scholar] [CrossRef] [PubMed]
- Ziller, M.J.; Gu, H.; Muller, F.; Donaghey, J.; Tsai, L.T.; Kohlbacher, O.; De Jager, P.L.; Rosen, E.D.; Bennett, D.A.; Bernstein, B.E.; et al. Charting a dynamic DNA methylation landscape of the human genome. Nature 2013, 500, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Mishra, G.; Nitsch, D.; Black, S.; De Stavola, B.; Kuh, D.; Hardy, R. A structured approach to modelling the effects of binary exposure variables over the life course. Int. J. Epidemiol. 2009, 38, 528–537. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Vocht, F.; Simpkin, A.J.; Richmond, R.C.; Relton, C.; Tilling, K. Assessment of Offspring DNA Methylation across the Lifecourse Associated with Prenatal Maternal Smoking Using Bayesian Mixture Modelling. Int. J. Environ. Res. Public Health 2015, 12, 14461-14476. https://0-doi-org.brum.beds.ac.uk/10.3390/ijerph121114461
De Vocht F, Simpkin AJ, Richmond RC, Relton C, Tilling K. Assessment of Offspring DNA Methylation across the Lifecourse Associated with Prenatal Maternal Smoking Using Bayesian Mixture Modelling. International Journal of Environmental Research and Public Health. 2015; 12(11):14461-14476. https://0-doi-org.brum.beds.ac.uk/10.3390/ijerph121114461
Chicago/Turabian StyleDe Vocht, Frank, Andrew J Simpkin, Rebecca C. Richmond, Caroline Relton, and Kate Tilling. 2015. "Assessment of Offspring DNA Methylation across the Lifecourse Associated with Prenatal Maternal Smoking Using Bayesian Mixture Modelling" International Journal of Environmental Research and Public Health 12, no. 11: 14461-14476. https://0-doi-org.brum.beds.ac.uk/10.3390/ijerph121114461