
Epigenetic Effect of Environmental Factors on Autism Spectrum Disorders

{kind=link}
Abstract
:1. Introduction
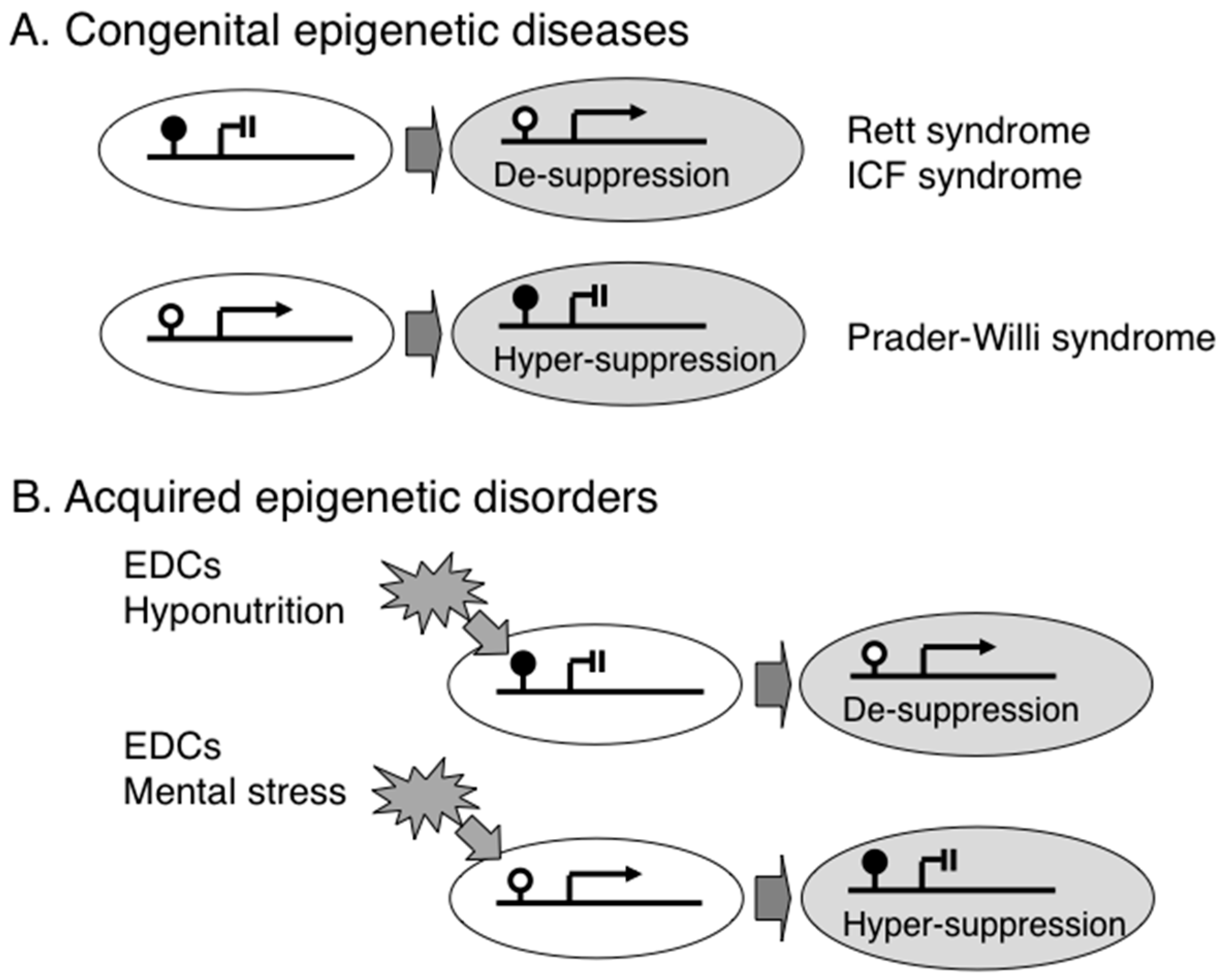
2. Congenital Epigenetic Diseases
3. Acquired Epigenetic Disorders
4. Transgenerational Epigenetic Inheritance
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Text Revision, 4th ed.; American Psychiatric Association: Washington, DC, USA, 2000. [Google Scholar]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Association: Arlington, VA, USA, 2013. [Google Scholar]
- American Psychiatric Association. Proposed Revision: A 05 Autism Spectrum Disorder. DSM-5 development website. Available online: http://web-beta.archive.org/web/20121115164727/http://www.dsm5.org/ProposedRevision/Pages/proposedrevision.aspx?rid=94 (accessed on 31 January 2016).
- Sealey, L.A.; Hughes, B.W.; Sriskanda, A.N.; Guest, J.R.; Gibson, A.D.; Johnson-Williams, L.; Pace, D.G.; Bagasra, O. Environmental factors in the development of autism spectrum disorders. Environ. Int. 2016, 88, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Larsson, H.J.; Eaton, W.W.; Madsen, K.M.; Vestergaard, M.; Olesen, A.V.; Agerbo, E.; Schendel, D.; Thorsen, P.; Mortensen, P.B. Risk factors for autism: Perinatal factors, parental psychiatric history, and socioeconomic status. Am. J. Epidemiol. 2005, 161, 916–925. [Google Scholar] [CrossRef] [PubMed]
- Jokiranta, E.; Brown, A.S.; Heinimaa, M.; Cheslack-Postava, K.; Suominen, A.; Sourander, A. Parental psychiatric disorders and autism spectrum disorders. Psychiatry Res. 2013, 207, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.L.; Lyall, K.; Rich-Edwards, J.W.; Ascherio, A.; Weisskopf, M.G. Association of maternal exposure to childhood abuse with elevated risk for autism in offspring. JAMA Psychiatry 2013, 70, 508–515. [Google Scholar] [CrossRef] [PubMed]
- Belluscio, L.M.; Berardino, B.G.; Ferroni, N.M.; Ceruti, J.M.; Cánepa, E.T. Early protein malnutrition negatively impacts physical growth and neurological reflexes and evokes anxiety and depressive-like behaviors. Physiol. Behav. 2014, 129, 237–254. [Google Scholar] [CrossRef] [PubMed]
- Berger, B.E.; Navar-Boggan, A.M.; Omer, S.B. Congenital rubella syndrome and autism spectrum disorder prevented by rubella vaccination—United States; 2001–2010. BMC Public Health 2011, 11, 340. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, A.; Moriuchi, H.; Matsuzaki, J.; Motoyama, K.; Moriuchi, M. Retrospective diagnosis of congenital cytomegalovirus infection in children with autism spectrum disorder but no other major neurologic deficit. Brain Dev. 2015, 37, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Heilbrun, L.P.; Palmer, R.F.; Jaen, C.R.; Svoboda, M.D.; Miller, C.S.; Perkins, J. Maternal Chemical and Drug Intolerances: Potential Risk Factors for Autism and Attention Deficit Hyperactivity Disorder (ADHD). J. Am. Board Fam. Med. 2015, 28, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.I.; Kern, J.K. Evidence of microglial activation in autism and its possible role in brain under connectivity. Neuron Glia Biol. 2011, 7, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Theoharides, T.C.; Asadi, S.; Patel, A.B. Focal brain inflammation and autism. J. Neuroinflamm. 2013, 10, 46. [Google Scholar] [CrossRef] [PubMed]
- McDougle, C.J. Toward an immune-mediated subtype of autism spectrum disorder. Brain Res. 2015, 1617, 72–92. [Google Scholar] [CrossRef] [PubMed]
- Atladóttir, H.Ó.; Henriksen, T.B.; Schendel, D.E.; Parner, E.T. Autism after infection, febrile episodes, and antibiotic use during pregnancy: An exploratory study. Pediatrics 2012, 130, e1447–e1454. [Google Scholar] [CrossRef] [PubMed]
- Kuo, H.C.; Wu, C.M.; Chang, W.P.; Kuo, C.N.; Yeter, D.; Lin, C.Y.; Pai, J.T.; Chi, Y.C.; Lin, C.H.; Wang, L.J.; Chang, W.C. Association between Kawasaki disease and autism: A population-based study in Taiwan. Int. J. Environ. Res. Public Health 2014, 11, 3705–3716. [Google Scholar] [CrossRef] [PubMed]
- Esper, F.; Shapiro, E.D.; Weibel, C.; Ferguson, D.; Landry, M.L.; Kahn, J.S. Association between a novel human coronavirus and Kawasaki disease. J. Infect. Dis. 2005, 191, 499–502. [Google Scholar] [CrossRef] [PubMed]
- Kalkbrenner, A.E.; Schmidt, R.J.; Penlesky, A.C. Environmental chemical exposures and autism spectrum disorders: A review of the epidemiological evidence. Curr. Probl. Pediatr. Adolesc. Health Care 2014, 44, 277–318. [Google Scholar] [CrossRef] [PubMed]
- Corradi, A.; Fadda, M.; Piton, A.; Patry, L.; Marte, A.; Rossi, P.; Cadieux-Dion, M.; Gauthier, J.; Lapointe, L.; Mottron, L.; et al. SYN2 is an autism predisposing gene: Loss-of-function mutations alter synaptic vesicle cycling and axon outgrowth. Hum. Mol. Genet. 2014, 23, 90–103. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, P.J.; Campbell, N.G.; Sharma, S.; Erreger, K.; Herborg Hansen, F.; Saunders, C.; Belovich, A.N.; NIH ARRA Autism Sequencing Consortium; Sahai, M.A.; Cook, E.H.; et al. De novo mutation in the dopamine transporter gene associates dopamine dysfunction with autism spectrum disorder. Mol. Psychiatry 2013, 18, 1315–1323. [Google Scholar] [CrossRef] [PubMed]
- Hill-Yardin, E.L.; Argyropoulos, A.; Hosie, S.; Rind, G.; Anderson, P.; Hannan, A.J.; O’Brien, T.J. Reduced susceptibility to induced seizures in the Neuroligin-3(R451C) mouse model of autism. Neurosci. Lett. 2015, 589, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Sala, C.; Vicidomini, C.; Bigi, I.; Mossa, A.; Verpelli, C. Shank synaptic scaffold proteins: Keys to understanding the pathogenesis of autism and other synaptic disorders. J. Neurochem. 2015, 135, 849–858. [Google Scholar] [CrossRef] [PubMed]
- Leblond, C.S.; Nava, C.; Polge, A.; Gauthier, J.; Huguet, G.; Lumbroso, S.; Giuliano, F.; Stordeur, C.; Depienne, C.; Mouzat, K.; et al. Meta-analysis of SHANK Mutations in Autism Spectrum Disorders: A gradient of severity in cognitive impairments. PLoS Genet. 2014, 10, e1004580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizuno, M.; Matsumoto, A.; Hamada, N.; Ito, H.; Miyauchi, A.; Jimbo, E.F.; Momoi, M.Y.; Tabata, H.; Yamagata, T.; Nagata, K. Role of an adaptor protein Lin-7B in brain development: Possible involvement in autism spectrum disorders. J. Neurochem. 2015, 132, 61–69. [Google Scholar] [CrossRef] [PubMed]
- De Rubeis, S.; He, X.; Goldberg, A.P.; Poultney, C.S.; Samocha, K.; Cicek, A.E.; Kou, Y.; Liu, L.; Fromer, M.; Walker, S.; et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014, 515, 209–215. [Google Scholar]
- Balan, S.; Iwayama, Y.; Maekawa, M.; Toyota, T.; Ohnishi, T.; Toyoshima, M.; Shimamoto, C.; Esaki, K.; Yamada, K.; Iwata, Y.; et al. Exon resequencing of H3K9 methyltransferase complex genes, EHMT1, EHTM2 and WIZ, in Japanese autism subjects. Mol. Autism 2014, 5, 49. [Google Scholar] [CrossRef] [PubMed]
- Barnard, R.A.; Pomaville, M.B.; O’Roak, B.J. Mutations and Modeling of the Chromatin Remodeler CHD8 Define an Emerging Autism Etiology. Front. Neurosci. 2015, 9, 477. [Google Scholar] [CrossRef] [PubMed]
- Zoghbi, H.Y. Postnatal neurodevelopmental disorders: Meeting at the synapse? Science 2003, 302, 826–830. [Google Scholar] [CrossRef] [PubMed]
- Boettiger, A.N.; Bintu, B.; Moffitt, J.R.; Wang, S.; Beliveau, B.J.; Fudenberg, G.; Imakaev, M.; Mirny, L.A.; Wu, C.T.; Zhuang, X. Super-resolution imaging reveals distinct chromatin folding for different epigenetic states. Nature 2016, 529, 418–422. [Google Scholar] [CrossRef] [PubMed]
- Hori, K.; Nagai, T.; Shan, W.; Sakamoto, A.; Taya, S.; Hashimoto, R.; Hayashi, T.; Abe, M.; Yamazaki, M.; Nakao, K.; et al. Cytoskeletal regulation by AUTS2 in neuronal migration and neuritogenesis. Cell Rep. 2014, 9, 2166–2179. [Google Scholar] [CrossRef] [PubMed]
- Kalscheuer, V.M.; FitzPatrick, D.; Tommerup, N.; Bugge, M.; Niebuhr, E.; Neumann, L.M.; Tzschach, A.; Shoichet, S.A.; Menzel, C.; Erdogan, F.; et al. Mutations in autism susceptibility candidate 2 (AUTS2) in patients with mental retardation. Hum. Genet. 2007, 121, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Beunders, G.; Voorhoeve, E.; Golzio, C.; Pardo, L.M.; Rosenfeld, J.A.; Talkowski, M.E.; Simonic, I.; Lionel, A.C.; Vergult, S.; Pyatt, R.E.; et al. Exonic deletions in AUTS2 cause a syndromic form of intellectual disability and suggest a critical role for the C terminus. Am. J. Hum. Genet. 2013, 92, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Lee, P.; Stafford, J.M.; von Schimmelmann, M.; Schaefer, A.; Reinberg, D. An AUTS2-Polycomb complex activates gene expression in the CNS. Nature 2014, 516, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Oksenberg, N.; Haliburton, G.D.; Eckalbar, W.L.; Oren, I.; Nishizaki, S.; Murphy, K.; Pollard, K.S.; Birnbaum, R.Y.; Ahituv, N. Genome-wide distribution of Auts2 binding localizes with active neurodevelopmental genes. Transl. Psychiatry 2014, 4, e431. [Google Scholar] [CrossRef] [PubMed]
- GeneReview-MeCP2 Related Disorders. Available online: http://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/books/NBK1497/ (accessed on 28 February 2016).
- Amir, R.E.; van den Veyver, I.B.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H.Y. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999, 23, 185–188. [Google Scholar] [PubMed]
- Ballestar, E.; Yusufzai, T.M.; Wolffe, A.P. Effects of Rett syndrome mutations of the methyl-CpG binding domain of the transcriptional repressor MeCP2 on selectivity for association with methylated DNA. Biochemistry 2000, 39, 7100–7106. [Google Scholar] [CrossRef] [PubMed]
- Wolffe, A.P. Histone deacetylase: A regulator of transcription. Science 1996, 272, 371–372. [Google Scholar] [CrossRef]
- Jones, P.L.; Veenstra, G.J.; Wade, P.A.; Vermaak, D.; Kass, S.U.; Landsbergerv, N.; Strouboulis, J.; Wolffe, A.P. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat. Genet. 1998, 19, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Nan, X.; Bird, A. The biological functions of the methyl-CpG-binding protein MeCP2 and its implication in Rett syndrome. Brain Dev. 2001, 23 (Suppl. 1), S32–S37. [Google Scholar] [CrossRef]
- Chen, W.G.; Chang, Q.; Lin, Y.; Meissner, A.; West, A.E.; Griffith, E.C.; Jaenisch, R.; Greenberg, M.E. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science 2003, 302, 885–889. [Google Scholar] [CrossRef] [PubMed]
- Martinowich, K.; Hattori, D.; Wu, H.; Fouse, S.; He, F.; Hu, Y.; Fanv, G.; Sun, Y.E. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science 2003, 302, 890–893. [Google Scholar] [CrossRef] [PubMed]
- Horike, S.; Cai, S.; Miyano, M.; Cheng, J.F.; Kohwi-Shigematsu, T. Loss of silent-chromatin looping and impaired imprinting of DLX5 in Rett syndrome. Nat. Genet. 2005, 37, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Peddada, S.; Yasui, D.H.; LaSalle, J.M. Inhibitors of differentiation (ID1, ID2, ID3 and ID4) genes are neuronal targets of MeCP2 that are elevated in Rett syndrome. Hum. Mol. Genet. 2006, 15, 2003–2014. [Google Scholar] [CrossRef] [PubMed]
- Itoh, M.; Ide, S.; Takashima, S.; Kudo, S.; Nomura, Y.; Segawa, M.; Kubota, T.; Mori, H.; Tanaka, S.; Horie, H.; Tanabe, Y.; Goto, Y. Methyl CpG-binding protein 2 (a mutation of which causes Rett syndrome) directly regulates insulin-like growth factor binding protein 3 in mouse and human brains. J. Neuropathol. Exp. Neurol. 2007, 66, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Carouge, D.; Host, L.; Aunis, D.; Zwiller, J.; Anglard, P. CDKL5 is a brain MeCP2 target gene regulated by DNA methylation. Neurobiol. Dis. 2010, 38, 414–424. [Google Scholar] [CrossRef] [PubMed]
- Miyake, K.; Hirasawa, T.; Soutome, M.; Itoh, M.; Goto, Y.; Endoh, K.; Takahashi, K.; Kudo, S.; Nakagawa, T.; Yokoi, S.; et al. The protocadherins, PCDHB1 and PCDH7, are regulated by MeCP2 in neuronal cells and brain tissues: Implication for pathogenesis of Rett syndrome. BMC Neurosci. 2011, 12, 81. [Google Scholar] [CrossRef] [PubMed]
- Chao, H.T.; Zoghbi, H.Y.; Rosenmund, C. MeCP2 controls excitatory synaptic strength by regulating glutamatergic synapse number. Neuron 2007, 56, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Marchetto, M.C.; Carromeu, C.; Acab, A.; Yu, D.; Yeo, G.W.; Mu, Y.; Chen, G.; Gage, F.H.; Muotri, A.R. A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell 2010, 143, 527–539. [Google Scholar] [CrossRef] [PubMed]
- Farra, N.; Zhang, W.B.; Pasceri, P.; Eubanks, J.H.; Salter, M.W.; Ellisv, J. Rett syndrome induced pluripotent stem cell-derived neurons reveal novel neurophysiological alterations. Mol. Psychiatry 2012, 17, 1261–1271. [Google Scholar] [CrossRef] [PubMed]
- Djuric, U.; Cheung, A.Y.; Zhang, W.; Mok, R.S.; Lai, W.; Piekna, A.; Hendry, J.A.; Ross, P.J.; Pasceri, P.; Kim, D.S.; et al. MECP2e1 isoform mutation affects the form and function of neurons derived from Rett syndrome patient iPS cells. Neurobiol. Dis. 2015, 76, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Andoh-Noda, T.; Akamatsu, W.; Miyake, K.; Matsumoto, T.; Yamaguchi, R.; Sanosaka, T.; Okada, Y.; Kobayashi, T.; Ohyama, M.; Nakashima, K.; et al. Differentiation of multipotent neural stem cells derived from Rett syndrome patients is biased toward the astrocytic lineage. Mol. Brain 2015, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Van Esch, H.; Bauters, M.; Ignatius, J.; Jansen, M.; Raynaud, M.; Hollanders, K.; Lugtenberg, D.; Bienvenu, T.; Jensen, LR.; Gecz, J.; et al. Duplication of the MECP2 region is a frequent cause of severe mental retardation and progressive neurological symptoms in males. Am. J. Hum. Genet. 2005, 77, 442–453. [Google Scholar] [CrossRef] [PubMed]
- Novara, F.; Simonati, A.; Sicca, F.; Battini, R.; Fiori, S.; Contaldo, A.; Criscuolo, L.; Zuffardi, O.; Ciccone, R. MECP2 duplication phenotype in symptomatic females: Report of three further cases. Mol. Cytogenet. 2014, 7, 10. [Google Scholar] [CrossRef] [PubMed]
- Na, E.S.; Nelson, E.D.; Adachi, M.; Autry, A.E.; Mahgoub, M.A.; Kavalali, E.T.; Monteggia, L.M. A mouse model for MeCP2 duplication syndrome: MeCP2 overexpression impairs learning and memory and synaptic transmission. J. Neurosci. 2012, 32, 3109–3117. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Li, X.; Zhang, J.T.; Cai, Y.J.; Cheng, T.L.; Cheng, C.; Wang, Y.; Zhang, C.C.; Nie, Y.H.; Chen, Z.F.; et al. Autism-like behaviours and germline transmission in transgenic monkeys overexpressing MeCP2. Nature 2016, 530, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Cyranoski, D. Monkeys genetically modified to show autism symptoms. Nature 2016, 529, 449. [Google Scholar] [CrossRef] [PubMed]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef]
- Kubota, T.; Furuumi, H.; Kamoda, T.; Iwasaki, N.; Tobita, N.; Fujiwara, N.; Goto, Y.; Matsui, A.; Sasaki, H.; Kajii, T. ICF syndrome in a girl with DNA hypomethylation but without detectable DNMT3B mutation. Am. J. Med. Genet. A 2004, 129A, 290–293. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Wu, Z.; Liu, Z.; Hu, G.; Yu, J.; Chang, K.H.; Kim, K.P.; Le, T.; Faull, K.F.; Rao, N.; et al. Selective demethylation and altered gene expression are associated with ICF syndrome in human-induced pluripotent stem cells and mesenchymal stem cells. Hum. Mol. Genet. 2014, 23, 6448–6457. [Google Scholar] [CrossRef] [PubMed]
- Simo-Riudalbas, L.; Diaz-Lagares, A.; Gatto, S.; Gagliardi, M.; Crujeiras, A.B.; Matarazzo, M.R.; Esteller, M.; Sandoval, J. Genome-Wide DNA Methylation Analysis Identifies Novel Hypomethylated Non-Pericentromeric Genes with Potential Clinical Implications in ICF Syndrome. PLoS ONE 2015, 10, e0132517. [Google Scholar]
- Tatton-Brown, K.; Seal, S.; Ruark, E.; Harmer, J.; Ramsay, E.; del Vecchio Duarte, S.; Zachariou, A.; Hanks, S.; O’Brien, E.; Aksglaede, L.; et al. Mutations in the DNA methyltransferase gene DNMT3A cause an overgrowth syndrome with intellectual disability. Nat. Genet. 2014, 46, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Kubota, T.; Das, S.; Christian, S.L.; Baylin, S.B.; Herman, J.G.; Ledbetter, D.H. Methylation-specific PCR simplifies imprinting analysis. Nat. Genet. 1997, 16, 16–17. [Google Scholar] [PubMed]
- Schaaf, C.P.; Gonzalez-Garay, M.L.; Xia, F.; Potocki, L.; Gripp, K.W.; Zhang, B.; Peters, B.A.; McElwain, M.A.; Drmanac, R.; Beaudet, A.L.; et al. Truncating mutations of MAGEL2 cause Prader-Willi phenotypes and autism. Nat. Genet. 2013, 45, 1405–1408. [Google Scholar] [CrossRef] [PubMed]
- Stelzer, Y.; Sagi, I.; Yanuka, O.; Eiges, R.; Benvenisty, N. The noncoding RNA IPW regulates the imprinted DLK1-DIO3 locus in an induced pluripotent stem cell model of Prader-Willi syndrome. Nat. Genet. 2014, 46, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Online Mendelian Inheritance of Men (OMIM) #176270. Available online: http://omim.org/entry/176270 (accessed on 29 February 2016).
- Rangasamy, S.; D’Mello, S.R.; Narayanan, V. Epigenetics, autism spectrum, and neurodevelopmental disorders. Neurotherapeutics 2013, 10, 742–756. [Google Scholar] [CrossRef] [PubMed]
- Scoles, H.A.; Urraca, N.; Chadwick, S.W.; Reiter, L.T.; Lasalle, J.M. Increased copy number for methylated maternal 15q duplications leads to changes in gene and protein expression in human cortical samples. Mol. Autism 2011, 2, 19. [Google Scholar] [CrossRef] [PubMed]
- Gregory, S.G.; Connelly, J.J.; Towers, A.J.; Johnson, J.; Biscocho, D.; Markunas, C.A.; Lintas, C.; Abramson, R.K.; Wright, H.H.; Ellis, P.; et al. Genomic and epigenetic evidence for oxytocin receptor deficiency in autism. BMC Med. 2009, 7, 62. [Google Scholar] [CrossRef] [PubMed]
- James, S.J.; Shpyleva, S.; Melnyk, S.; Pavliv, O.; Pogribny, I.P. Complex epigenetic regulation of engrailed-2 (EN-2) homeobox gene in the autism cerebellum. Transl. Psychiatry 2013, 3, e232. [Google Scholar] [CrossRef] [PubMed]
- Zhubi, A.; Chen, Y.; Dong, E.; Cook, E.H.; Guidotti, A.; Grayson, D.R. Increased binding of MeCP2 to the GAD1 and RELN promoters may be mediated by an enrichment of 5-hmC in autism spectrum disorder (ASD) cerebellum. Transl. Psychiatry 2014, 4, e349. [Google Scholar] [CrossRef] [PubMed]
- Ladd-Acosta, C.; Hansen, K.D.; Briem, E.; Fallin, M.D.; Kaufmann, W.E.; Feinberg, A.P. Common DNA methylation alterations in multiple brain regions in autism. Mol. Psychiatry 2014, 19, 862–871. [Google Scholar] [CrossRef] [PubMed]
- Nardone, S.; Sams, D.S.; Reuveni, E.; Getselter, D.; Oron, O.; Karpuj, M.; Elliott, E. DNA methylation analysis of the autistic brain reveals multiple dysregulated biological pathways. Transl. Psychiatry 2014, 4, e433. [Google Scholar] [CrossRef] [PubMed]
- Castro, K.; Klein, L.D.; Baronio, D.; Gottfried, C.; Riesgo, R.; Perry, I.S. Folic acid and autism: What do we know? Nutr. Neurosci. 2014. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Fang, Y.; Zhang, F.; Xu, M.; Zhang, J.; Yan, J.; Ju, W.; Brown, W.T.; Zhong, N. Hypermethylation of the enolase gene (ENO2) in autism. Eur. J. Pediatr. 2014, 173, 1233–1244. [Google Scholar] [CrossRef] [PubMed]
- Mikeska, T.; Craig, J.M. DNA methylation biomarkers: Cancer and beyond. Genes 2014, 5, 821–864. [Google Scholar] [CrossRef] [PubMed]
- Loke, Y.J.; Hannan, A.J.; Craig, J.M. The role of epigenetic change in autism spectrum disorders. Front. Neurol. 2015, 6, 107. [Google Scholar] [CrossRef] [PubMed]
- Crews, D.; Gillette, R.; Miller-Crews, I.; Gore, A.C.; Skinner, M.K. Nature, nurture and epigenetics. Mol. Cell Endocrinol. 2014, 398, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Yaoi, T.; Itoh, K.; Nakamura, K.; Ogi, H.; Fujiwara, Y.; Fushiki, S. Genome-wide analysis of epigenomic alterations in fetal mouse forebrain after exposure to low doses of bisphenol A. Biochem. Biophys. Res. Commun. 2008, 376, 563–567. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, E.; Dolinoy, D.C.; Mancuso, P. Perinatal bisphenol A exposures increase production of pro-inflammatory mediators in bone marrow-derived mast cells of adult mice. J. Immunotoxicol. 2014, 11, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Weinhouse, C.; Bergin, I.L.; Harris, C.; Dolinoy, D.C. Stat3 is a candidate epigenetic biomarker of perinatal Bisphenol A exposure associated with murine hepatic tumors with implications for human health. Epigenetics 2015, 10, 1099–1110. [Google Scholar] [CrossRef] [PubMed]
- Dao, T.; Hong, X.; Wang, X.; Tang, W.Y. Aberrant 5’-CpG Methylation of Cord Blood TNFα Associated with Maternal Exposure to Polybrominated Diphenyl Ethers. PLoS ONE 2015, 10, e0138815. [Google Scholar] [CrossRef] [PubMed]
- Richmond, R.C.; Simpkin, A.J.; Woodward, G.; Gaunt, T.R.; Lyttleton, O.; McArdle, W.L.; Ring, S.M.; Smith, A.D.; Timpson, N.J.; Tilling, K.; et al. Prenatal exposure to maternal smoking and offspring DNA methylation across the lifecourse: Findings from the Avon Longitudinal Study of Parents and Children (ALSPAC). Hum. Mol. Genet. 2015, 24, 2201–2217. [Google Scholar] [CrossRef] [PubMed]
- Lillycrop, K.A.; Phillips, E.S.; Torrens, C.; Hanson, M.A.; Jackson, A.A.; Burdge, G.C. Feeding pregnant rats a protein-restricted diet persistently alters the methylation of specific cytosines in the hepatic PPARα promoter of the offspring. Br. J. Nutr. 2008, 100, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Demetriou, C.A.; van Veldhoven, K.; Relton, C.; Stringhini, S.; Kyriacou, K.; Vineis, P. Biological embedding of early-life exposures and disease risk in humans: A role for DNA methylation. Eur. J. Clin. Invest. 2015, 45, 303–332. [Google Scholar] [CrossRef] [PubMed]
- Gluckman, P.D.; Seng, C.Y.; Fukuoka, H.; Beedle, A.S.; Hanson, M.A. Low birthweight and subsequent obesity in Japan. Lancet 2007, 369, 1081–1082. [Google Scholar] [CrossRef]
- St Clair, D.; Xu, M.; Wang, P.; Yu, Y.; Fang, Y.; Zhang, F.; Zheng, X.; Gu, N.; Feng, G.; Sham, P.; et al. Rates of adult schizophrenia following prenatal exposure to the Chinese famine of 1959–1961. JAMA 2005, 294, 557–562. [Google Scholar] [CrossRef] [PubMed]
- Painter, R.C.; de Rooij, S.R.; Bossuyt, P.M.; Simmers, T.A.; Osmond, C.; Barker, D.J.; Bleker, O.P.; Roseboom, T.J. Early onset of coronary artery disease after prenatal exposure to the Dutch famine. Am. J. Clin. Nutr. 2006, 84, 322–327. [Google Scholar] [PubMed]
- Silveira, P.P.; Manfro, G.G. Retrospective studies. Adv. Neurobiol. 2015, 10, 251–267. [Google Scholar] [PubMed]
- Benyshek, D.C. The “early life” origins of obesity-related health disorders: New discoveries regarding the intergenerational transmission of developmentally programmed traits in the global cardiometabolic health crisis. Am. J. Phys. Anthropol. 2013, 152 (Suppl. 57), 79–93. [Google Scholar] [CrossRef] [PubMed]
- Weaver, I.C.; Cervoni, N.; Champagne, F.A.; D’Alessio, A.C.; Sharma, S.; Seckl, J.R.; Dymov, S.; Szyf, M.; Meaney, M.J. Epigenetic programming by maternal behavior. Nat. Neurosci. 2004, 7, 847–854. [Google Scholar] [CrossRef] [PubMed]
- Cao-Lei, L.; Massart, R.; Suderman, M.J.; Machnes, Z.; Elgbeili, G.; Laplante, D.P.; Szyf, M.; King, S. DNA methylation signatures triggered by prenatal maternal stress exposure to a natural disaster: Project Ice Storm. PLoS ONE 2014, 9, e107653. [Google Scholar] [CrossRef] [PubMed]
- Vangeel, E.B.; Izzi, B.; Hompes, T.; Vansteelandt, K.; Lambrechts, D.; Freson, K.; Claes, S. DNA methylation in imprinted genes IGF2 and GNASXL is associated with prenatal maternal stress. Genes Brain Behav. 2015, 14, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Braithwaite, E.C.; Kundakovic, M.; Ramchandani, P.G.; Murphy, S.E.; Champagne, F.A. Maternal prenatal depressive symptoms predict infant NR3C1 1F and BDNF IV DNA methylation. Epigenetics 2015, 10, 408–417. [Google Scholar] [CrossRef] [PubMed]
- Palma-Gudiel, H.; Córdova-Palomera, A.; Eixarch, E.; Deuschle, M.; Fañanás, L. Maternal psychosocial stress during pregnancy alters the epigenetic signature of the glucocorticoid receptor gene promoter in their offspring: A meta-analysis. Epigenetics 2015, 10, 893–902. [Google Scholar] [CrossRef] [PubMed]
- Skinner, M.K.; Manikkam, M.; Guerrero-Bosagna, C. Epigenetic transgenerational actions of environmental factors in disease etiology. Trends Endocrinol. Metab. 2010, 21, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Whitelaw, E. Disputing Lamarckian epigenetic inheritance in mammals. Genome Biol. 2015, 16, 60. [Google Scholar] [CrossRef] [PubMed]
- Daxinger, L.; Whitelaw, E. Understanding transgenerational epigenetic inheritance via the gametes in mammals. Nat. Rev. Genet. 2012, 13, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Franklin, T.B.; Russig, H.; Weiss, I.C.; Gräff, J.; Linder, N.; Michalon, A.; Vizi, S.; Mansuy, I.M. Epigenetic transmission of the impact of early stress across generations. Biol. Psychiatry 2010, 68, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Babenko, O.; Kovalchuk, I.; Metz, G.A. Stress-induced perinatal and transgenerational epigenetic programming of brain development and mental health. Neurosci. Biobehav. Rev. 2015, 48, 70–91. [Google Scholar] [CrossRef] [PubMed]
- Anway, M.D.; Skinner, M.K. Epigenetic programming of the germ line: Effects of endocrine disruptors on the development of transgenerational disease. Reprod. Biomed. Online 2008, 16, 23–25. [Google Scholar] [CrossRef]
- Mao, Z.; Xia, W.; Chang, H.; Huo, W.; Li, Y.; Xu, S. Paternal BPA exposure in early life alters IGF2 epigenetic status in sperm and induces pancreatic impairment in rat offspring. Toxicol. Lett. 2015, 238, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Dias, B.G.; Ressler, K.J. Experimental evidence needed to demonstrate inter- and trans-generational effects of ancestral experiences in mammals. Bioessays 2014, 36, 919–923. [Google Scholar] [CrossRef] [PubMed]
- Heard, E.; Martienssen, R.A. Transgenerational epigenetic inheritance: Myths and mechanisms. Cell 2014, 157, 95–109. [Google Scholar] [CrossRef] [PubMed]
- Stouder, C.; Paoloni-Giacobino, A. Specific transgenerational imprinting effects of the endocrine disruptor methoxychlor on male gametes. Reproduction 2011, 141, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Hanson, M.A.; Low, F.M.; Gluckman, P.D. Epigenetic epidemiology: The rebirth of soft inheritance. Ann. Nutr. Metab. 2011, 58 (Suppl. 2), 8–15. [Google Scholar] [CrossRef] [PubMed]
- Dickins, T.E.; Rahman, Q. The extended evolutionary synthesis and the role of soft inheritance in evolution. Proc. Biol. Sci. 2012, 279, 2913–2921. [Google Scholar] [PubMed]
- Jessberger, S.; Nakashima, K.; Clemenson, G.D., Jr.; Mejia, E.; Mathews, E.; Ure, K.; Ogawa, S.; Sinton, C.M.; Gage, F.H.; Hsieh, J. Epigenetic modulation of seizure-induced neurogenesis and cognitive decline. J. Neurosci. 2007, 27, 5967–5975. [Google Scholar] [CrossRef] [PubMed]
- Tsankova, N.M.; Berton, O.; Renthal, W.; Kumar, A.; Neve, R.L.; Nestler, E.J. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat. Neurosci. 2006, 9, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Nghia, N.A.; Hirasawa, T.; Kasai, H.; Obata, C.; Moriishi, K.; Mochizuki, K.; Koizumi, S.; Kubota, T. Long-term imipramine treatment increases N-methyl-d-aspartate receptor activity and expression via epigenetic mechanisms. Eur. J. Pharmacol. 2015, 752, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Surén, P.; Roth, C.; Bresnahan, M.; Haugen, M.; Hornig, M.; Hirtz, D.; Lie, K.K.; Lipkin, W.I.; Magnus, P.; Reichborn-Kjennerud, T.; et al. Association between maternal use of folic acid supplements and risk of autism spectrum disorders in children. JAMA 2013, 309, 570–577. [Google Scholar] [CrossRef] [PubMed]
- Guy, J.; Gan, J.; Selfridge, J.; Cobb, S.; Bird, A. Reversal of neurological defects in a mouse model of Rett syndrome. Science 2007, 315, 1143–1147. [Google Scholar] [CrossRef] [PubMed]
- Derecki, N.C.; Cronk, J.C.; Lu, Z.; Xu, E.; Abbott, S.B.; Guyenet, P.G.; Kipnis, J. Wild-type microglia arrest pathology in a mouse model of Rett syndrome. Nature 2012, 484, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Sztainberg, Y.; Chen, H.M.; Swann, J.W.; Hao, S.; Tang, B.; Wu, Z.; Tang, J.; Wan, Y.W.; Liu, Z.; Rigo, F.; Zoghbi, H.Y. Reversal of phenotypes in MECP2 duplication mice using genetic rescue or antisense oligonucleotides. Nature 2015, 528, 123–126. [Google Scholar] [CrossRef] [PubMed]

© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kubota, T.; Mochizuki, K. Epigenetic Effect of Environmental Factors on Autism Spectrum Disorders. Int. J. Environ. Res. Public Health 2016, 13, 504. https://0-doi-org.brum.beds.ac.uk/10.3390/ijerph13050504
Kubota T, Mochizuki K. Epigenetic Effect of Environmental Factors on Autism Spectrum Disorders. International Journal of Environmental Research and Public Health. 2016; 13(5):504. https://0-doi-org.brum.beds.ac.uk/10.3390/ijerph13050504
Chicago/Turabian StyleKubota, Takeo, and Kazuki Mochizuki. 2016. "Epigenetic Effect of Environmental Factors on Autism Spectrum Disorders" International Journal of Environmental Research and Public Health 13, no. 5: 504. https://0-doi-org.brum.beds.ac.uk/10.3390/ijerph13050504