Single-Cell Analysis Reveals that Chronic Silver Nanoparticle Exposure Induces Cell Division Defects in Human Epithelial Cells

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Silver Nanoparticle Characterization
2.2. Cell Culture and Experimental Design
2.3. ICP-MS
2.4. Transmission Electron Microscopy
2.5. Growth Curves
2.6. Live Cell Imaging and Analysis
2.7. Trypan Blue Assay for Cell Viability
2.8. Immunostaining, Fluorescence Microscopy, and Fixed-Cell Analysis
3. Results
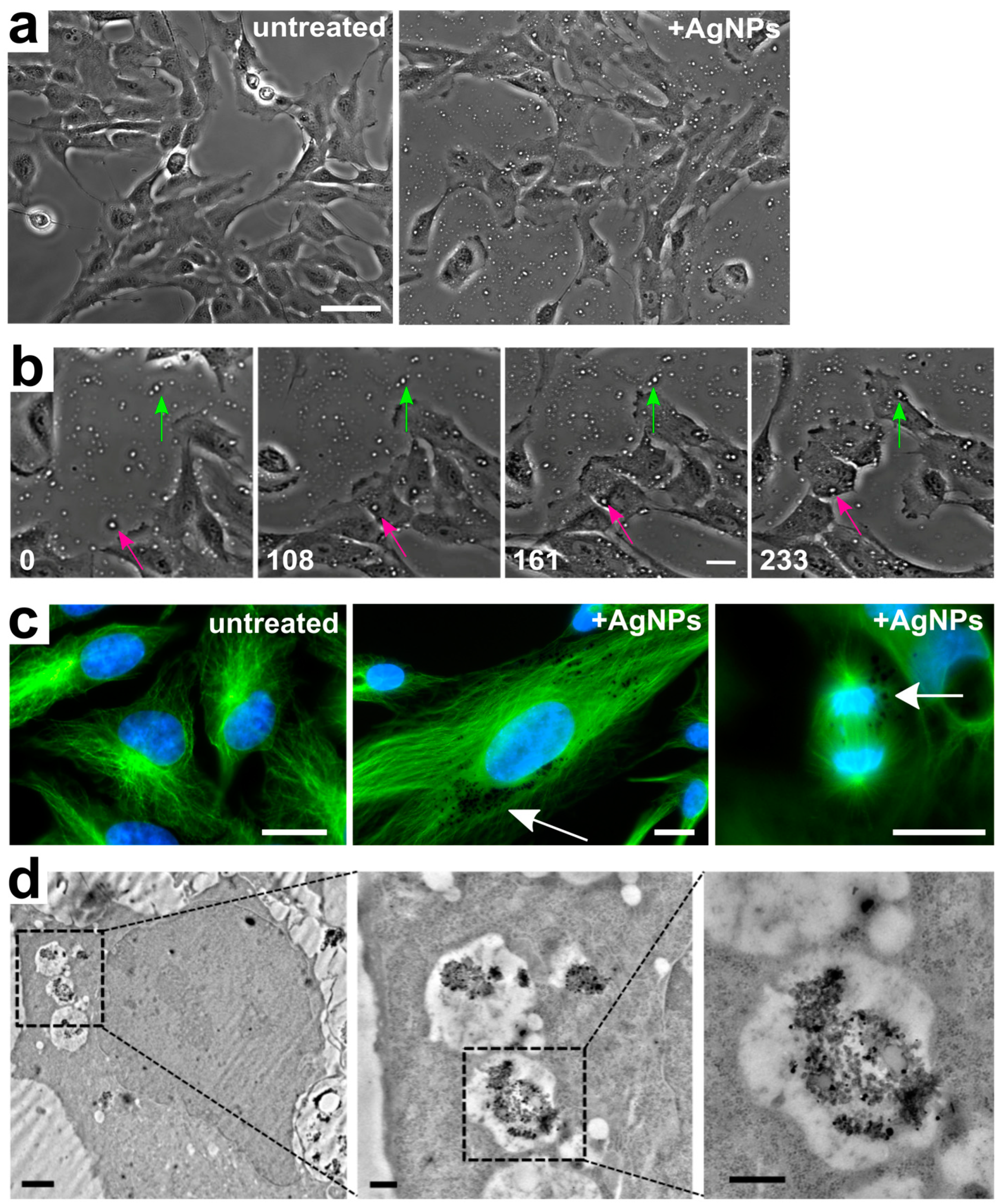
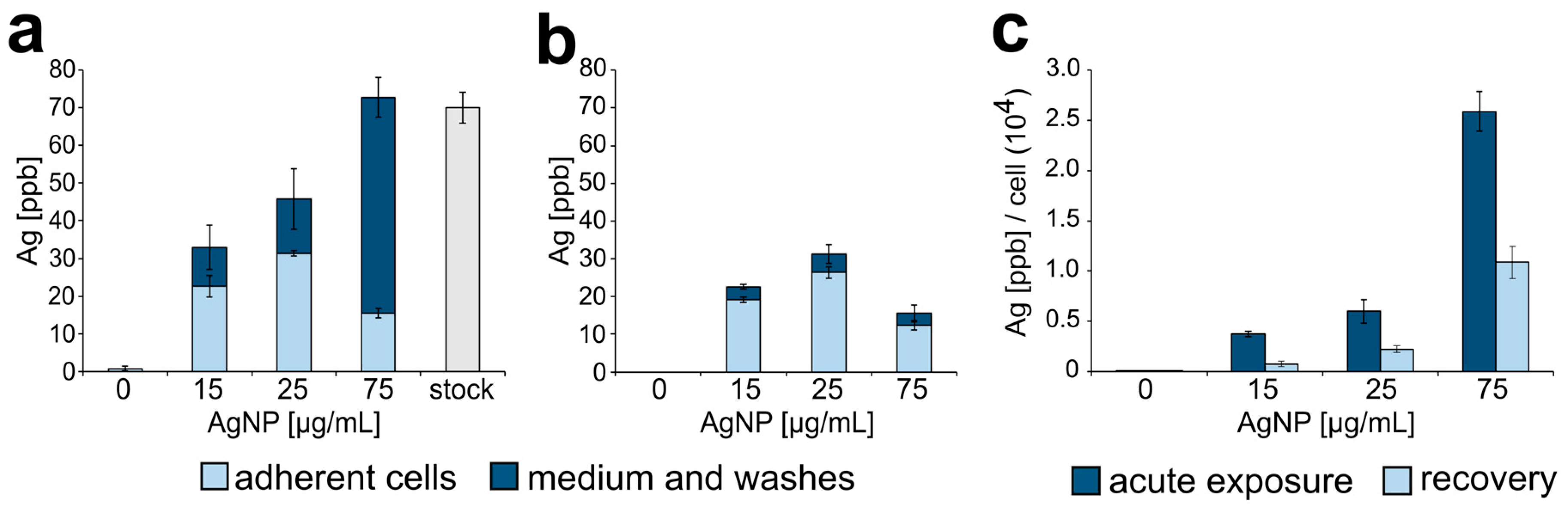
3.1. Silver Nanoparticle Internalization and Accumulation
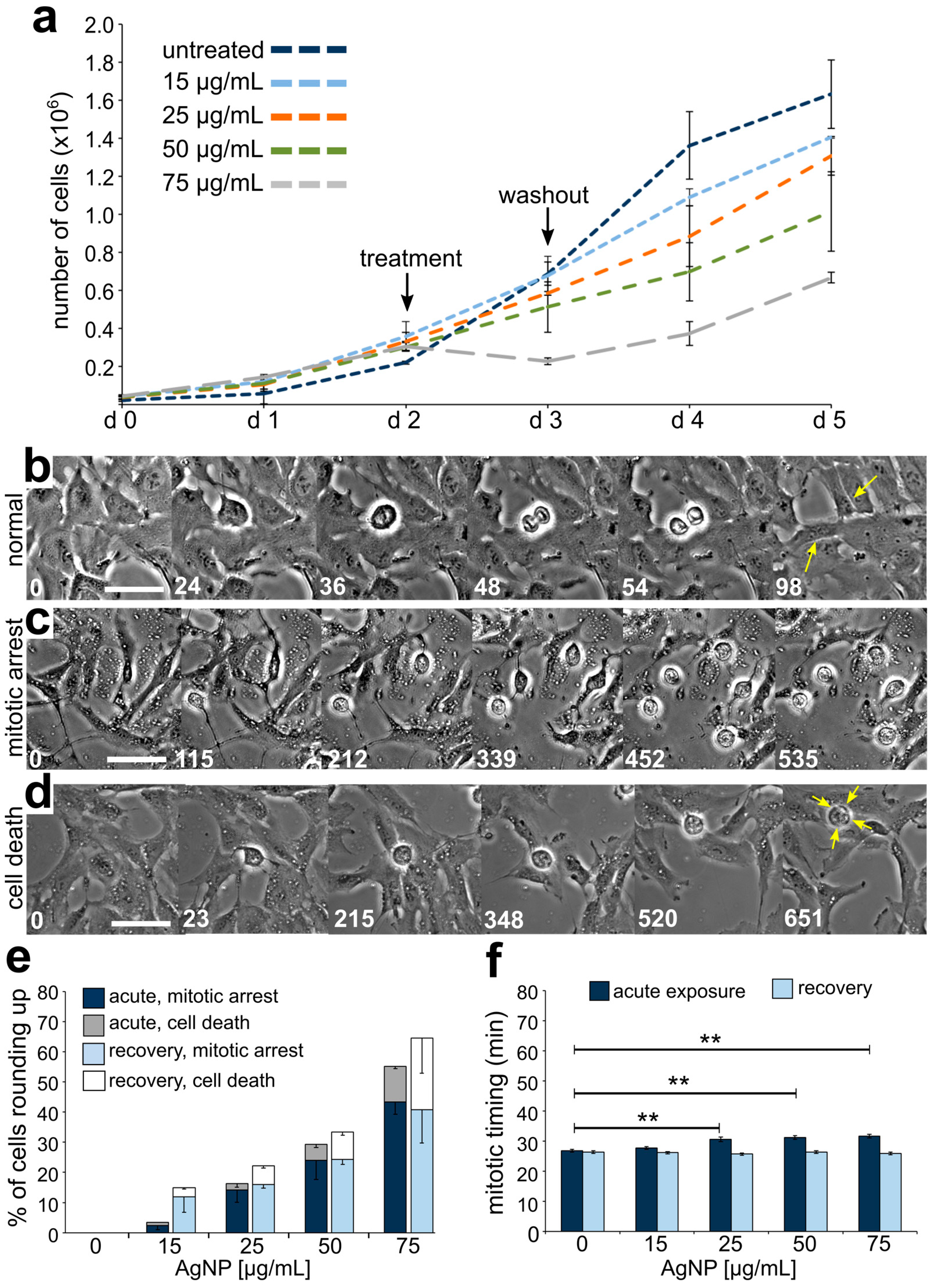
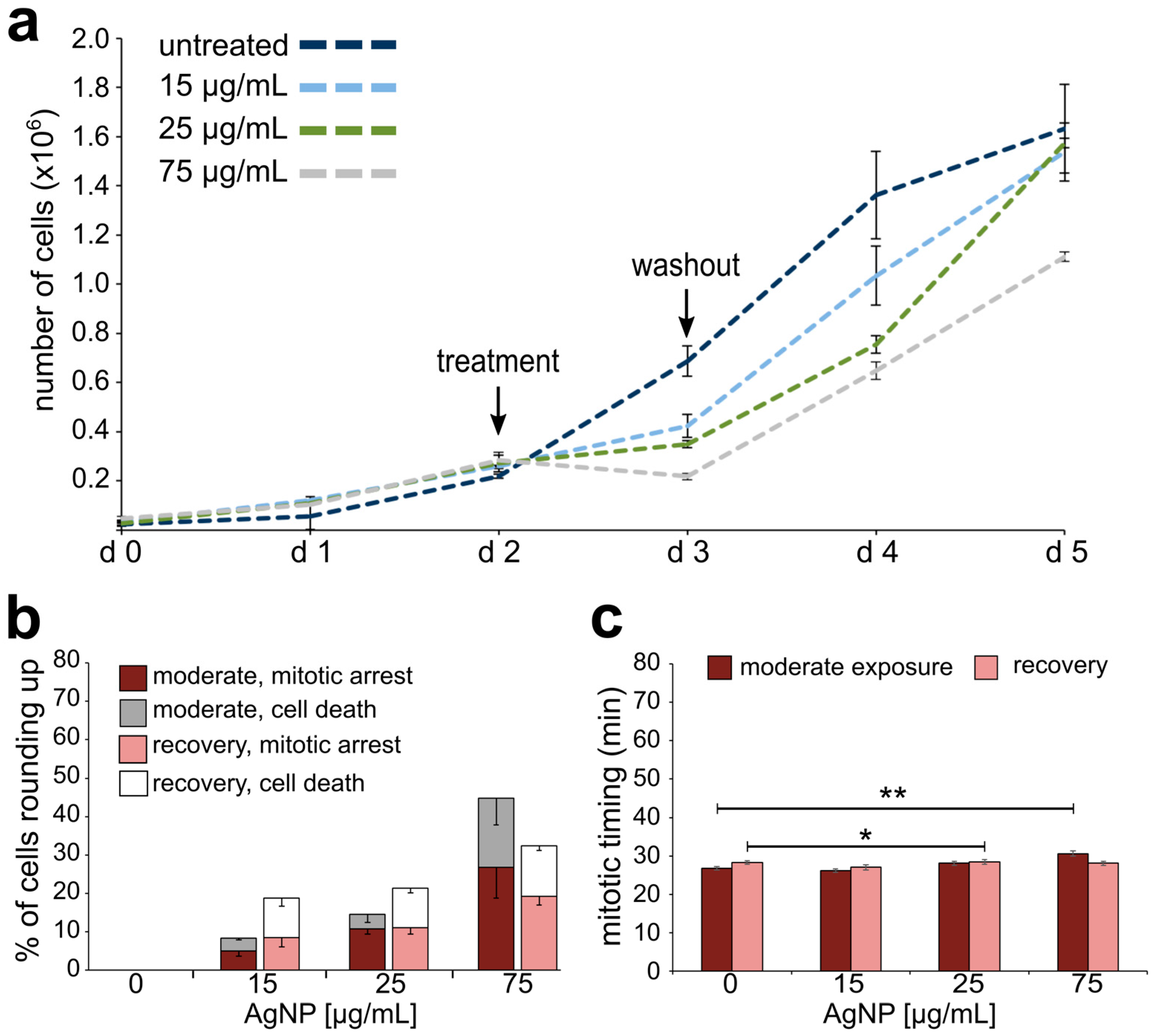
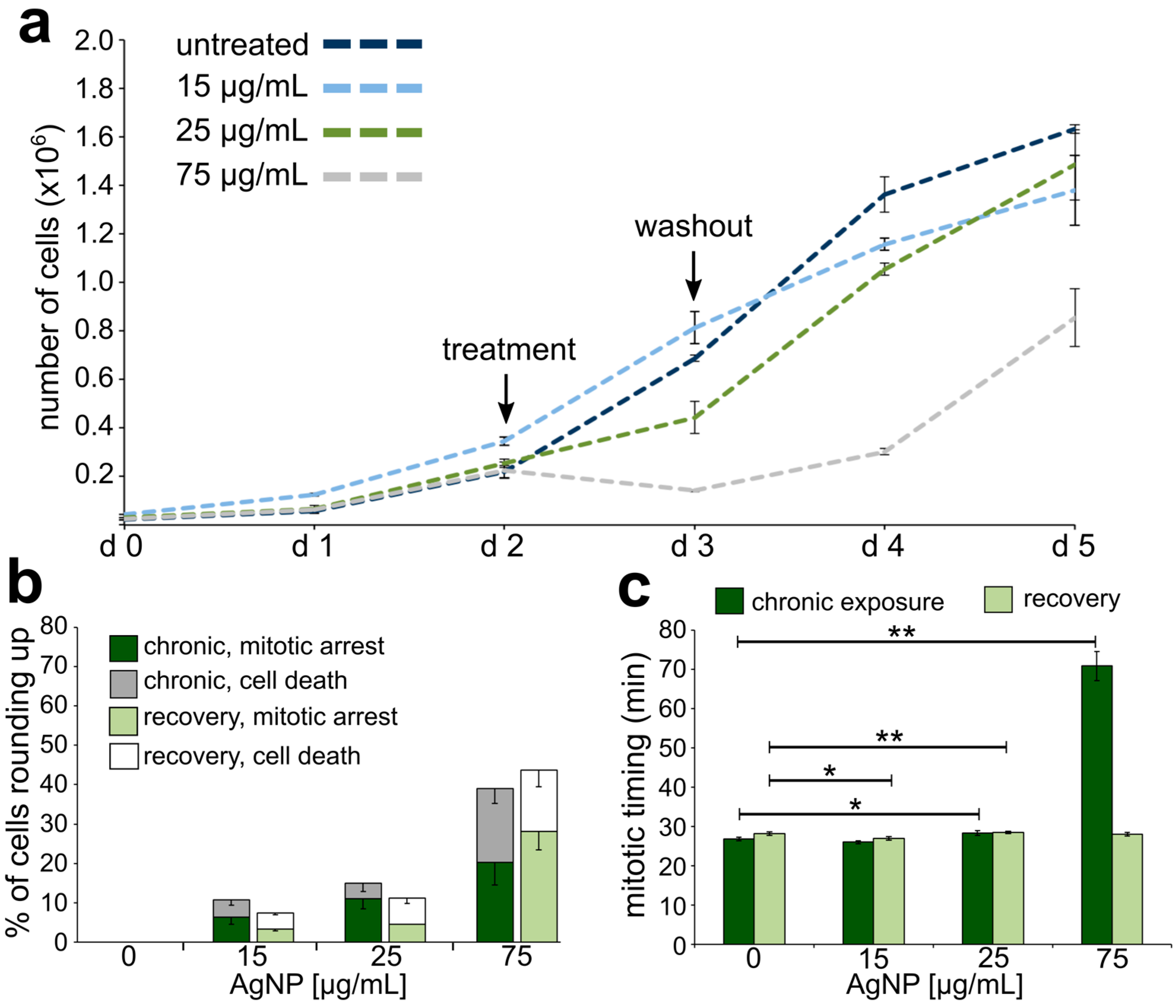
3.2. Reduced Cell Viability and Mitotic Delay from AgNP Exposure
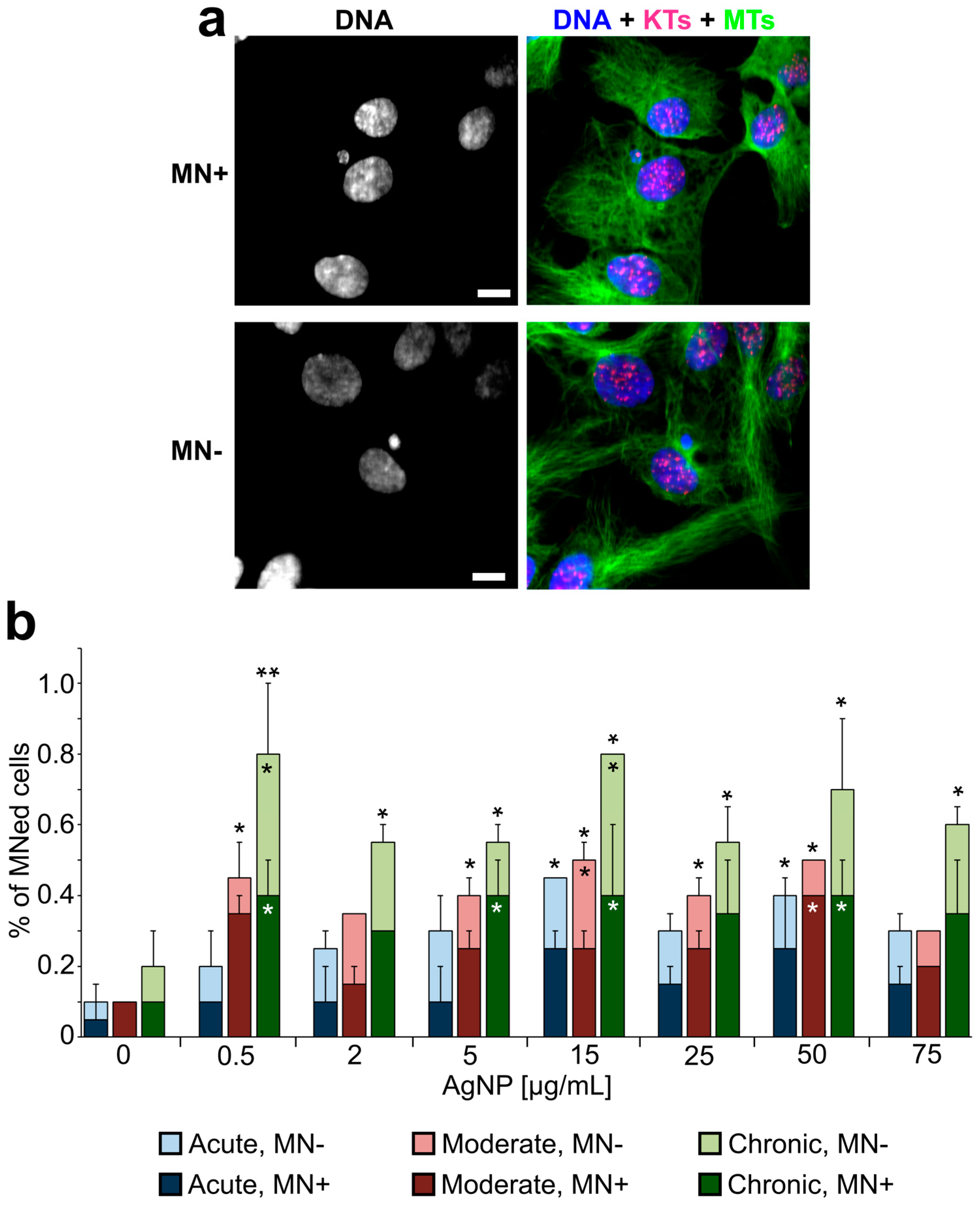
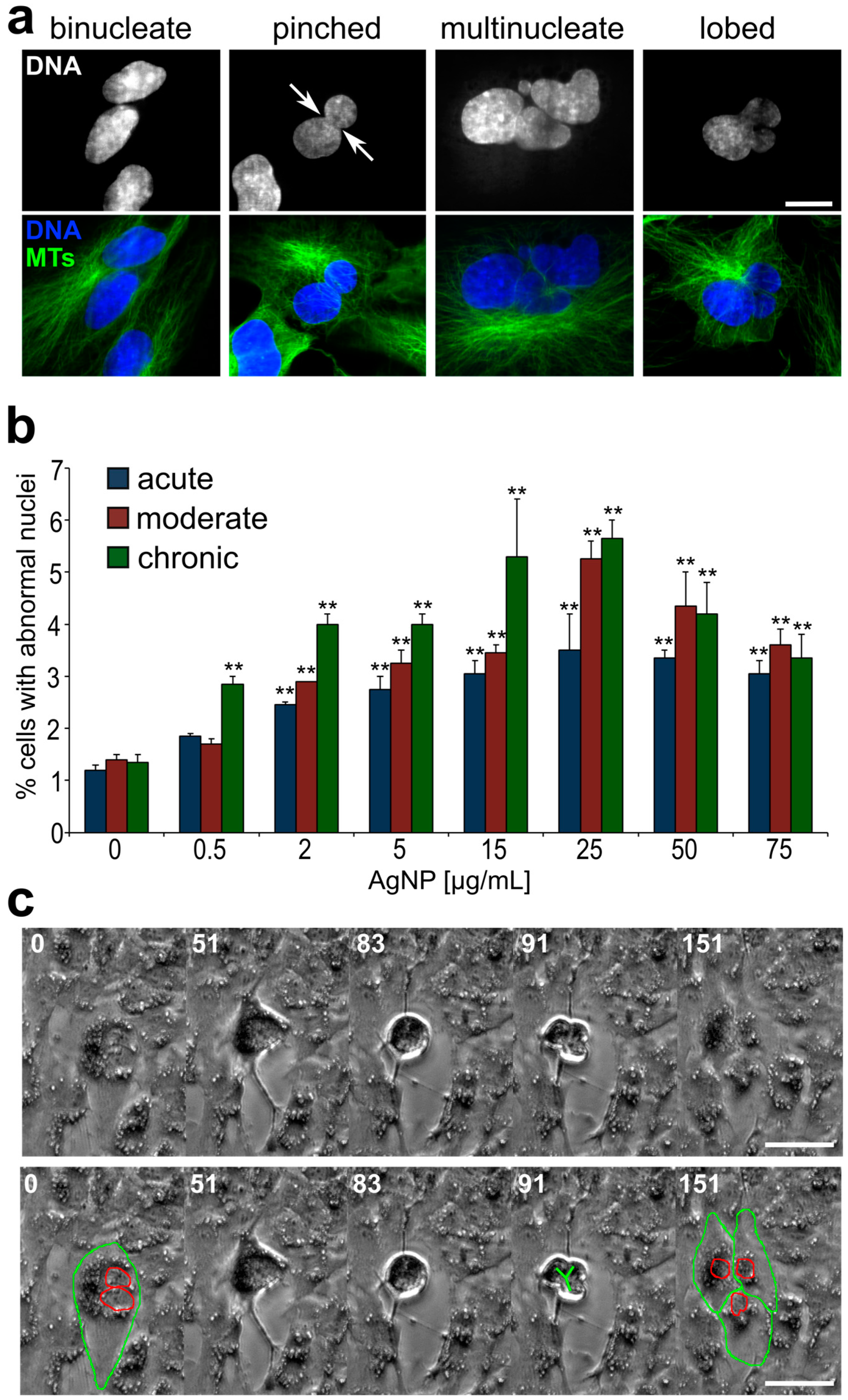
3.3. Emergence of Abnormal Nuclear Phenotypes in AgNP-Treated Cell Populations
4. Discussion
4.1. Long-Term in vitro Exposure to AgNPs, Even at Very Low Doses, Leads to the Accumulation of Cells with Abnormal DNA Content
4.2. Cells with Abnormal DNA Content Emerge as a Result of AgNP-Induced Cell Division Defects
4.3. The Health Risk Associated with Cell Division-Dependent Defects
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- United States Enviornmental Protection Agency. Chemical Data Report 2016; EPA: Washington, DC, USA, 2016.
- Hudson-Shore, M. Statistics of Scientific Procedures on Living Animals in Great Britain 2015- highlighting an ongoing upward trend in animal use and missed opportunities. Altern. Lab. Anim. 2016, 44, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Krewski, D.; Acosta, D., Jr.; Andersen, M.; Anderson, H.; Bailar, J., III; Boekelheide, K.; Kelsey, K. Toxicity testing in the 21st century: A vision and a strategy. J. Toxicol. Environ. Health B Crit. Rev. 2010, 13, 51–138. [Google Scholar] [CrossRef] [PubMed]
- Taylor, K.; Gordon, N.; Langley, G.; Higgins, W. Estimates for worldwide laboratory animal use in 2005. Altern. Lab. Anim. 2008, 36, 327–342. [Google Scholar] [CrossRef] [PubMed]
- Zainzinger, V. Animal tests surge under new US chemical law. Science 2018, 360, 585–586. [Google Scholar] [CrossRef] [PubMed]
- Van Vliet, E. Current standing and future prospects for the technologies proposed to transform toxicity testing in the 21st century. Altern. Anim. Exp. 2011, 28, 17–44. [Google Scholar]
- Burden, N.; Sewell, F.; Chapman, K. Testing chemical safety: What is needed to ensure the widespread application of non-animal approaches? PLoS Biol. 2015, 13, e1002156. [Google Scholar] [CrossRef] [PubMed]
- Prior, H.; Casey, W.; Kimber, I.; Whelan, M.; Sewell, F. Reflections on the progress towards non-animal methods for acute toxicity testing of chemicals. Regul. Toxicol. Pharmacol. 2018, 102, 30–33. [Google Scholar] [CrossRef]
- Burden, N.; Aschberger, K.; Chaudhry, Q.; Clift, M.; Doak, S.; Fowler, P.; Johnston, H.; Landsiedel, R.; Rowland, J.; Stone, V. The 3Rs as a framework to support a 21st century approach for nanosafety assessment. Nano Today 2017, 12, 10–13. [Google Scholar] [CrossRef]
- Alves, V.; Muratov, E.; Zakharov, A.; Muratov, N.; Andrade, C.; Tropsha, A. Chemical toxicity prediction for major classes of industrial chemicals: Is it possible to develop universal models covering cosmetics, drugs, and pesticides? Food Chem. Toxicol. 2018, 112, 526–534. [Google Scholar] [CrossRef]
- Jain, A.; Singh, D.; Dubey, K.; Maurya, R.; Mittal, S.; Pandey, A. Models and Methods for In Vitro Toxicity. In In Vitro Toxicology; Academic Press: Cambridge, MA, USA, 2018; pp. 45–65. [Google Scholar]
- Eskes, C.; Boström, A.; Bowe, G.; Coecke, S.; Hartung, T.; Hendriks, G.; Pamies, D.; Piton, A.; Rovida, C. Good cell culture practices & in vitro toxicology. Toxicol. In Vitro 2017, 45, 272–277. [Google Scholar]
- Coleman, J.; Liu, R.; Wang, K.; Kumar, A. Detecting Apoptosis, Autophagy, and Necrosis. In Apoptosis Methods in Toxicology; Humana Press: New York, NY, USA, 2016. [Google Scholar]
- Smart, R.; Hodgson, E. Molecular and Biochemical Toxicology; John Wiley & Sons: Hoboken, NJ, USA, 2018. [Google Scholar]
- Dhawan, A.; Anderson, D. The Comet Assay in Toxicology; Royal Society of Chemistry: London, UK, 2016. [Google Scholar]
- Corton, J.; Williams, A.; Yauk, C. Using a gene expression biomarker to identify DNA damage-inducing agents in microarray profiles. Environ. Mol. Mutagen. 2018, 59, 772–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pozarowski, P.; Darzynkiewicz, Z. Analysis of cell cycle by flow cytometry. Checkp. Control. Cancer 2010, 281, 301–311. [Google Scholar]
- Fenech, M. The in vitro micronucleus technique. Mutat. Res. 2000, 455, 81–95. [Google Scholar] [CrossRef]
- Smith-Roe, S.; Swartz, C.; Shepard, K.; Bryce, S.; Dertinger, S.; Waidyanatha, S.; Kissling, G.; Auerbach, S.; Witt, K.L. Black cohosh extracts and powders induce micronuclei, a biomarker of genetic damage, in human cells. Environ. Mol. Mutagen. 2018, 59, 416–426. [Google Scholar] [CrossRef]
- Ly, P.; Cleveland, D. Rebuilding Chromosomes After Catastrophe: Emerging Mechanisms of Chromothripsis. Trends Cell Biol. 2017, 27, 917–930. [Google Scholar] [CrossRef] [PubMed]
- Russo, A.; Pacchierotti, F.; Cimini, D.; Ganem, N.; Genescà, A.; Natarajan, A.; Degrassi, F. Genomic instability: Crossing pathways at the origin of structural and numerical chromosome changes. Environ. Mol. Mutagen. 2015, 56, 563–580. [Google Scholar] [CrossRef] [PubMed]
- Benameur, L.; Orsière, T.; Rose, J.; Botta, A. Detection of environmental clastogens and aneugens in human fibroblasts by cytokinesis-blocked micronucleus assay associated with immunofluorescent staining of CENP-A in micronuclei. Chemosphere 2011, 84, 676–680. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Durán, N.; Durán, M.; de Jesus, M.; Seabra, A.; Fávaro, W.; Nakazato, G. Silver nanoparticles: A new view on mechanistic aspects on antimicrobial activity. Biol. Med. 2016, 12, 789–799. [Google Scholar] [CrossRef]
- Souza, L.; da Silva, V.; Franchi, L.; de Souza, T. Toxic and Beneficial Potential of Silver Nanoparticles: The Two Sides of the Same Coin. In Cellular and Molecular Toxicology of Nanoparticles; Springer: Cham, Switzerland, 2018; pp. 251–262. [Google Scholar]
- Chernousova, S.; Epple, M. Silver as Antibacterial Agent: Ion, Nanoparticle, and Metal. Angew. Chem. Int. Ed. Engl. 2013, 52, 1636–1653. [Google Scholar] [CrossRef]
- Vance, M.; Kuiken, T.; Vejerano, E.; Mcginnis, S.; Hochella, M.; Rejeski, D.; Hull, M. Nanotechnology in the real world: Redeveloping the nanomaterial consumer products inventory. Beilstein J. Nanotechnol. 2015, 6, 1769–1780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majeed, A.; Ullah, W.; Anwar, A.; Shuaib, A.; Ilyas, U.; Khalid, P.; Mustafa, G.; Junaid, M.; Faheem, B.; Ali, S. Cost-effective biosynthesis of silver nanoparticles using different organs of plants and their antimicrobial applications: A review. Mater. Technol. 2018, 33, 313–320. [Google Scholar] [CrossRef]
- Franci, G.; Falanga, A.; Galdiero, S.; Palomba, L.; Rai, M.; Morelli, G.; Galdiero, M. Silver nanoparticles as potential antibacterial agents. Molecules 2015, 20, 8856–8874. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, S.; Bera, T.; Roy, A.; Singh, G.; Ramachandrarao, P.; Dash, D. Characterization of enhanced antibacterial effects of novel silver nanoparticles. Nanotechnology 2007, 18, 225103. [Google Scholar] [CrossRef]
- Karthiga, P.; Rajeshkumar, S.; Annadurai, G. Mechanism of Larvicidal Activity of Antimicrobial Silver Nanoparticles Synthesized Using Garcinia mangostana Bark Extract. J. Clust. Sci. 2018, 29, 1233–1241. [Google Scholar] [CrossRef]
- Foldbjerg, R.; Autrup, H. Mechanisms of Silver Nanoparticle Toxicity. Arch. Basic Appl. Med. 2013, 1, 5–15. [Google Scholar]
- Hadrup, N.; Sharma, A.; Loeschner, K. Toxicity of silver ions, metallic silver, and silver nanoparticle materials after in vivo dermal and mucosal surface exposure: A review. Regul. Toxicol. Pharmacol. 2018, 98, 257–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naasz, S.; Altenburger, R.; Kühnel, D. Environmental mixtures of nanomaterials and chemicals: The Trojan-horse phenomenon and its relevance for ecotoxicity. Sci. Total Environ. 2018, 635, 1170–1181. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, I.; Hsieh, Y.; Wang, C.; Chen, I.; Huang, Y. Trojan-horse mechanism in the cellular uptake of silver nanoparticles verified by direct intra-and extracellular silver speciation analysis. Environ. Sci. Technol. 2015, 49, 3813–3821. [Google Scholar] [CrossRef]
- You, F.; Tang, W.; Yung, L.Y. Real-time monitoring of the Trojan-horse effect of silver nanoparticles by using a genetically encoded flourescent cell sensor. Nanoscale 2018, 10, 7726–7735. [Google Scholar] [CrossRef]
- Ahamed, M.; Alsalhi, M.; Siddiqui, M. Silver nanoparticle applications and human health. Clin. Chim. Acta 2010, 411, 1841–1848. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Ryu, D. Silver nanoparticle-induced oxidative stress, genotoxicity and apoptosis in cultured cells and animal tissues. J. Appl. Toxicol. 2013, 33, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Asharani, P.; Low, G.; Mun, K.; Hande, M.; Valiyaveettil, S. Cytotoxicity and Genotoxicity of Silver. ACS Nano 2009, 3, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Schneider, T.; Westermann, M.; Glei, M. In vitro uptake and toxicity studies of metal nanoparticles and metal oxide nanoparticles in human HT29 cells. Arch. Toxicol. 2017, 91, 3517–3527. [Google Scholar] [CrossRef] [PubMed]
- Harvanova, M.; Jiravova, J.; Malohlava, J.; Tomankova, K.; Jirova, D.; Kolarova, H. Ramen imaging of cellular uptake and studies of silver nanoparticles effect in BJ human fibroblasts cell lines. Int. J. Pharm. 2017, 528, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Wang, P.; Wu, J. Effect of exposure of osteoblast-like cells to low-dose silver nanoparticles: Uptake, retention and osteogenic activity. Artif. Cells Nanomed. Biotechnol. 2018, 47, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Castro-Gamboa, S.; Garcia-Garcia, M.R.; PiñonZarate, G.; Rojas-Lemus, M.; Jarquin-Yañez, K.; Herrera-Enriquez, M.; Fortoul, T.; Toledano-Magaña, Y.; Garcia-Iglesias, T.; Pestryakov, A.; et al. Toxicity of silver nanoparticles in mouse bone marrow-derived dendritic cells: Implications for phenotype. J. Immunotoxicol. 2019. [Google Scholar] [CrossRef]
- Tang, J.; Lu, X.; Chen, B.; Cai, E.; Liu, W.; Jiang, J.; Chen, F.; Shan, X.; Zhang, H. Mechanisms of silver nanoparticles-induced cytotoxicity and apoptosis in rat tracheal epithelial cells. J. Toxicol. Sci. 2019, 44, 155–165. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Li, T.; Su, X.; Li, J.; Li, W.; Gan, J.; Wu, T.; Kong, L.; Zhang, T.; Tang, M.; et al. Genotoxic effects of silver nanoparticles with/without coating in human liver HepG2 cells and in mice. J. Appl. Toxicol. 2019, 39, 908–918. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, Y.; Gao, M.; Wang, Z.; Liu, R.; Xia, T.; Liu, S. Silver Nanoparticles Compromise Female Embryonic Stem Cell Differentiation through Disturbing X Chromosome Inactivation. ACS Nano 2019, 13, 2050–2061. [Google Scholar] [CrossRef]
- Piao, M.; Kang, K.; Lee, I.; Kim, H.; Kim, S.; Choi, J.; Hyun, J. Silver nanoparticles induce oxidative cell damage in human liver cells through inhibition of reduced glutathione and induction of mitochondria-involved apoptosis. Toxicol. Lett. 2011, 201, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Park, E.; Yi, J.; Kim, Y.; Choi, K.; Park, K. Silver nanoparticles induce cytotoxicity by a Trojan-horse type mechanism. Toxicol. In Vitro 2010, 24, 872–878. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Wu, Y.; Wang, C.; Li, H.; Wang, T.; Liao, C.; Jiang, G. Impact of silver nanoparticles on human cells: Effect of particle size. Nanotoxicology 2010, 4, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Chairuangkitti, P.; Lawanprasert, S.; Roytrakul, S.; Aueviriyavit, S.; Phummiratch, D.; Kulthong, K.; Chanvorachote, P.; Maniratanachote, R. Silver nanoparticles induce toxicity in A549 cells via ROS-dependent and ROS-independent pathways. Toxicol. In Vitro 2013, 27, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Eom, H.; Choi, J. p38 MAPK Activation, DNA Damage, Cell Cycle Arrest and Apoptosis As Mechanisms of Toxicity of Silver Nanoparticles in Jurkat T Cells. Environ. Sci. Technol. 2010, 44, 8337–8342. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Tang, J.; Zhang, Z.; Chen, Y.; Zhou, G.; Xi, T. Investigation of the cytotoxicity mechanism of silver nanoparticles in vitro. Biomed. Mater. 2010, 5, 044103. [Google Scholar] [CrossRef] [PubMed]
- Bastos, V.; Duarte, I.; Santos, C.; Oliveira, H. Genotoxicity of citrate-coated silver nanoparticles to human keratinocytes assessed by the comet assay and cytokinesis blocked micronucleus assay. Environ. Sci. Pollut. Res. 2017, 24, 5039–5048. [Google Scholar] [CrossRef]
- Luther, E.; Koehler, Y.; Diendorf, J.; Epple, M.; Dringen, R. Accumulation of silver nanoparticles by cultured primary brain astrocytes. Nanotechnology 2011, 22, 375101. [Google Scholar] [CrossRef]
- Arora, S.; Tyagi, N.; Bhardwaj, A.; Rusu, L.; Palanki, R.; Vig, K.; Singh, S. Silver nanoparticles protect human keratinocytes against UVB radiation-induced DNA damage and apoptosis: Potential for prevention of skin carcinogenesis. Nanomedicine 2015, 11, 1265–1275. [Google Scholar] [CrossRef]
- Baudoin, N.; Cimini, D. A guide to classifying mitotic stages and mitotic defects in fixed cells. Chromosoma 2018, 127, 215–227. [Google Scholar] [CrossRef]
- Krystek, P.; Kettler, K.; van der Wagt, B.; de Jong, W. Exploring influences on the cellular uptake of medium-sized silver nanoparticles into THP-1 cells. Microchem. J. 2015, 120, 45–50. [Google Scholar] [CrossRef]
- Maiorano, G.; Sabella, S.; Sorce, B.; Brunetti, V.; Malvindi, M.; Cingolani, R.; Pompa, P. Effects of cell culture media on the dynamic formation of protein−nanoparticle complexes and influence on the cellular response. ACS Nano 2010, 4, 7481–7491. [Google Scholar] [CrossRef] [PubMed]
- Müller, L.; Steiner, S.; Rodriguez-Lorenzo, L.; Petri-Fink, A.; Rothen-Rutishauser, B.; Latzin, P. Exposure to silver nanoparticles affects viability and function of natural killer cells, mostly via the release of ions. Cell Biol. Toxicol. 2018, 34, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Degrassi, F.; Tanzarella, C. Immunofluorescent staining of kinetochores in micronuclei: A new assay for the detection of aneuploidy. Mutat. Res. 1988, 203, 339–345. [Google Scholar] [CrossRef]
- Sung, J.; Lee, E.; Lee, J.; Yu, I. Lung Function Changes in Sprague-Dawley Rats After Prolonged Inhalation Exposure to Silver Nanoparticles. Inhal. Toxicol. 2008, 20, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Santoro, C.; Duchsherer, N.; Grainger, D. Minimal In Vitro Antimicrobial Efficacy and Ocular Cell Toxicity from Silver Nanoparticles. Nanobiotechnology 2007, 3, 55–65. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Kim, M.; Lee, S.; Oh, S.; Chung, K. Genotoxic effects of silver nanoparticles stimulated by oxidative stress in human normal bronchial epithelial (BEAS-2B) cells. Mutat. Res. 2011, 726, 129–135. [Google Scholar] [CrossRef]
- Bakhoum, S.; Kabeche, L.; Wood, M.; Laucius, C.; Qu, D.; Laughney, A.; Zaki, B. Numerical chromosomal instability mediates susceptibility to radiation treatment. Nat. Commun. 2015, 6, 5990. [Google Scholar] [CrossRef]
- Hodgson, J.; Darnton, A. The quantitative risks of mesothelioma and lung cancer in relation to asbestos exposure. Ann. Occup. Hyg. 2000, 44, 565–601. [Google Scholar] [CrossRef]
- Selikoff, I.; Churg, J.; Hammond, E. Relation between exposure to asbestos and mesothelioma. N. Engl. J. Med. 1965, 272, 560–565. [Google Scholar] [CrossRef]
- Oshimura, M.; Hesterberg, T.W.; Tsutsui, T.; Barrett, J.C. Correlation of asbestos-induced cytogenetic effects with cell transformation of Syrian hamster embryo cells in culture. Cancer Res. 1984, 1, 5017–5022. [Google Scholar]
- Walker, C.; Everitt, J.; Barrett, J. Possible cellular and molecular mechanisms for asbestos carcinogenicity. Am. J. Ind. Med. 1992, 21, 253–273. [Google Scholar] [CrossRef] [PubMed]
- Ault, J.; Cole, R.; Jensen, C.; Jensen, L.; Bachert, L.; Rieder, C. Behavior of crocidolite asbestos during mitosis in living vertebrate lung epithelial cells. Cancer Res. 1995, 55, 792–798. [Google Scholar] [PubMed]
- Jensen, C.; Jensen, L.; Rieder, C.; Cole, R.; Ault, J. Long crocidolite asbestos fibers cause polyploidy by sterically blocking cytokinesis. Carcinogenesis 1996, 17, 2013–2021. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Chen, Y.; Wang, Y.; Yin, Y.; Qu, G.; Song, M.; Wang, H. Ultra-long silver nanowires induced mitotic abnormalities and cytokinetic failure in A549 cells. Nanotoxicology 2019, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Johnston, H.; Hutchison, G.; Christensen, F.; Peters, S.; Hankin, S.; Aschberger, K.; Stone, V. A critical review of the biological mechanisms underlying the in vivo and in vitro toxicity of carbon nanotubes: The contribution of physico-chemical characteristics. Nanotoxicology 2010, 4, 207–246. [Google Scholar] [CrossRef] [PubMed]
- Sargent, L.; Hubbs, A.; Young, S.; Kashon, M.; Dinu, C.; Salisbury, J.; Benkovic, S.; Lowry, D.; Murray, A.; Kisin, E.; et al. Single-walled carbon nanotube-induced mitotic disruption. Mutat. Res. 2012, 745, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, A.; Kumar, Y. Cancer cell micronucleus: An update on clinical and diagnostic applications. APMIS 2013, 121, 569–581. [Google Scholar] [CrossRef]
- Bonassi, S.; Znaor, A.; Ceppi, M.; Lando, C.; Chang, W.; Holland, N.; Kirsch-Volders, M.; Zeiger, E.; Ban, S.; Barale, R.; et al. An increased micronucleus frequency in peripheral blood lymphocytes predicts the risk of cancer in humans. Carcinogenesis 2007, 28, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Bakhoum, S.; Silkworth, W.; Nardi, I.; Nicholson, J.; Compton, D.; Cimini, D. The mitotic origin of chromosomal instability. Curr. Biol. 2014, 24, 148–149. [Google Scholar] [CrossRef]
- Thompson, S.; Compton, D. Examining the link between chromosomal instability and aneuploidy in human cells. J. Cell Biol. 2008, 180, 665–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cimini, D.; Fioravanti, D.; Salmon, E.; Degrassi, F. Merotelic kinetochore orientation versus chromosome mono-orientation in the origin of lagging chromosomes in human primary cells. J. Cell Sci. 2002, 115, 507–515. [Google Scholar] [PubMed]
- Hoffelder, D.; Luo, L.; Burke, N.; Watkins, S.; Gollin, S.; Saunders, W. Resolution of anaphase bridges in cancer cells. Chromosoma 2004, 112, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Terradas, M.; Martin, M.; Hernandez, L.; Tusell, L.; Genesca, A. Nuclear envelope defects impede a proper response to micronuclear DNA lesions. Mutat. Res. 2012, 729, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Crasta, K.; Ganem, N.; Dagher, R.; Lantermann, A.; Ivanova, E.; Pan, Y.; Nezi, L.; Protopopov, A.; Chowdhury, D.; Pellman, D. DNA breaks and chromosome pulverization from errors in mitosis. Nature 2012, 482, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Leibowitz, M.; Zhang, C.; Pellman, D. Chromothripsis: A New Mechanism for Rapid Karyotype Evolution. Annu. Rev. Genet. 2015, 49, 183–1047. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, T.; Bandi, M.; Nitta, M.; Ivanova, E.; Bronson, R.; Pellman, D. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature 2005, 437, 1043–1047. [Google Scholar] [CrossRef] [PubMed]
- Zack, T.; Schumacher, S.; Carter, S.; Cherniack, A.; Saksena, G.; Tabak, B.; Lawrence, M.; Zhsng, C.; Wala, J.; Mermel, C.; et al. Pan-cancer patterns of somatic copy number alteration. Nat. Genet. 2013, 45, 1134–1140. [Google Scholar] [CrossRef] [Green Version]
- Wangsa, D.; Quintanilla, I.; Torabi, K.; Vila- Casadesus, M.; Ercilla, A.; Lus, G.; Yuce, Z.; Galofre, C.; Cuatrecasas, M.; Lozano, J.; et al. Near-tetraploid cancer cells show chromosome instability triggered by replication stress and exhibit enhanced invasiveness. FASEB J. 2018, 32, 3502–3517. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garcia, E.B.; Alms, C.; Hinman, A.W.; Kelly, C.; Smith, A.; Vance, M.; Loncarek, J.; Marr, L.C.; Cimini, D. Single-Cell Analysis Reveals that Chronic Silver Nanoparticle Exposure Induces Cell Division Defects in Human Epithelial Cells. Int. J. Environ. Res. Public Health 2019, 16, 2061. https://0-doi-org.brum.beds.ac.uk/10.3390/ijerph16112061
Garcia EB, Alms C, Hinman AW, Kelly C, Smith A, Vance M, Loncarek J, Marr LC, Cimini D. Single-Cell Analysis Reveals that Chronic Silver Nanoparticle Exposure Induces Cell Division Defects in Human Epithelial Cells. International Journal of Environmental Research and Public Health. 2019; 16(11):2061. https://0-doi-org.brum.beds.ac.uk/10.3390/ijerph16112061
Chicago/Turabian StyleGarcia, Ellen B., Cynthia Alms, Albert W. Hinman, Conor Kelly, Adam Smith, Marina Vance, Jadranka Loncarek, Linsey C. Marr, and Daniela Cimini. 2019. "Single-Cell Analysis Reveals that Chronic Silver Nanoparticle Exposure Induces Cell Division Defects in Human Epithelial Cells" International Journal of Environmental Research and Public Health 16, no. 11: 2061. https://0-doi-org.brum.beds.ac.uk/10.3390/ijerph16112061