Comprehensive Fungal Community Analysis of House Dust Using Next-Generation Sequencing


 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Culture Method for the Isolation of Fungi
2.3. DNA Extraction, PCR Amplification, Library Preparation and Sequencing
2.4. Sequencing Data Analysis
2.5. Alpha and Beta Diversity Analysis
2.6. Calculating the Correlation between the Culture and NGS Methods
3. Results and Discussion
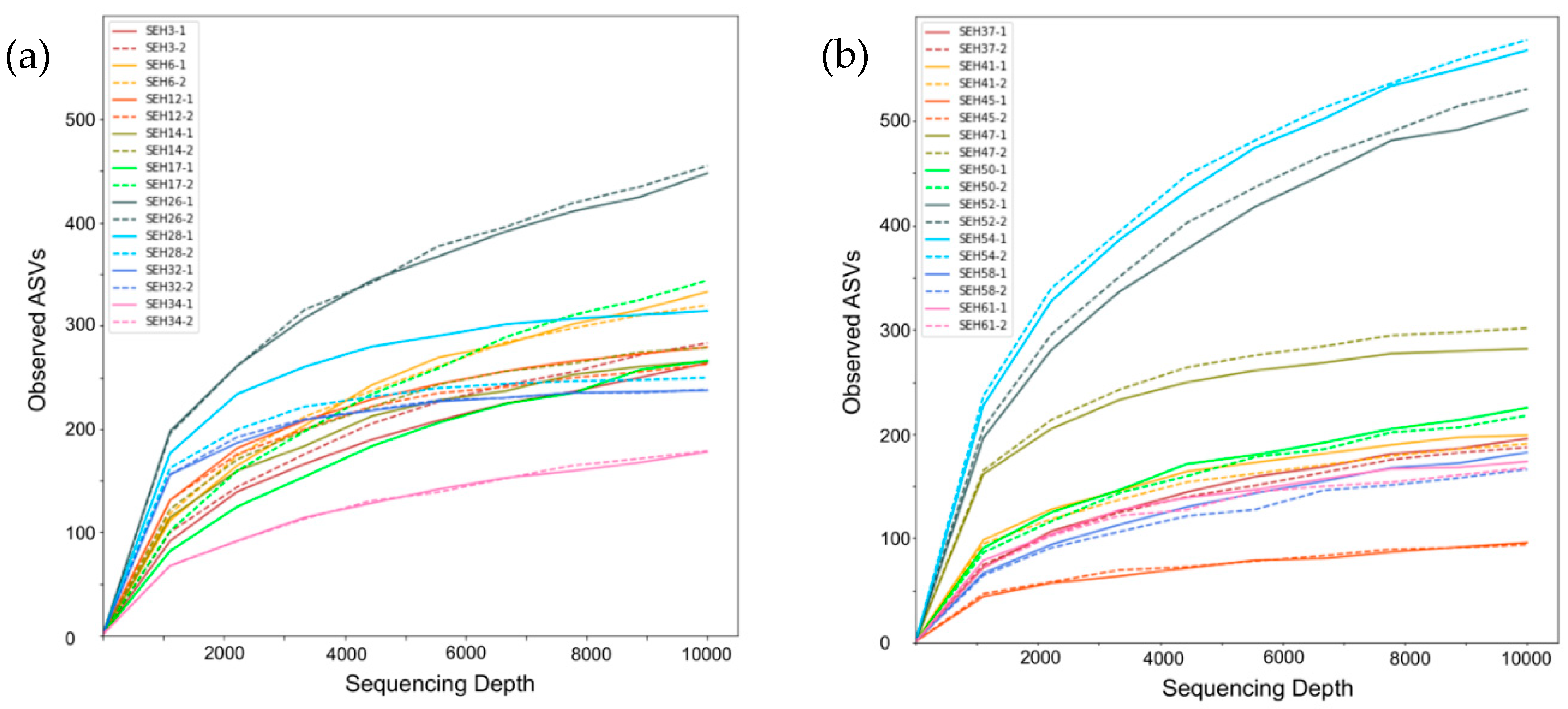
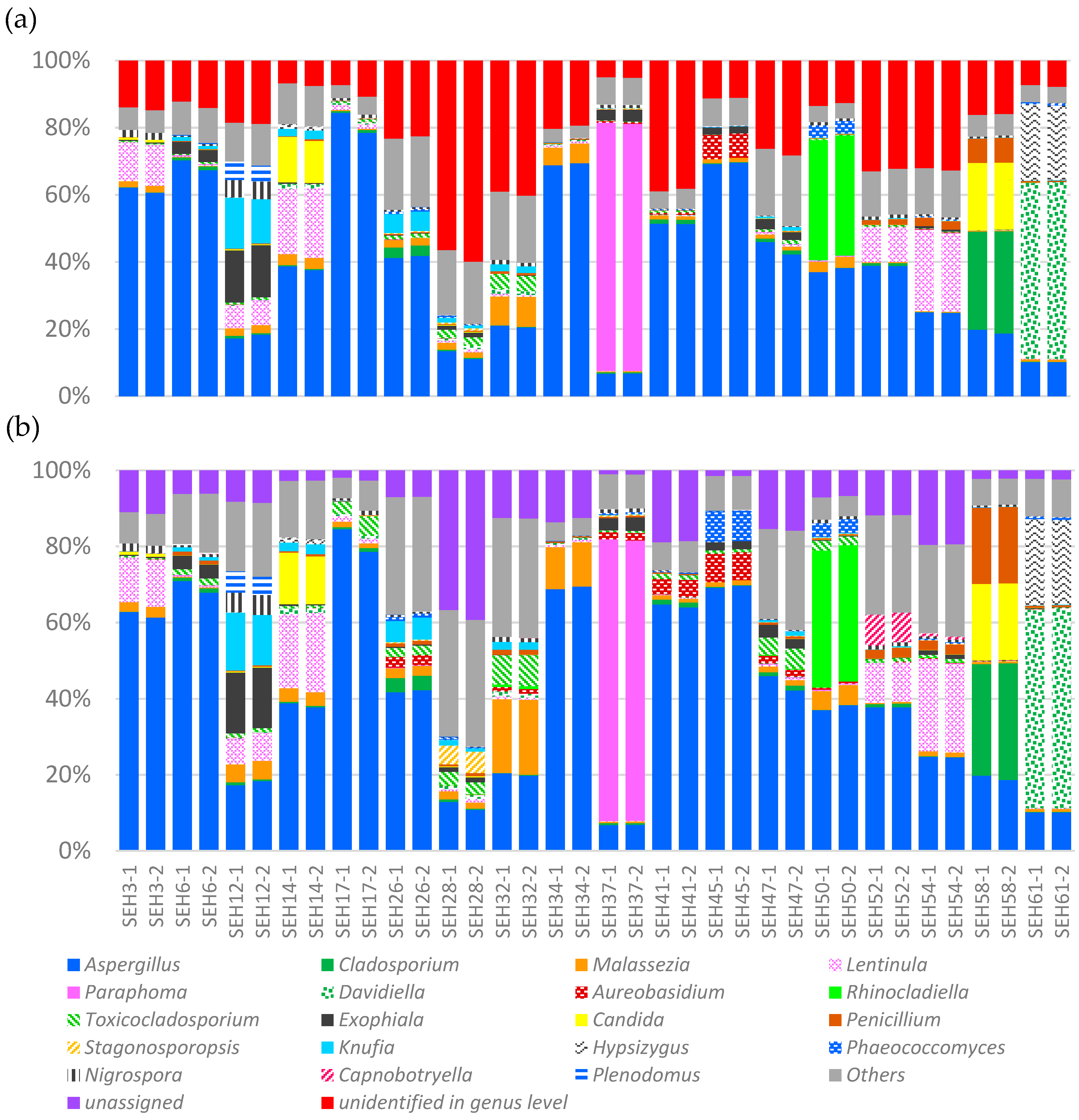
3.1. Sampling Results for the NGS Method
3.2. The Robustness of the PCR Step during the NGS Method
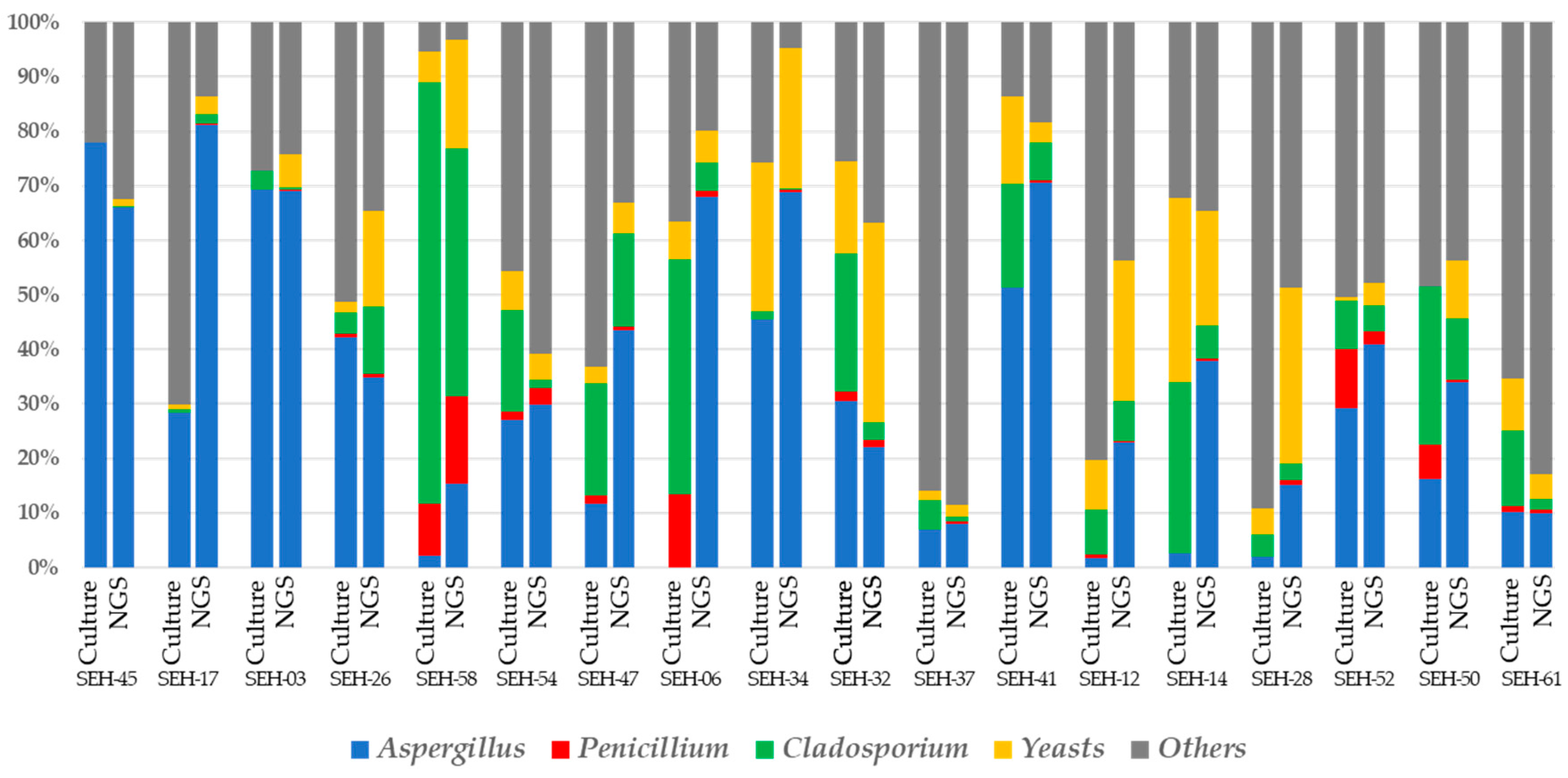
3.3. Fungal Community Structure Comparison between the Culture and NGS Methods
3.4. Correlations between the Culture and NGS-based Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Johansson, E.; Reponen, T.; Meller, J.; Vesper, S.; Yadav, J. Association of Streptomyces community composition determined by PCR-denaturing gradient gel electrophoresis with indoor mold status. Environ. Monit. Assess. 2014, 186, 8773–8783. [Google Scholar] [CrossRef] [PubMed]
- Day, D.B.; Basavanna, U. Real-time PCR detection of Listeria monocytogenes in infant formula and lettuce following macrophage-based isolation and enrichment. J. Appl. Microbiol. 2015, 118, 233–244. [Google Scholar] [CrossRef] [PubMed]
- The Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louca, S.; Mazel, F.; Doebeli, M.; Parfrey, L.W. A census-based estimate of Earth’s bacterial and archaeal diversity. PLoS ONE 2019, 17, e3000106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baxi, S.N.; Portnoy, J.M.; Larenas-Linnemann, D.; Phipatanakul, W.; Barnes, C.; Grimes, C.; Horner, W.E.; Kennedy, K.; Levetin, E.; Miller, J.D.; et al. Exposure and Health Effects of Fungi on Humans. J. Allergy Clin. Immunol. Pract. 2016, 4, 396–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukutomi, Y.; Taniguchi, M. Sensitization to fungal allergens: Resolved and unresolved issues. Allergol. Int. 2015, 64, 321–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oshikata, C.; Watanabe, M.; Saito, A.; Ishida, M.; Kobayashi, S.; Konuma, R.; Kamata, Y.; Terajima, J.; Cho, J.; Yanai, M.; et al. Allergic bronchopulmonary mycosis due to exposure to Eurotium herbariorum after the great east Japan earthquake. Prehosp. Disaster Med. 2017, 32, 688–690. [Google Scholar] [CrossRef] [PubMed]
- Deepak, D.; Rajput, M.S.; Sharma, B.; Chowdhary, A. Allergic bronchopulmonary mycosis fue to fungi other than Aspergillus. Eur. Ann. Allergy Clin. Immunol. Actions 2019, 51, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.-R.; Chen, Y.-H.; Lee, M.-F.; Hsu, L.-Y.; Tien, C.-J.; Shih, F.-M.; Hsiao, S.-C.; Wang, P.-H. Does Spore Count Matter in Fungal Allergy? The Role of Allergenic Fungal Species. Allergy Asthma Immunol. Res. 2016, 8, 404–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dallongeville, A.; Le Cann, P.; Zmirou-Navier, D.; Chevrier, C.; Costet, N.; Annesi-Maesano, I.; Blanchard, O. Concentration and determinants of molds and allergens in indoor air and house dust of French dwellings. Sci. Total. Environ. 2015, 536, 964–972. [Google Scholar] [CrossRef] [PubMed]
- Crawford, J.A.; Rosenbaum, P.F.; Anagnost, S.E.; Hunt, A.; Abraham, J.L. Indicators of airborne fungal concentrations in urban homes: Understanding the conditions that affect indoor fungal exposures. Sci. Total Environ. 2015, 517, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Núñez, A.; Amo de Paz, G.; Rastrojo, A.; García, A.M.; Alcamí, A.; Gutiérrez-Bustillo, A.M.; Moreno, D.A. Monitoring of airborne biological particles in outdoor atmosphere. Part 2: Metagenomics applied to urban environments. Int. Microbiol. 2016, 19, 69–80. [Google Scholar] [PubMed]
- Samson, R.A.; Hoekstra, E.S.; Frisvad, J.C. Introduction to Food—And Airborne Fungi, 7th ed.; Centraalbureau Voor Schimmelcultures: Utrecht, The Netherlands, 2004. [Google Scholar]
- Toju, H.; Tanabe, A.S.; Yamamoto, S.; Sato, H. High-coverage ITS primers for the DNA-based identification of Ascomycetes and Basidiomycetes in environmental samples. PLoS ONE 2012, 7, e40863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, R.H.; Larsson, K.H.; Taylor, A.F.S.; Bengtsson-Palme, J.; Jeppesen, T.S.; Schigel, D.; Kennedy, P.; Picard, K.; Glöckner, F.O.; Tedersoo, L.; et al. The UNITE database for molecular identification of fungi: Handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res. 2019, 47, D259–D264. [Google Scholar] [CrossRef] [PubMed]
- Motooka, D.; Fujimoto, K.; Tanaka, R.; Yaguchi, T.; Gotoh, K.; Maeda, Y.; Furuta, Y.; Kurakawa, T.; Goto, N.; Yasunaga, T.; et al. Fungal ITS1 deep-sequencing strategies to reconstruct the composition of a 26-species community and evaluation of the gut mycobiota of healthy Japanese individuals. Front. Microbiol. 2017, 8, 238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Difonzo, E.M.; Faggi, E.; Bassi, A.; Campisi, E.; Arunachalam, M.; Pini, G.; Scarfì, F.; Galeone, M. Malassezia skin diseases in humans. G. Ital. Dermatol. Venereol. 2013, 148, 609–619. [Google Scholar] [PubMed]
- Kamei, K.; Unno, H.; Nagao, K.; Kuriyama, T.; Nishimura, K.; Miyaji, M. Allergic Bronchopulmonary Mycosis Caused by the Basidiomycetous Fungus Schizophyllum commune. Clin. Infect. Dis. 1994, 18, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Emmons, C.W.; Binford, C.H.; Utz, J.P.; Kwon-Chung, K.J. Medical Mycology, 3rd ed.; Lea & Febiger: Philadelhia, PA, USA, 1977. [Google Scholar]
- Larone, D.H. Medically Important Fungi, 5th ed.; ASM Press: Wachington, DC, USA, 2011. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| House Dust Sample | Rate Difference between with Intact DB and with Modified DB *1(%) | |||
|---|---|---|---|---|
| Unassigned Reads with Genus Level Classification *2 | Other Genera in Figure 3 | Maximum in a Sample | ||
| Genus | Difference (%) | |||
| SHE-3 | −3.1 | 1.5 | Malassezia | 0.8 |
| SHE-6 | −7.0 | 4.2 | Toxicocladosporium | 1.2 |
| SHE-12 | −10.3 | 6.9 | Malassezia | 2.5 |
| SHE-14 | −4.4 | 3.0 | Toxicocladosporium | 0.6 |
| SHE-17 | −6.7 | 2.1 | Toxicocladosporium | 3.5 |
| SHE-26 | −15.9 | 9.4 | Aureobasidium | 2.7 |
| SHE-28 | −20.2 | 14.4 | Stagonosporopsis | 4.8 |
| SHE-32 | −27.1 | 11.2 | Malassezia | 10.7 |
| SHE-34 | −6.8 | 0.9 | Malassezia | 5.8 |
| SHE-37 | −4.0 | 0.9 | Aureobasidium | 1.4 |
| SHE-41 | −19.8 | 2.2 | Aspergillus | 13.0 |
| SHE-45 | −9.6 | 0.7 | Phaeococcomyces | 7.9 |
| SHE-47 | −11.6 | 4.4 | Toxicocladosporium | 4.0 |
| SHE-50 | −6.2 | 0.9 | Toxicocladosporium | 2.2 |
| SHE-52 | −20.8 | 12.3 | Capnobotryella | 8.0 |
| SHE-54 | −12.9 | 9.9 | Malassezia | 1.0 |
| SHE-58 | −13.9 | 0.5 | Penicillium | 12.8 |
| SHE-61 | −5.2 | 4.9 | Penicillium | 0.1 |
| Fungal Group | Correlation Coefficients | |
|---|---|---|
| Total Fungal Count <106 cfu | Total Fungal Count >106 cfu | |
| Aspergillus | 0.29 | 0.66 |
| Penicillium | 0.57 | 0.94 |
| Cladosporium | 0.38 | 0.37 |
| Yeasts | 0.41 | 0.46 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Izawa, K.; Kubosaki, A.; Kobayashi, N.; Akiyama, Y.; Yamazaki, A.; Hashimoto, K.; Konuma, R.; Kamata, Y.; Hara-Kudo, Y.; Hasegawa, K.; et al. Comprehensive Fungal Community Analysis of House Dust Using Next-Generation Sequencing. Int. J. Environ. Res. Public Health 2020, 17, 5842. https://0-doi-org.brum.beds.ac.uk/10.3390/ijerph17165842
Izawa K, Kubosaki A, Kobayashi N, Akiyama Y, Yamazaki A, Hashimoto K, Konuma R, Kamata Y, Hara-Kudo Y, Hasegawa K, et al. Comprehensive Fungal Community Analysis of House Dust Using Next-Generation Sequencing. International Journal of Environmental Research and Public Health. 2020; 17(16):5842. https://0-doi-org.brum.beds.ac.uk/10.3390/ijerph17165842
Chicago/Turabian StyleIzawa, Kazuki, Atsutaka Kubosaki, Naoki Kobayashi, Yutaka Akiyama, Akiko Yamazaki, Kazuhiro Hashimoto, Rumi Konuma, Yoichi Kamata, Yukiko Hara-Kudo, Kenichi Hasegawa, and et al. 2020. "Comprehensive Fungal Community Analysis of House Dust Using Next-Generation Sequencing" International Journal of Environmental Research and Public Health 17, no. 16: 5842. https://0-doi-org.brum.beds.ac.uk/10.3390/ijerph17165842