
A Review of the Role of Endo/Sarcoplasmic Reticulum-Mitochondria Ca2+ Transport in Diseases and Skeletal Muscle Function

 , ,
, , {kind=link}
{kind=link}
Abstract
:1. Introduction
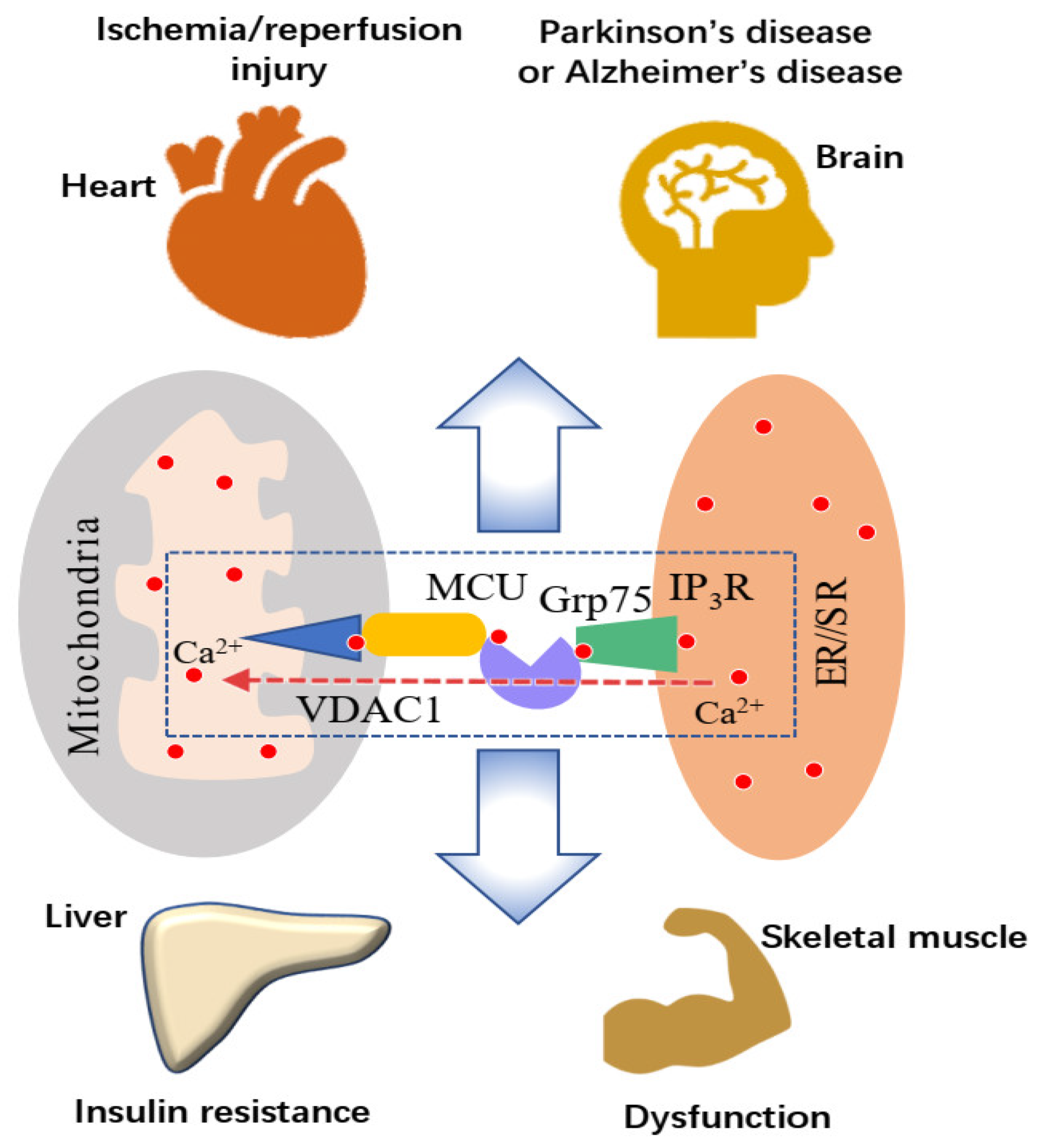
2. Mitochondria-Associated Membrane: A Platform for ER-Mitochondria Ca2+ Transport
2.1. High Ca2+ Microdomain between the ER and Mitochondria
2.2. Endoplasmic Reticulum-Mitochondrial Ca2+ Transport
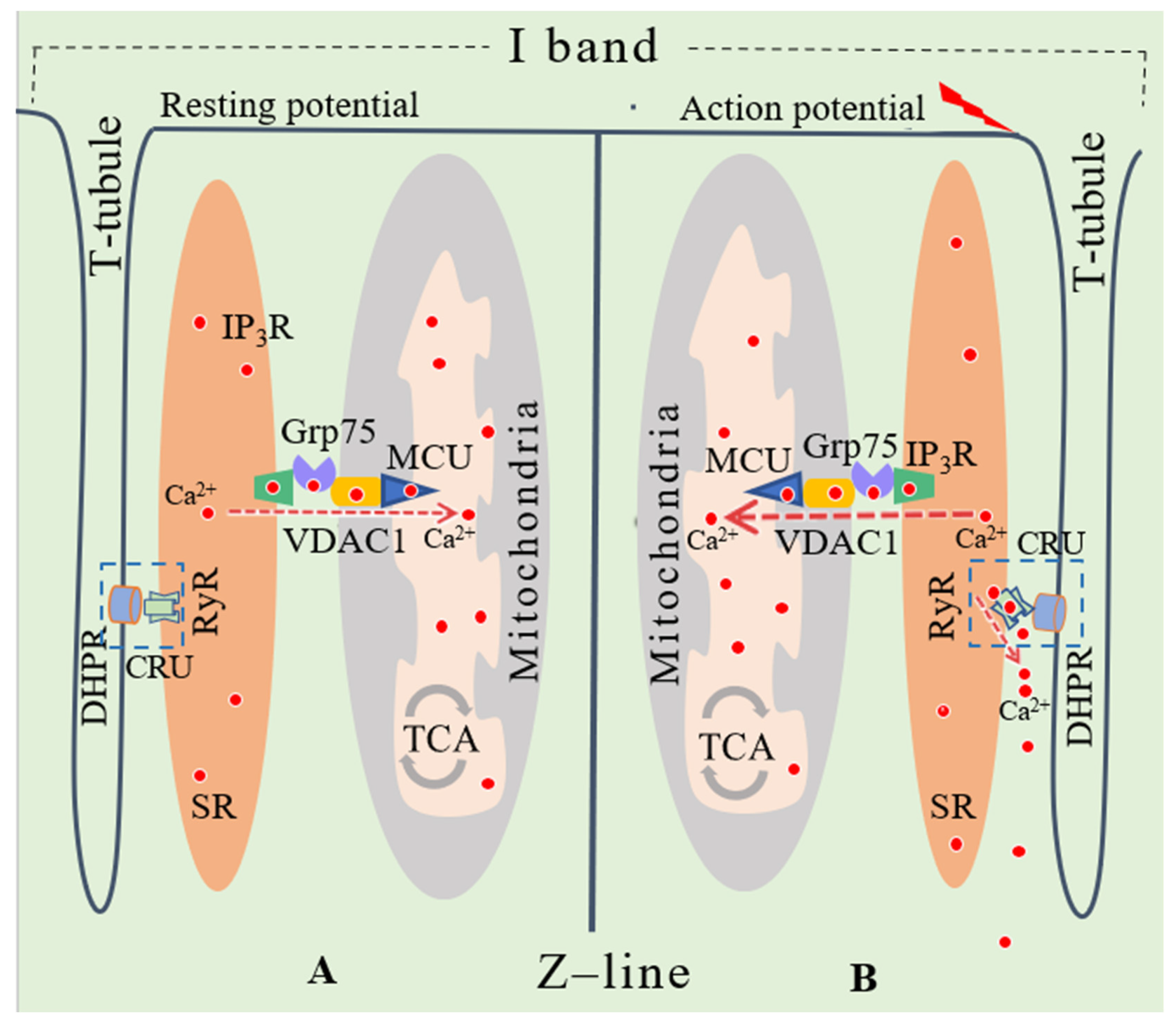
3. Effects of the MAM and SR-Mitochondria Ca2+ Transport on Skeletal Muscle Function
3.1. Mitochondria-Associated Membrane in Cardiac and Skeletal Muscle
3.2. Ca2+ Release by SR in Skeletal Muscle Fibres
4. Sarcoplasmic Reticulum-Mitochondrial Ca2+ Transport in Relation to Skeletal Muscle Dysfunction
5. Sarcoplasmic Reticulum-Mitochondria Ca2+ Transport and Mitochondrial Ca2+ Overload
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Rowland, A.A.; Voeltz, G.K. Endoplasmic reticulum-mitochondria contacts: Function of the junction. Nat. Rev. Mol. Cell Biol. 2012, 13, 607–625. [Google Scholar] [CrossRef] [Green Version]
- Simmen, T.; Herrera-Cruz, M.S. Plastic mitochondria-endoplasmic reticulum (ER) contacts use chaperones and tethers to mould their structure and signaling. Curr. Opin. Cell Biol. 2018, 53, 61–69. [Google Scholar] [CrossRef]
- Rieusset, J. The role of endoplasmic reticulum-mitochondria contact sites in the control of glucose homeostasis: An update. Cell Death Dis. 2018, 9, 388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Guan, N.; Ren, Y.-L.; Wei, Q.-J.; Tao, Y.-H.; Yang, G.-S.; Liu, X.-Y.; Bu, D.-F.; Zhang, Y.; Zhu, S.-N. IP3R-Grp75-VDAC1-MCU calcium regulation axis antagonists protect podocytes from apoptosis and decrease proteinuria in an Adriamycin nephropathy rat model. BMC Nephrol. 2018, 19, 140. [Google Scholar] [CrossRef]
- Gomez, L.; Thiebaut, P.A.; Paillard, M.; Ducreux, S.; Abrial, M.; Crola Da Silva, C.; Durand, A.; Alam, M.R.; Van Coppenolle, F.; Sheu, S.S.; et al. The SR/ER-mitochondria calcium crosstalk is regulated by GSK3β during reperfusion injury. Cell Death Differ. 2015, 22, 1890. [Google Scholar] [CrossRef] [Green Version]
- Zulian, A.; Schiavone, M.; Giorgio, V.; Bernardi, P. Forty years later: Mitochondria as therapeutic targets in muscle diseases. Pharmacol. Res. 2016, 113 Pt A, 563–573. [Google Scholar] [CrossRef]
- Rattray, B.; Thompson, M.; Ruell, P.; Caillaud, C. Specific training improves skeletal muscle mitochondrial calcium homeostasis after eccentric exercise. Eur. J. Appl. Physiol. 2013, 113, 427–436. [Google Scholar] [CrossRef]
- Rizo-Roca, D.; Rios-Kristjansson, J.G.; Nunez-Espinosa, C.; Santos-Alves, E.; Magalhaes, J.; Ascensao, A.; Pages, T.; Viscor, G.; Torrella, J.R. Modulation of mitochondrial biomarkers by intermittent hypobaric hypoxia and aerobic exercise after eccentric exercise in trained rats. Appl. Physiol. Nutr. Metab. 2017, 42, 683–693. [Google Scholar] [CrossRef]
- Yoo, S.Z.; No, M.H.; Heo, J.W.; Park, D.H.; Kang, J.H.; Kim, J.H.; Seo, D.Y.; Han, J.; Jung, S.J.; Kwak, H.B. Effects of acute exercise on mitochondrial function, dynamics, and mitophagy in rat cardiac and skeletal muscles. Int. Neurourol. J. 2019, 23 (Suppl. 1), S22–S31. [Google Scholar] [CrossRef]
- Csordás, G.; Várnai, P.; Golenár, T.; Roy, S.; Purkins, G.; Schneider, T.G.; Balla, T.; Hajnóczky, G. Imaging interorganelle contacts and local calcium dynamics at the ER-mitochondrial interface. Mol. Cell 2010, 39, 121–132. [Google Scholar] [CrossRef]
- Rizzuto, R.; Brini, M.; Murgia, M.; Pozzan, T. Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science 1993, 262, 744–747. [Google Scholar] [CrossRef]
- Rizzuto, R.; Pinton, P.; Carrington, W.; Fay, F.S.; Fogarty, K.E.; Lifshitz, L.M.; Tuft, R.A.; Pozzan, T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 1998, 280, 1763–1766. [Google Scholar] [CrossRef] [PubMed]
- Pauly, M.; Angebault-Prouteau, C.; Dridi, H.; Notarnicola, C.; Scheuermann, V.; Lacampagne, A.; Matecki, S.; Fauconnier, J. ER stress disturbs SR/ER-mitochondria Ca2+ transfer: Implications in Duchenne muscular dystrophy. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2229–2239. [Google Scholar] [CrossRef] [PubMed]
- Pietrangelo, L.; D’Incecco, A.; Ainbinder, A.; Michelucci, A.; Kern, H.; Dirksen, R.T.; Boncompagni, S.; Protasi, F. Age-dependent uncoupling of mitochondria from Ca2+ release units in skeletal muscle. Oncotarget 2015, 6, 35358–35371. [Google Scholar] [CrossRef] [Green Version]
- Thoudam, T.; Ha, C.-M.; Leem, J.; Chanda, D.; Park, J.-S.; Kim, H.-J.; Jeon, J.-H.; Choi, Y.-K.; Liangpunsakul, S.; Huh, Y.H.; et al. PDK4 augments ER-mitochondria contact to dampen skeletal muscle insulin signaling during obesity. Diabetes 2019, 68, 571–586. [Google Scholar] [CrossRef]
- Liu, X.; Yu, Y.; Wang, Y.; Yan, S.; Wang, R.; Li, J. Effect of High-load Exercise on the Mitochondrial Fusion and Fission Proteins in Rat Skeletal muscle. J. Cap. Univ. Phys. Educ. Sports 2016, 28, 167–171. [Google Scholar] [CrossRef]
- Shang, H.; Bai, S.; Xia, Z.; Zhou, Y.; Wang, R. Effects of acupuncture on mitochondrial structure and function of skeletal muscle in rats with heavy load exercise. Chin. J. Rehabil. Med. 2018, 33, 901–908. [Google Scholar] [CrossRef]
- Bravo, R.; Vicencio, J.M.; Parra, V.; Troncoso, R.; Munoz, J.P.; Bui, M.; Quiroga, C.; Rodriguez, A.E.; Verdejo, H.E.; Ferreira, J.; et al. Increased ER-mitochondrial coupling promotes mitochondrial respiration and bioenergetics during early phases of ER stress. J. Cell Sci. 2011, 124 Pt 13, 2143–2152. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Lu, Q.; Wang, Q.; Ding, Y.; Ma, Z.; Mao, X.; Huang, K.; Xie, Z.; Zou, M.-H. Binding of FUN14 domain containing 1 with inositol 1,4,5-trisphosphate receptor in mitochondria-associated endoplasmic reticulum membranes maintains mitochondrial dynamics and function in hearts in vivo. Circulation 2017, 136, 2248–2266. [Google Scholar] [CrossRef]
- Fernström, M.; Tonkonogi, M.; Sahlin, K. Effects of acute and chronic endurance exercise on mitochondrial uncoupling in human skeletal muscle. J. Physiol. 2004, 554 Pt 3, 755–763. [Google Scholar] [CrossRef]
- Mammucari, C.; Raffaello, A.; Vecellio Reane, D.; Gherardi, G.; De Mario, A.; Rizzuto, R. Mitochondrial calcium uptake in organ physiology: From molecular mechanism to animal models. Pflug. Arch. Eur. J. Physiol. 2018, 470, 1165–1179. [Google Scholar] [CrossRef] [Green Version]
- Xue, J.; Li, R.; Zhao, X.; Ma, C.; Lv, X.; Liu, L.; Liu, P. Morusin induces paraptosis-like cell death through mitochondrial calcium overload and dysfunction in epithelial ovarian cancer. Chem. Biol. Interact. 2018, 283, 59–74. [Google Scholar] [CrossRef]
- Missiroli, S.; Danese, A.; Iannitti, T.; Patergnani, S.; Perrone, M.; Previati, M.; Giorgi, C.; Pinton, P. Endoplasmic reticulum-mitochondria Ca2+ crosstalk in the control of the tumor cell fate. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 858–864. [Google Scholar] [CrossRef]
- Hurst, S.; Hoek, J.; Sheu, S.-S. Mitochondrial Ca2+ and regulation of the permeability transition pore. J. Bioenerg. Biomembr. 2017, 49, 27–47. [Google Scholar] [CrossRef] [Green Version]
- Shoshan-Barmatz, V.; Shteinfer-Kuzmine, A.; Verma, A. VDAC1 at the Intersection of Cell Metabolism, Apoptosis, and Diseases. Biomolecules 2020, 10, 1485. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Krelin, Y.; Shteinfer-Kuzmine, A. VDAC1 functions in Ca2+ homeostasis and cell life and death in health and disease. Cell Calcium 2018, 69, 81–100. [Google Scholar] [CrossRef]
- Xu, Z.; Zhang, D.; He, X.; Huang, Y.; Shao, H. Transport of calcium ions into mitochondria. Curr. Genom. 2016, 17, 215–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penna, E.; Espino, J.; De Stefani, D.; Rizzuto, R. The MCU complex in cell death. Cell Calcium 2018, 69, 73–80. [Google Scholar] [CrossRef]
- Giacomello, M.; Drago, I.; Bortolozzi, M.; Scorzeto, M.; Gianelle, A.; Pizzo, P.; Pozzan, T. Ca2+ hot spots on the mitochondrial surface are generated by Ca2+ mobilization from stores, but not by activation of store-operated Ca2+ channels. Mol. Cell 2010, 38, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Giacomello, M.; Pellegrini, L. The coming of age of the mitochondria-ER contact: A matter of thickness. Cell Death Differ. 2016, 23, 1417–1427. [Google Scholar] [CrossRef]
- Csordás, G.; Thomas, A.P.; Hajnóczky, G. Quasi-synaptic calcium signal transmission between endoplasmic reticulum and mitochondria. EMBO J. 1999, 18, 96–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ríos, E.; Gillespie, D.; Franzini-Armstrong, C. The binding interactions that maintain excitation-contraction coupling junctions in skeletal muscle. J. Gen. Physiol. 2019, 151, 593–605. [Google Scholar] [CrossRef] [Green Version]
- Katona, M.; Alzayady, K.; Yule, D.I.; Hajnoczky, G. IP3 receptor isoform dependence of the ER-mitochondrial Ca2+-transfer in mammalian cells. Biophys. J. 2017, 112, 130a. [Google Scholar] [CrossRef] [Green Version]
- Rapizzi, E.; Pinton, P.; Szabadkai, G.; Wieckowski, M.R.; Vandecasteele, G.; Baird, G.; Tuft, R.A.; Fogarty, K.E.; Rizzuto, R. Recombinant expression of the voltage-dependent anion channel enhances the transfer of Ca2+ microdomains to mitochondria. J. Cell Biol. 2002, 159, 613–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Eletto, M.; Rossin, F.; Occhigrossi, L.; Farrace, M.G.; Faccenda, D.; Desai, R.; Marchi, S.; Refolo, G.; Falasca, L.; Antonioli, M.; et al. Transglutaminase Type 2 Regulates ER-Mitochondria Contact Sites by Interacting with GRP75. Cell Rep. 2018, 25, 3573–3581. [Google Scholar] [CrossRef] [Green Version]
- Szabadkai, G.; Bianchi, K.; Várnai, P.; De Stefani, D.; Wieckowski, M.R.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 2006, 175, 901–911. [Google Scholar] [CrossRef] [Green Version]
- Csordás, G.; Renken, C.; Várnai, P.; Walter, L.; Weaver, D.; Buttle, K.F.; Balla, T.; Mannella, C.A.; Hajnóczky, G. Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell Biol. 2006, 174, 915–921. [Google Scholar] [CrossRef] [Green Version]
- Van Vliet, A.R.; Verfaillie, T.; Agostinis, P. New functions of mitochondria associated membranes in cellular signaling. Biochim. Biophys. Acta 2014, 1843, 2253–2262. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Ma, X.; Fujioka, H.; Liu, J.; Chen, S.; Zhu, X. DJ-1 regulates the integrity and function of ER-mitochondria association through interaction with IP3R3-Grp75-VDAC1. Proc. Natl. Acad. Sci. USA 2019, 116, 25322–25328. [Google Scholar] [CrossRef]
- Cárdenas-Pérez, R.E.; Camacho, A. Roles of calcium and mitochondria-associated membranes in the development of obesity and diabetes. Med. Univ. 2016, 18, 23–33. [Google Scholar] [CrossRef] [Green Version]
- Bononi, A.; Giorgi, C.; Patergnani, S.; Larson, D.; Verbruggen, K.; Tanji, M.; Pellegrini, L.; Signorato, V.; Olivetto, F.; Pastorino, S.; et al. BAP1 regulates IP3R3-mediated Ca2+ flux to mitochondria suppressing cell transformation. Nature 2017, 546, 549–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanova, H.; Kerkhofs, M.; La Rovere, R.M.; Bultynck, G. Endoplasmic reticulum-mitochondrial Ca2+ fluxes underlying cancer cell survival. Front. Oncol. 2017, 7, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Area-Gomez, E.; Schon, E.A. Alzheimer disease. Adv. Exp. Med. Biol. 2017, 997, 149–156. [Google Scholar] [CrossRef]
- Bravo-Sagua, R.; Parra, V.; López-Crisosto, C.; Díaz, P.; Quest, A.F.G.; Lavandero, S. Calcium transport and signaling in mitochondria. Compr. Physiol. 2017, 7, 623–634. [Google Scholar] [CrossRef] [PubMed]
- Ogata, T.; Yamasaki, Y. High-resolution scanning electron-microscopic studies on the three-dimensional structure of mitochondria and sarcoplasmic reticulum in the different twitch muscle fibers of the frog. Cell Tissue Res. 1987, 250, 489–497. [Google Scholar] [CrossRef]
- Ainbinder, A.; Boncompagni, S.; Protasi, F.; Dirksen, R.T. Role of Mitofusin-2 in mitochondrial localization and calcium uptake in skeletal muscle. Cell Calcium 2015, 57, 14–24. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Du, X.; Yin, C.; Chang, Z. Shotgun proteomic analysis of sarcoplasmic reticulum preparations from rabbit skeletal muscle. Proteomics 2013, 13, 2335–2338. [Google Scholar] [CrossRef] [PubMed]
- Eisner, V.; Csordás, G.; Hajnóczky, G. Interactions between sarco-endoplasmic reticulum and mitochondria in cardiac and skeletal muscle-pivotal roles in Ca2+ and reactive oxygen species signaling. J. Cell Sci. 2013, 126 Pt 14, 2965–2978. [Google Scholar] [CrossRef] [Green Version]
- Elgass, K.D.; Smith, E.A.; LeGros, M.A.; Larabell, C.A.; Ryan, M.T. Analysis of ER-mitochondria contacts using correlative fluorescence microscopy and soft X-ray tomography of mammalian cells. J. Cell Sci. 2015, 128, 2795–2804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrera-Cruz, M.S.; Simmen, T. Of yeast, mice and men: MAMs come in two flavors. Biol. Direct 2017, 12, 3. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Du, X.; Deng, J.; Gu, M.; Hu, H.; Gui, M.; Yin, C.-C.; Chang, Z. The interactions between mitochondria and sarcoplasmic reticulum and the proteome characterization of mitochondrion-associated membrane from rabbit skeletal muscle. Proteomics 2015, 15, 2701–2704. [Google Scholar] [CrossRef]
- Wu, C.X.; Liu, R.; Gao, M.; Zhao, G.; Wu, S.; Wu, C.F.; Du, G.H. Pinocembrin protects brain against ischemia/reperfusion injury by attenuating endoplasmic reticulum stress induced apoptosis. Neurosci. Lett. 2013, 546, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Sood, A.; Jeyaraju, D.V.; Prudent, J.; Caron, A.; Lemieux, P.; McBride, H.M.; Laplante, M.; Tóth, K.; Pellegrini, L. A Mitofusin-2-dependent inactivating cleavage of Opa1 links changes in mitochondria cristae and ER contacts in the postprandial liver. Proc. Natl. Acad. Sci. USA 2014, 111, 16017–16022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Brito, O.M.; Scorrano, L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 2008, 456, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Filadi, R.; Pozzan, T. Generation and functions of second messengers microdomains. Cell Calcium 2015, 58, 405–414. [Google Scholar] [CrossRef]
- Tubbs, E.; Axelsson, A.S.; Vial, G.; Wollheim, C.B.; Rieusset, J.; Rosengren, A.H. Sulforaphane improves disrupted ER-mitochondria interactions and suppresses exaggerated hepatic glucose production. Mol. Cell. Endocrinol. 2018, 461, 205–214. [Google Scholar] [CrossRef]
- Takekura, H.; Fujinami, N.; Nishizawa, T.; Ogasawara, H.; Kasuga, N. Eccentric exercise-induced morphological changes in the membrane systems involved in excitation-contraction coupling in rat skeletal muscle. J. Physiol. 2001, 533 Pt 2, 571–583. [Google Scholar] [CrossRef]
- Paolini, C.; Protasi, F.; Franzini-Armstrong, C. The relative position of RyR feet and DHPR tetrads in skeletal muscle. J. Mol. Biol. 2004, 342, 145–153. [Google Scholar] [CrossRef]
- Romberg, C.F.; Beqollari, D.; Meza, U.; Bannister, R.A. RGK protein-mediated impairment of slow depolarization- dependent Ca2+ entry into developing myotubes. Channels 2014, 8, 243–248. [Google Scholar] [CrossRef] [Green Version]
- Csordás, G.; Thomas, A.P.; Hajnóczky, G. Calcium signal transmission between ryanodine receptors and mitochondria in cardiac muscle. Trends Cardiovasc. Med. 2001, 11, 269–275. [Google Scholar] [CrossRef]
- Zhou, J.; Dhakal, K.; Yi, J. Mitochondrial Ca2+ uptake in skeletal muscle health and disease. Sci. China Life Sci. 2016, 59, 770–776. [Google Scholar] [CrossRef] [Green Version]
- Hajnóczky, G.; Robb-Gaspers, L.; Seitz, M.B.; Thomas, A. Decoding of cytosolic calcium oscillations in the mitochondria. Cell 1995, 82, 415–424. [Google Scholar] [CrossRef] [Green Version]
- Díaz-Vegas, A.R.; Alex, C.; Denisse, V.; Paola, L.; Cecilia, H.; Gaia, G.; Diego, D.S.; Cristina, M.; Rosario, R.; Ariel, C.-F. Mitochondrial Calcium Increase Induced by RyR1 and IP3R Channel Activation After Membrane Depolarization Regulates Skeletal Muscle Metabolism. Front. Physiol. 2018, 9, 791–805. [Google Scholar] [CrossRef] [Green Version]
- Shkryl, V.M.; Shirokova, N. Transfer and tunneling of Ca2+ from sarcoplasmic reticulum to mitochondria in skeletal muscle. J. Biol. Chem. 2006, 281, 1547–1554. [Google Scholar] [CrossRef] [Green Version]
- Simona, B.; Ann, E.R.; Micaroni, M.; Beznoussenko, G.V.; Polishchuk, R.S.; Dirksen, R.T.; Protasi, F. Mitochondria are linked to calcium stores in striated muscle by developmentally regulated tethering structures. Mol. Biol. Cell 2009, 20, 1058–1067. [Google Scholar] [CrossRef]
- Rieusset, J. Mitochondria-associated membranes (MAMs): An emerging platform connecting energy and immune sensing to metabolic flexibility. Biochem. Biophys. Res. Commun. 2018, 500, 35–44. [Google Scholar] [CrossRef]
- Del Campo, A.; Contreras-Hernández, I.; Castro-Sepúlveda, M.; Campos, C.A.; Figueroa, R.; Tevy, M.F.; Eisner, V.; Casas, M.; Enrique, J. Muscle function decline and mitochondria changes in middle age precede sarcopenia in mice. Aging 2018, 10, 34–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theurey, P.; Tubbs, E.; Vial, G.; Jacquemetton, J.; Bendridi, N.; Chauvin, M.-A.; Alam, M.R.; Le Romancer, M.; Vidal, H.; Rieusset, J. Mitochondria-associated endoplasmic reticulum membranes allow adaptation of mitochondrial metabolism to glucose availability in the liver. J. Mol. Cell Biol. 2016, 8, 129–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.-H.; Chen, Y.-F.; Wu, C.-Y.; Wu, P.-C.; Huang, Y.-L.; Kao, C.-H.; Lin, C.-H.; Kao, L.-S.; Tsai, T.-F.; Wei, Y.-H. Cisd2 modulates the differentiation and functioning of adipocytes by regulating intracellular Ca2+ homeostasis. Hum. Mol. Genet. 2014, 23, 4770–4785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tubbs, E.; Chanon, S.; Robert, M.; Bendridi, N.; Bidaux, G.; Chauvin, M.-A.; Ji-Cao, J.; Durand, C.; Gauvrit-Ramette, D.; Vidal, H.; et al. Disruption of mitochondria-associated endoplasmic reticulum membrane (MAM) integrity contributes to muscle insulin resistance in mice and humans. Diabetes 2018, 67, 636–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arruda, A.P.; Pers, B.M.; Parlakgül, G.; Güney, E.; Inouye, K.; Hotamisligil, G.S. Chronic enrichment of hepatic endoplasmic reticulum-mitochondria contact leads to mitochondrial dysfunction in obesity. Nat. Med. 2014, 20, 1427–1435. [Google Scholar] [CrossRef] [Green Version]
- Shinjo, S.; Jiang, S.; Nameta, M.; Suzuki, T.; Kanai, M.; Nomura, Y.; Goda, N. Disruption of the mitochondria-associated ER membrane (MAM) plays a central role in palmitic acid-induced insulin resistance. Exp. Cell Res. 2017, 359, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Wright, L.E.; Vecellio Reane, D.; Milan, G.; Terrin, A.; Di Bello, G.; Belligoli, A.; Sanna, M.; Foletto, M.; Favaretto, F.; Raffaello, A.; et al. Increased mitochondrial calcium uniporter in adipocytes underlies mitochondrial alterations associated with insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2017, 313, E641–E650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, L.-F.; Chen, P.-J.; Xiao, W.-H. Roles and mechanism of microRNAs in the regulation of skeletal muscle insulin resistance. Sheng Li Xue Bao [Acta Physiol. Sin.] 2019, 71, 497–504. [Google Scholar] [PubMed]
- Zhang, Z.; Di Cui, T.Z.; Sun, Y.; Ding, S. Swimming differentially affects T2DM-induced skeletal muscle ER stress and mitochondrial dysfunction related to MAM. Diabetes Metab. Syndr. Obes. Targets Ther. 2020, 13, 1417–1428. [Google Scholar] [CrossRef]
- Taddeo, E.P.; Laker, R.C.; Breen, D.S.; Akhtar, Y.N.; Kenwood, B.M.; Liao, J.A.; Zhang, M.; Fazakerley, D.J.; Tomsig, J.L.; Harris, T.E.; et al. Opening of the mitochondrial permeability transition pore links mitochondrial dysfunction to insulin resistance in skeletal muscle. Mol. Metab. 2014, 3, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Timpani, C.A.; Hayes, A.; Rybalka, E. Revisiting the dystrophin-ATP connection: How half a century of research still implicates mitochondrial dysfunction in Duchenne Muscular Dystrophy aetiology. Med. Hypotheses 2015, 85, 1021–1033. [Google Scholar] [CrossRef]
- Davies, K.; Guiraud, S. Regenerative biomarkers for Duchenne muscular dystrophy. Neural Regen. Res. 2019, 14, 1317–1320. [Google Scholar] [CrossRef]
- Farini, A.; Sitzia, C.; Cassinelli, L.; Colleoni, F.; Parolini, D.; Giovanella, U.; Maciotta, S.; Colombo, A.; Meregalli, M.; Torrente, Y. Inositol 1,4,5-trisphosphate (IP3)-dependent Ca2+ signaling mediates delayed myogenesis in Duchenne muscular dystrophy fetal muscle. Development 2016, 143, 658–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valladares, D.; Utreras-Mendoza, Y.; Campos, C.; Morales, C.; Diaz-Vegas, A.; Contreras-Ferrat, A.; Westermeier, F.; Jaimovich, E.; Marchi, S.; Pinton, P.; et al. IP3 receptor blockade restores autophagy and mitochondrial function in skeletal muscle fibers of dystrophic mice. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3685–3695. [Google Scholar] [CrossRef]
- Gill, J.F.; Delezie, J.; Santos, G.; McGuirk, S.; Schnyder, S.; Frank, S.; Rausch, M.; St-Pierre, J.; Handschin, C. Peroxisome proliferator-activated receptor gamma coactivator 1alpha regulates mitochondrial calcium homeostasis, sarcoplasmic reticulum stress, and cell death to mitigate skeletal muscle aging. Aging Cell 2019, 18, e12993. [Google Scholar] [CrossRef] [Green Version]
- Umanskaya, A.; Santulli, G.; Xie, W.; Andersson, D.C.; Reiken, S.R.; Marks, A.R. Genetically enhancing mitochondrial antioxidant activity improves muscle function in aging. Proc. Natl. Acad. Sci. USA 2014, 111, 15250–15255. [Google Scholar] [CrossRef] [Green Version]
- Zampieri, S.; Mammucari, C.; Romanello, V.; Barberi, L.; Pietrangelo, L.; Fusella, A.; Mosole, S.; Gherardi, G.; Höfer, C.; Löfler, S.; et al. Physical exercise in aging human skeletal muscle increases mitochondrial calcium uniporter expression levels and affects mitochondria dynamics. Physiol. Rep. 2016, 4. [Google Scholar] [CrossRef]
- Feliciano, P. Mitochondria association to Calcium Release Units is controlled by age and muscle activity. Eur. J. Transl. Myol. 2014, 25, 257–262. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Z.; Shang, Y.; Tao, J.; Zhang, J.; Sha, B. Endoplasmic reticulum stress signaling pathways: Activation and diseases. Curr. Protein Pept. Sci. 2019. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.R.; Ma, H.; Yang, F.; Belcher, J.; Ding, X.Q. Endoplasmic reticulum (ER) Ca2+-channel activity contributes to ER stress and cone death in cyclic nucleotide-gated channel deficiency. J. Biol. Chem. 2017, 292, 11189–11205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, D.R.; Wang, R. Calcium and energy: Making the cake and eating it too? Cell 2010, 142, 200–202. [Google Scholar] [CrossRef] [Green Version]
- Chami, M.; Oulès, B.; Szabadkai, G.; Tacine, R.; Rizzuto, R.; Paterlini-Bréchot, P. Role of SERCA1 truncated isoform in the proapoptotic calcium transfer from ER to mitochondria during ER stress. Mol. Cell 2008, 32, 641–651. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, S.-S.; Zhou, S.; Crowley-McHattan, Z.J.; Wang, R.-Y.; Li, J.-P. A Review of the Role of Endo/Sarcoplasmic Reticulum-Mitochondria Ca2+ Transport in Diseases and Skeletal Muscle Function. Int. J. Environ. Res. Public Health 2021, 18, 3874. https://0-doi-org.brum.beds.ac.uk/10.3390/ijerph18083874
Zhang S-S, Zhou S, Crowley-McHattan ZJ, Wang R-Y, Li J-P. A Review of the Role of Endo/Sarcoplasmic Reticulum-Mitochondria Ca2+ Transport in Diseases and Skeletal Muscle Function. International Journal of Environmental Research and Public Health. 2021; 18(8):3874. https://0-doi-org.brum.beds.ac.uk/10.3390/ijerph18083874
Chicago/Turabian StyleZhang, Shuang-Shuang, Shi Zhou, Zachary J. Crowley-McHattan, Rui-Yuan Wang, and Jun-Ping Li. 2021. "A Review of the Role of Endo/Sarcoplasmic Reticulum-Mitochondria Ca2+ Transport in Diseases and Skeletal Muscle Function" International Journal of Environmental Research and Public Health 18, no. 8: 3874. https://0-doi-org.brum.beds.ac.uk/10.3390/ijerph18083874