Ozone Enhances Diesel Exhaust Particles (DEP)-Induced Interleukin-8 (IL-8) Gene Expression in Human Airway Epithelial Cells through Activation of Nuclear Factors- κB (NF-κB) and IL-6 (NF-IL6)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Introduction
Materials and Methods
Materials
Cell Culture
Measurement of Lactate Dehydrogenase Levels
In vitro Ozone Exposure System
Measurement of IL-8 Protein
Cell Fractionation
Nuclear Factors -κB (NF-κB) and IL6 (NF-IL6) Activation Assays
Gel Mobility Assays
Statistical Analysis
Results
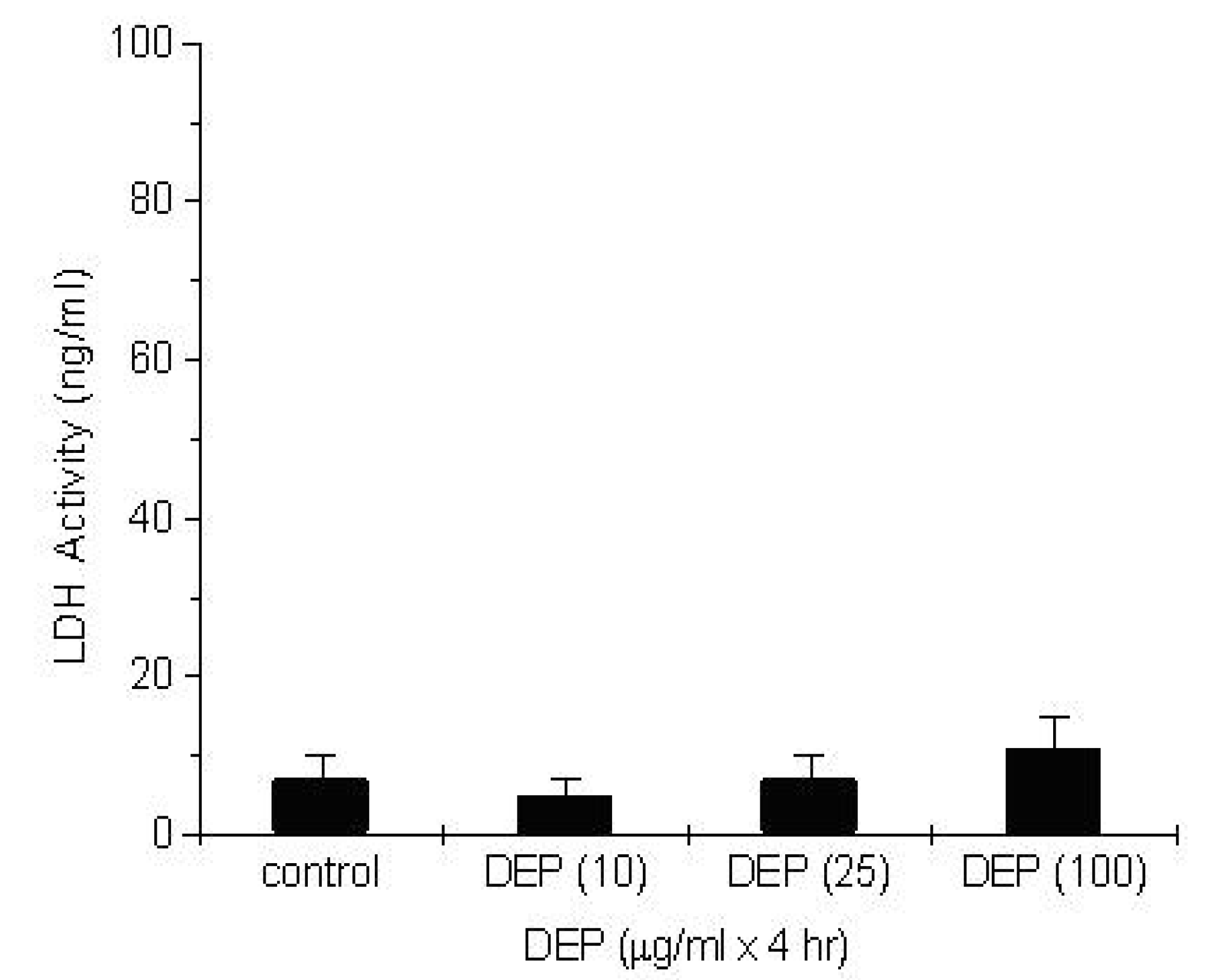
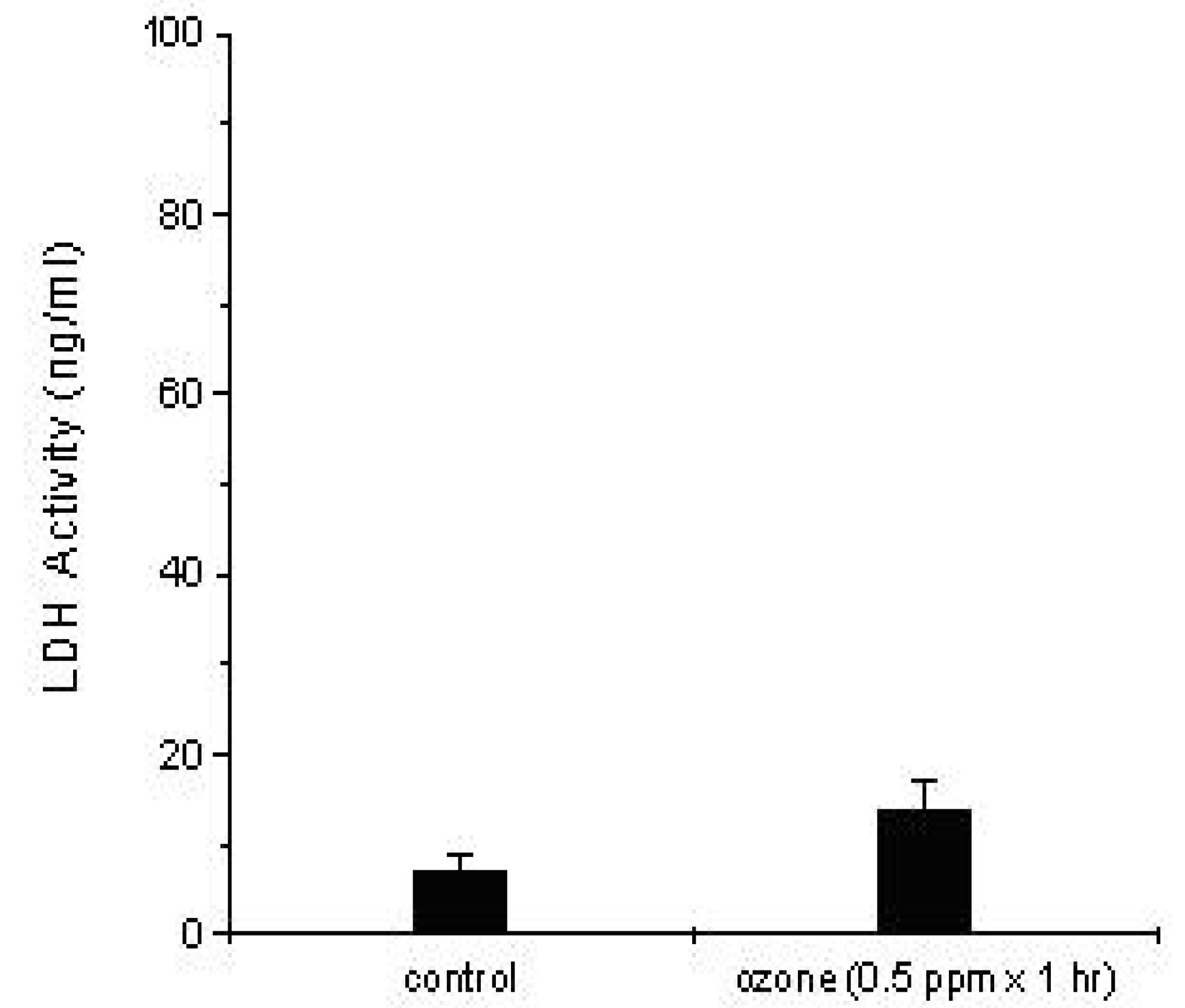
Measurement of the Cytotoxic Effect of DEP and Ozone Exposure



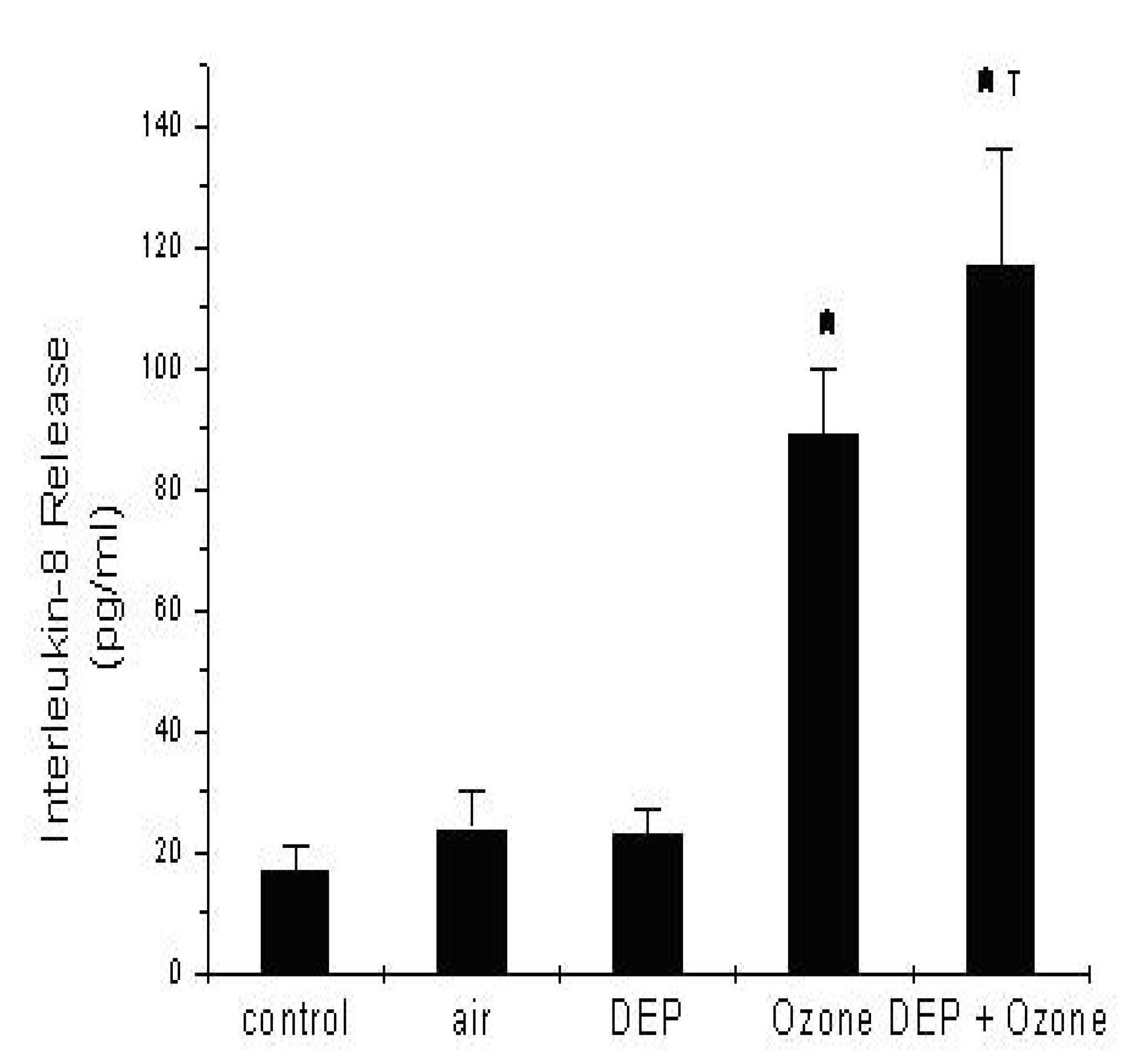
Effect of DEP on IL-8 Release
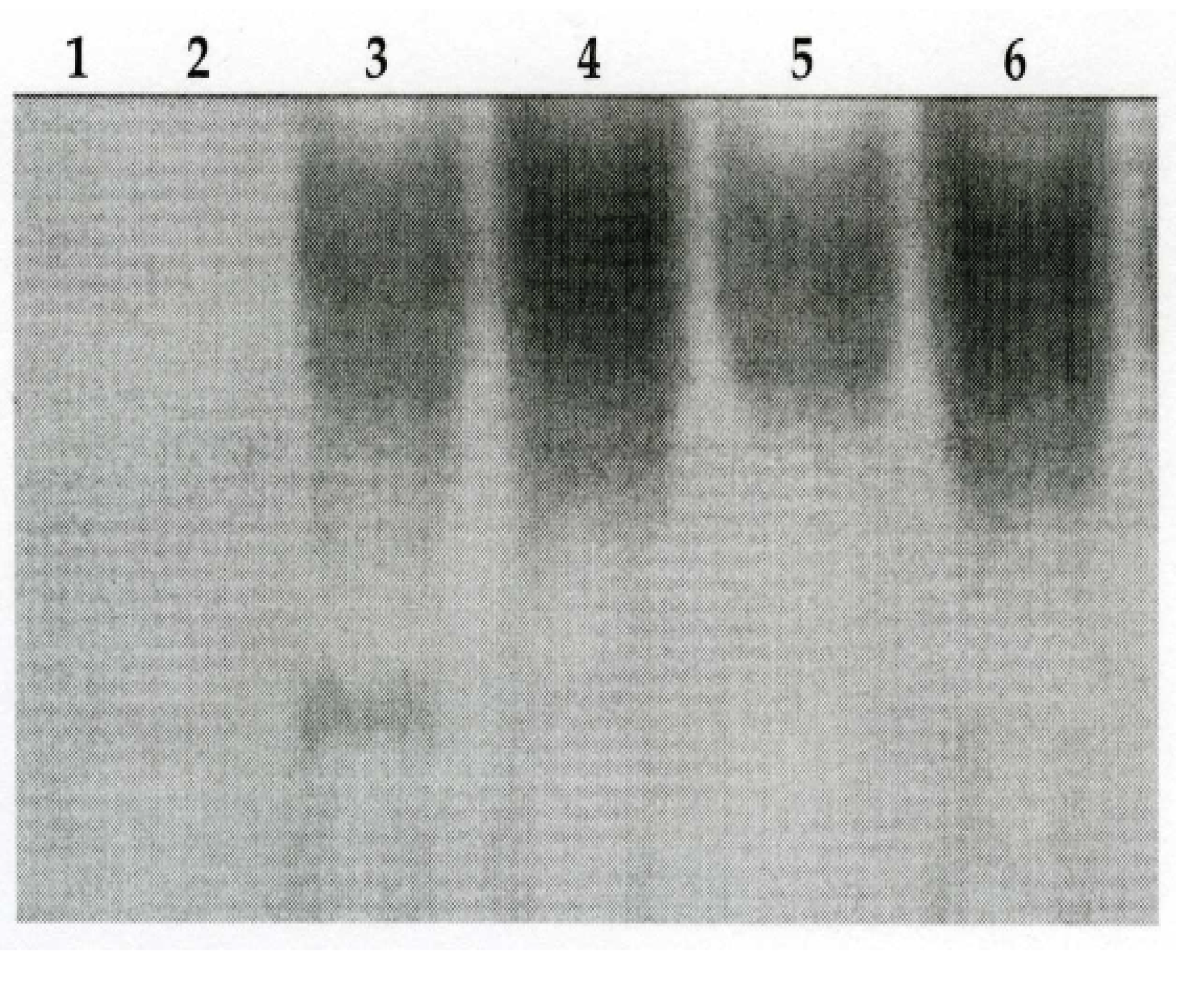
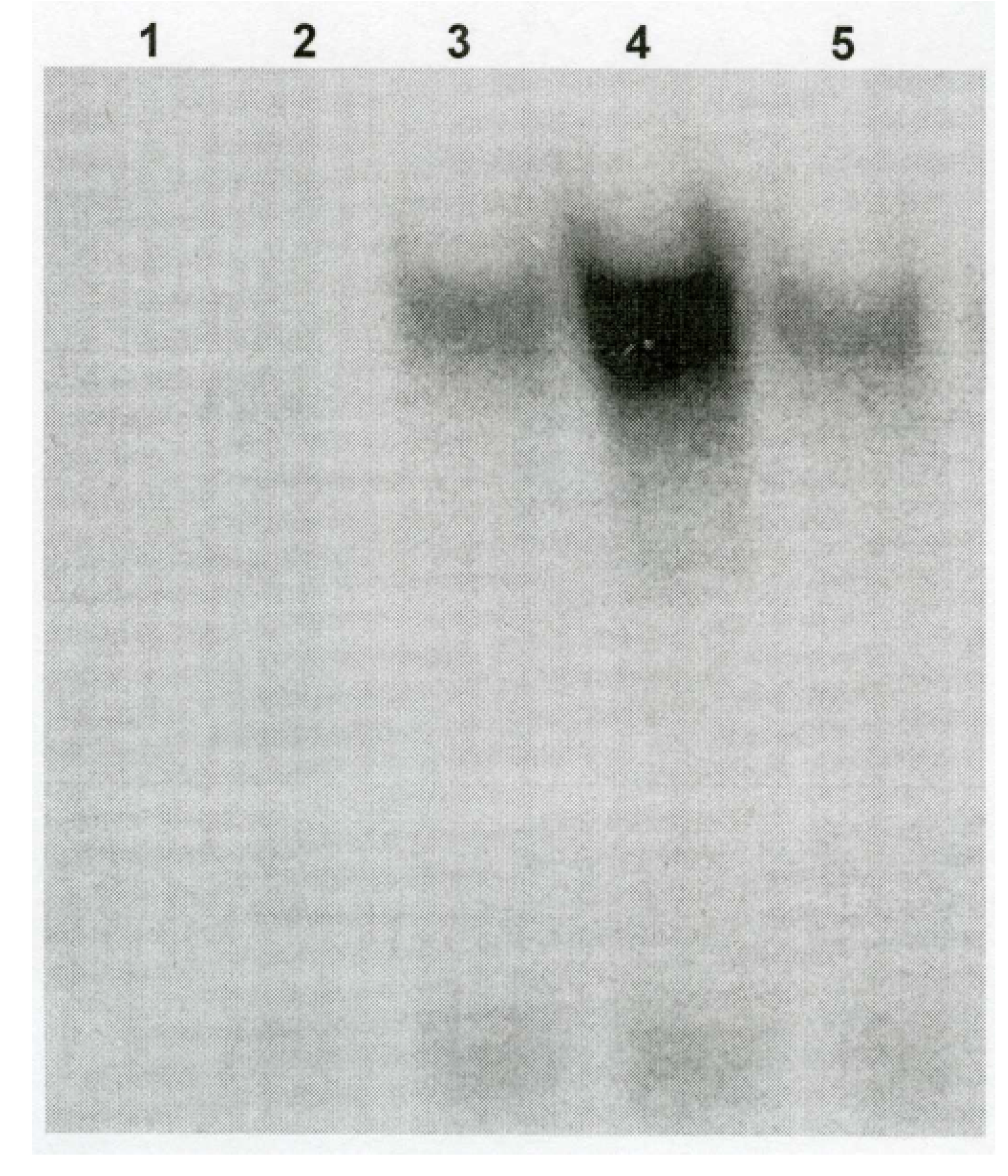
Ozone Exposure Increases DEP-Induced NF-κB Binding Activity

Ozone Exposure Increases NF-IL6-DNA Binding Activity

Discussion
Acknowledgments
References
- U.S. Environmental Protection Agency. Air Quality Criteria for Ozone and Other Photochemical Oxidants. National Technical Information Service: Springfield, VA, Report No. EPA/600/8-84/020df; p. 1986.
- Lippmann, M. Health effects of ozone: a critical review. J. Air Poll. Control Assoc. 1989, 39, 672–695. [Google Scholar]
- Koren, H.S.; Devlin, R.B.; Graham, D.E.; Mann, R.; McGee, M.P.; Horstman, D.H.; Kozumbo, W.J.; Becker, S.; House, D.E.; McDonnell, W.F.; Bromberg, P.A. Ozone-induced inflammation in the lower airways of human subjects. Am. Rev. Respir. Dis. 1989, 139, 407–415. [Google Scholar] [CrossRef]
- Devlin, R.B.; McDonnell, W.F.; Becker, S.; Madden, M.C.; McGee, M.P.; Perez, R.; Hatch, G.; Hourse, D.E.; Koren, H.S. Time-dependent changes of inflammatory mediators in the lungs of humans exposed to 0.4 ppm ozone for 2 hr: a comparison of mediators found in bronchoalveolar lavage fluid 1 and 18 hr after exposure. Toxicol. Appl. Pharmacol. 1996, 138, 176–185. [Google Scholar] [CrossRef]
- Pino, M.V.; Levin, J.R.; Stovall, M.Y.; Hyde, D.M. Pulmonary Inflammation and epithelial injury in response to acute ozone exposure in the rat. Toxicol. Appl. Pharmacol. 1992, 112, 64–72. [Google Scholar] [CrossRef]
- Basha, M.A.; Gross, K.B.; Gwizdala, C.J.; Haidar, A.H.; Popovich, J., Jr. Bronchoalveolar lavage neutrophilia in asthmatics and healthy volunteers after controlled exposure to ozone and filtered purified air. Chest 1994, 106, 1757–1765. [Google Scholar] [CrossRef]
- Balmes, J.R.; Chen, L.L.; Scannell, C.; Tager, I.; Christian, D.; Hearne, P.Q.; Kelly, T.; Aris, R. Ozone-induced decrements in FEV1 and FVC do not correlate with measures of inflammation. Am. J. Respir. Crit. Care Med. 1996, 153, 904–909. [Google Scholar] [CrossRef]
- Health Effects Institute. Effects of ozone on normal and potentially sensitive human subjects. Part I: airway inflammation and responsiveness to ozone in normal and asthmatic subjects. Research Report No. 78; Cambridge, MA, 1997. [Google Scholar]
- Dockery, D.W.; POPE, C.A., 3rd. Acute respiratory effects of particulate air pollution. Annu. Rev. Public Health. 1994, 15, 107–132. [Google Scholar] [CrossRef]
- Norris, G.; Youn-Pong, S.N.; Koenig, J.Q.; Larson, T.V.; Sheppard, L.; Stout, J.W. An association between fine particles and asthma emergency department visits for children in Seattle. Environ. Health Perspect. 1999, 107, 489–493. [Google Scholar]
- Ghio, A.J. Biological effects of Utah Valley ambient air particles in humans: review. J. Aerosol Med. 2004, 17, 157–164. [Google Scholar] [CrossRef]
- Timblin, C.; Berube, K.; Churg, A.; Driscoll, K.; Gordon, T.; Hemenway, D.; Walsh, E.; Cummins, A.B.; Vacek, P.; Mossman, B. Ambient particulate matter causes activation of the c-jun kinase/stress-activated protein kinase cascade and DNA synthesis in lung epithelial cells. Cancer Res. 1998, 58, 4543–4547. [Google Scholar]
- Meng, Z.; Dabdub, D.; Seinfeld, J.H. Chemical coupling between atmospheric ozone and particulate matter. Science 1997, 277, 116–119. [Google Scholar] [CrossRef]
- Kafoury, R.M.; Pryor, W.A.; Squadrito, G.L.; Salgo, M.G.; Zou, X.; Friedman, M. Lipid ozonation products activate phospholipases A2, C, and D. Toxicol. Appl. Pharmacol. 1998, 150, 338–349. [Google Scholar] [CrossRef]
- Kafoury, R.M.; Pryor, W.A.; Squadrito, G.L.; Salgo, M.G.; Zou, X.; and Friedman, M. Induction of inflammatory mediators in human airway epithelial cells by lipid ozonation products. Am. J. Respir. Crit. Care Med. 1999, 160, 1934–1942. [Google Scholar]
- Pryor, W.A.; Squadrito, G.L.; Friedman, M. The cascade mechanism to explain ozone toxicity: the role of lipid ozonation products. Free Rad. Biol. Med. 1995, 19, 935–941. [Google Scholar] [CrossRef]
- Devlin, R.B.; McKinnon, K.P.; Noah, T.; Becker, S.; and Koren, H.S. Ozone-induced release of cytokines and fibronectin production by alveolar macrophages and airway epithelial cells. Am. J. Physiol. 1994, 266, L612–L619. [Google Scholar]
- Balmes, J.R.; Chen, L.L.; Scannell, C.; Tager, I.; Christian, D.; Hearne, P.Q.; Kelly, T.; and Aris, R. Ozone-induced decrements in FEV1 and FVC do not correlate with measures of inflammation. Am. J. Respir. Dis. Crit. Care Med. 1996, 153, 904–909. [Google Scholar]
- Seltzer, J.; Bigby, B.G.; Stulbarg, M.; Holtzman, M.J.; Nadel, J.A.; Ueki, I.F.; Leikauf, G.D.; Goetzel, E.J.; Boushey, H.A. O3-induced change in bronchial reactivity to methacholine and airway inflammation in humans. J. Appl. Physiol. 1986, 60, 1321–1326. [Google Scholar]
- Koren, H.S.; Devlin, R.B.; Becker, S.; Perez, R.; McDonnell, W.F. Time-dependent changes of markers associated with inflammation in the lungs of humans exposed to ambient levels of ozone. Toxicol. Pathol. 1991, 19, 406–411. [Google Scholar]
- Ohtoshi, T.; Tazikawa, H.; Okazaki, H.; Kawasaki, S.; Takeuchi, N.; Ohta, K.; Ito, K. Diesel exhaust particles (DEP) stimulate human airway epithelial cells to produce cytokines relevant to airway inflammation in vitro. J. Allergy Clin. Immunol. 1998, 101, 778–785. [Google Scholar] [CrossRef]
- Sagai, M.; Furuyama, A.; Ichinose, T. Biological effects of diesel exhaust particles (DEP) III. Pathogenesis of asthma-like symptoms in mice. Free Rad. Biol. Med. 1996, 21, 199. [Google Scholar] [CrossRef]
- Nel, A.E.; Diaz-Sanchez, D.; Ng, D.; Hiura, J.; Saxon, A. Enhancement of allergic inflammation by the interaction between diesel exhaust particles and the immune system. J. Allergy Clin. Immunol. 1998, 102, 539–554. [Google Scholar] [CrossRef]
- Peterson, B.A.; Saxon, A. Global increases in allergic respiratory disease: The possible roles of diesel exhaust particles. Ann. Allergy Asthma Immunol. 1996, 77((4)), 263–268. [Google Scholar] [CrossRef]
- Diaz-Sanchez, D. The roles of diesel exhaust particles and their associated polyaromatic hydrocarbons in the induction of allergic airway disease. Allergy 1997, 52((38 s)), 52–56. [Google Scholar] [CrossRef]
- Berry, J.; Henoc, P.; Galle, P. Phagocytosis by cells of the pulmonary alveoli. Am. J. Pathol. 1978, 93, 27–44. [Google Scholar]
- Sen, R.; Baltimore, D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell 1986, 46((5)), 705–716. [Google Scholar]
- Baldwin, A.S., Jr. Series introduction: The transcription factor NF-κB and human disease. J. Clin. Invest. 2001, 107((1)), 3–6. [Google Scholar] [CrossRef]
- Stein, B.; Baldwin, A.S. Distinct mechanisms for the regulation of the interleukin-8 gene involve synergism and cooperativity between CIEBP and NF-κB. Mol. Cell Biol. 1993, 13, 7191–7198. [Google Scholar]
- Christman, J.W.; Sadikot, R.T.; Blackwell, T.S. The role of nuclear factor-κβ in pulmonary disease. Chest 2000, 117, 1482–1487. [Google Scholar] [CrossRef]
- Graziano, F.M.; Cook, E.B.; Stahl, J.L. Cytokines, chemokines, RANTES, and eotaxin. Allergy Asthma Proc. 1999, 20, 141–146. [Google Scholar] [CrossRef]
- Anderson, J.P. Resolution of chronic inflammation by therapeutic induction of apoptosis. Trends Pharmacol. Sci. 1996, 17, 438–442. [Google Scholar] [CrossRef]
- Samet, J.M.; Friedman, M. Effect of ozone on platelet activating factor metabolism in phorbol-differentiated HL60 cells. Toxicol. Appl. Pharmacol. 1992, 117, 19–25. [Google Scholar] [CrossRef]
- Wright, D.T.; Adler, K.B.; Akley, N.J.; Dailey, L.A.; Friedman, M. Ozone stimulates release of platelet activating factor and activates phospholipases in guinea pig tracheal epithelial cells in primary culture. Toxicol. Appl. Pharmacol. 1994, 127, 27–36. [Google Scholar] [CrossRef]
- McKinnon, K.P.; Madden, M.C.; Noah, T.L.; Devlin, R.B. In vitro ozone exposure increases release of arachidonic acid products from a human bronchial epithelial cell line. Toxicol. Appl. Pharmacol. 1993, 118, 215–223. [Google Scholar]
- Daniel, W.W. Biostatistics: a foundation for analysis in health sciences, 5th ed.John Wiley: New York, 1991; pp. 274–366. [Google Scholar]
- Fujimoto, K.; Yasuo, M.; Urushibata, K.; Hanaoka, M.; Koizumi, T.; Kubo, K. Airway inflammation during stable and acutely exacerbated chronic obstructive pulmonary disease. Eur. Respir. J. 2005, 25, 640–646. [Google Scholar] [CrossRef]
- Oudijk, E.J.; Nijhuis, E.H.; Zwank, M.D.; van de Graaf, E.A.; Mager, H.J.; Coffer, P.J.; Lammers, J.W.; Koendermann, L. Systemic inflammation in COPD visualized by gene profiling in peripheral blood neutrophils. Thorax 2005, 60, 538–544. [Google Scholar] [CrossRef]
- Driscoll, K.E.; Carter, J.M.; Hassenbein, D.G.; Howard, B. Cytokines and particle-induced inflammatory cell recruitment. Environ. Health Perspect. 1997, 105, 1159–1164. [Google Scholar] [CrossRef]
- Watt, A.P.; Schock, B.C.; Ennis, M. Neutrophils and eosiphils: clinical implications of their appearance, presence and disappearance in asthma and COPD. Curr. Drug Targets Inflamm. Allergy 2005, 4, 415–423. [Google Scholar] [CrossRef]
- Arinir, U.; Klein, W.; Rohde, G.; Stemmler, S.; Epplen, J.T.; Schultze-Werninghaus, G. Polymorphisms in the interleukin-8 gene in patients with chronic obstructive pulmonary disease. Electrophoresis 2005, 26, 2888–2891. [Google Scholar] [CrossRef]
- Vachier, I.; Bonnans, C.; Chavis, C.; Farce, M.; Godard, P.; Bousquet, J.; Chanez, P. Severe asthma is associated with a loss of LX4, an endogenous anti-inflammatory compound. J. Allergy Clin. Immunol. 2005, 115, 55–60. [Google Scholar] [CrossRef]
- Barnes, P.J. Mediators of chronic obstructive pulmonary disease. Pharmacol. Rev. 2004, 56, 515–548. [Google Scholar] [CrossRef]
- Oppenheim, J.J.; Zachariae, C.O.C.; Mukaida, N.; Matsushima, K. Properties of the novel proinflammatory supergene “intercrine” cytokine family. Annu. Rev. Immunol. 1991, 9, 617–648. [Google Scholar] [CrossRef]
- Bagglioni, M.; Dewald, B.; Moser, B. Interleukin-8 and related chemotactic cytokines-CXC and CC chemokines. Adv. Immunol. 1994, 55, 97–179. [Google Scholar]
- Taub, D.D.; Oppenheim, J.J. Chemokines, inflammation, and the immune system. Ther. Immunol. 1994, 1, 229–242. [Google Scholar]
- Taub, D.D.; Anver, M.; Oppenheim, J.J.; Longo, D.L.; Murphy, W.J. T lymphocyte recruitment by inerleukin-8 (IL-8):IL-8-induced degranulation of neutrophils releases potent chemoattractants for human T lymphocytes both in vitro and in vivo. J. Clin. Invest. 1996, 97, 1931–1941. [Google Scholar] [CrossRef]
- Sadowska, A.M.; Manuel-y-Keenoy, B.; Backer, W.A. Inhibition of in vitro neutrophil migration through a bilayer of endothelial and epithelial cells using beta-2-agonists: concomitant effects on IL-8 and elastase secretion and impact of gluccocorticosteroids. Pulm. Pharmacol. Ther. 2005, 18, 354–362. [Google Scholar] [CrossRef]
- Takizawa, H.; Ohtoshi, T.; Kawasaki, S.; Abe, S.; Sugawara, I.; Nakahara, K.; Matsushima, K.; Kudoh, S. Diesel exhaust particles activate human bronchial epithelial cells to express inflammatory mediators in the airways: a review. Respirology 2000, 5, 197–203. [Google Scholar] [CrossRef]
- Steerenberg, P.A.; Zonnenberg, J.A.; Dormans, J.A.; Joon, P.N.; Wouters, I.M.; van Bree, L.; Scheepers, P.T.; van Lveren, H. Diesel exhaust particles induced release of interleukin 6 and 8 by (primed) human bronchial epithelial cells (BEAS-2B) in vitro. Exp. Lung Res. 1998, 24, 85–100. [Google Scholar] [CrossRef]
- Boland, S.; Baez-Squiban, A.; Fournier, T.; Houcine, O.; Gendron, M.-C.; Checrier, M.; Jouvenot, G.; Coste, A.; Abier, M.; Francelyne, M. Diesel exhaust particles are taken up by human airway epithelial cells in vitro and alter cytokine production. Am. J. Physiol. 1999, 276, L604–L613. [Google Scholar]
- Jaspers, I.; Flescher, E.; Chen, L.C. Ozone-induced IL-8 expression and transcription factor binding in respiratory epithelial cell. Am. J. Physiol. 1997, 272, L504–L511. [Google Scholar]
- Kafoury, R.M.; Gozal, E.; Squadrito, G.L.; Zou, X.; Pryor, W.A.; Friedman, M. Lipid ozonation products activate nuclear factor-κB (NF-κB) in human bronchial epithelial cells (BEAS-2B). Am. J. Respir. Crit. Care Med. 1999, 159, A463. [Google Scholar]
© 2005 MDPI. All rights reserved.
Share and Cite
Kafoury, R.M.; Kelley, J. Ozone Enhances Diesel Exhaust Particles (DEP)-Induced Interleukin-8 (IL-8) Gene Expression in Human Airway Epithelial Cells through Activation of Nuclear Factors- κB (NF-κB) and IL-6 (NF-IL6). Int. J. Environ. Res. Public Health 2005, 2, 403-410. https://0-doi-org.brum.beds.ac.uk/10.3390/ijerph2005030004
Kafoury RM, Kelley J. Ozone Enhances Diesel Exhaust Particles (DEP)-Induced Interleukin-8 (IL-8) Gene Expression in Human Airway Epithelial Cells through Activation of Nuclear Factors- κB (NF-κB) and IL-6 (NF-IL6). International Journal of Environmental Research and Public Health. 2005; 2(3):403-410. https://0-doi-org.brum.beds.ac.uk/10.3390/ijerph2005030004
Chicago/Turabian StyleKafoury, Ramzi M., and James Kelley. 2005. "Ozone Enhances Diesel Exhaust Particles (DEP)-Induced Interleukin-8 (IL-8) Gene Expression in Human Airway Epithelial Cells through Activation of Nuclear Factors- κB (NF-κB) and IL-6 (NF-IL6)" International Journal of Environmental Research and Public Health 2, no. 3: 403-410. https://0-doi-org.brum.beds.ac.uk/10.3390/ijerph2005030004