Investigation of Nano-Bio Interactions within a Pancreatic Tumor Microenvironment for the Advancement of Nanomedicine in Cancer Treatment

 , ,
, ,
Abstract
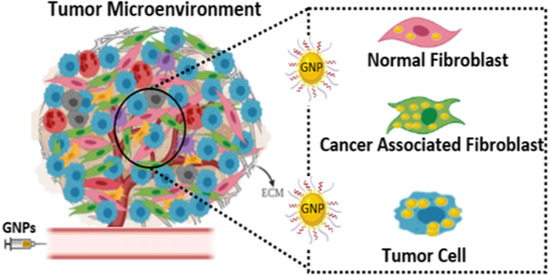
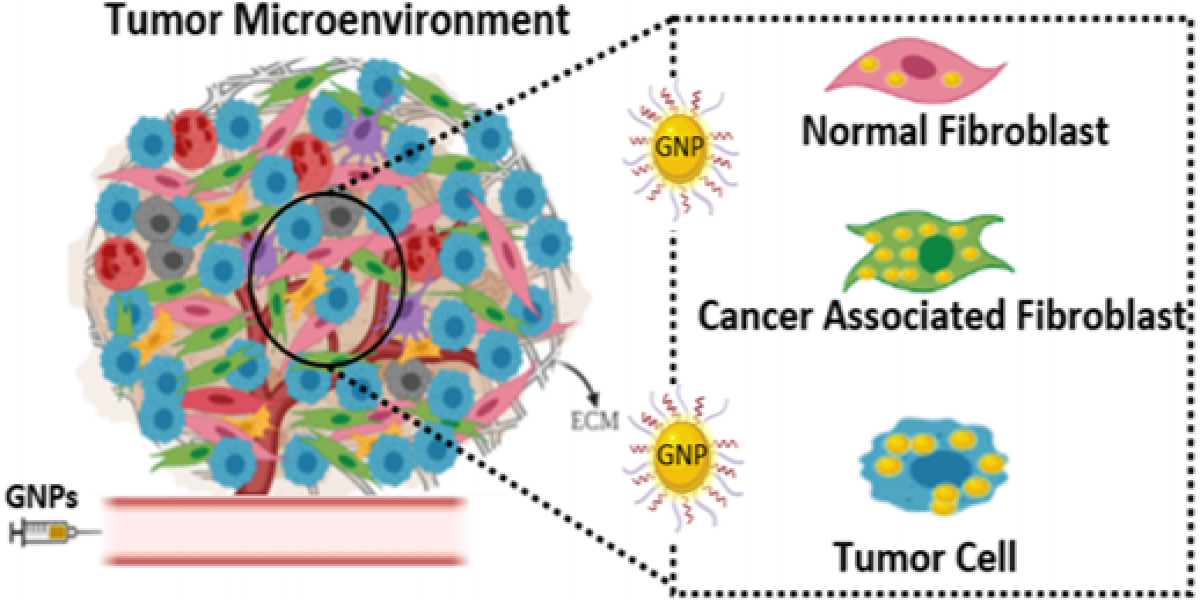
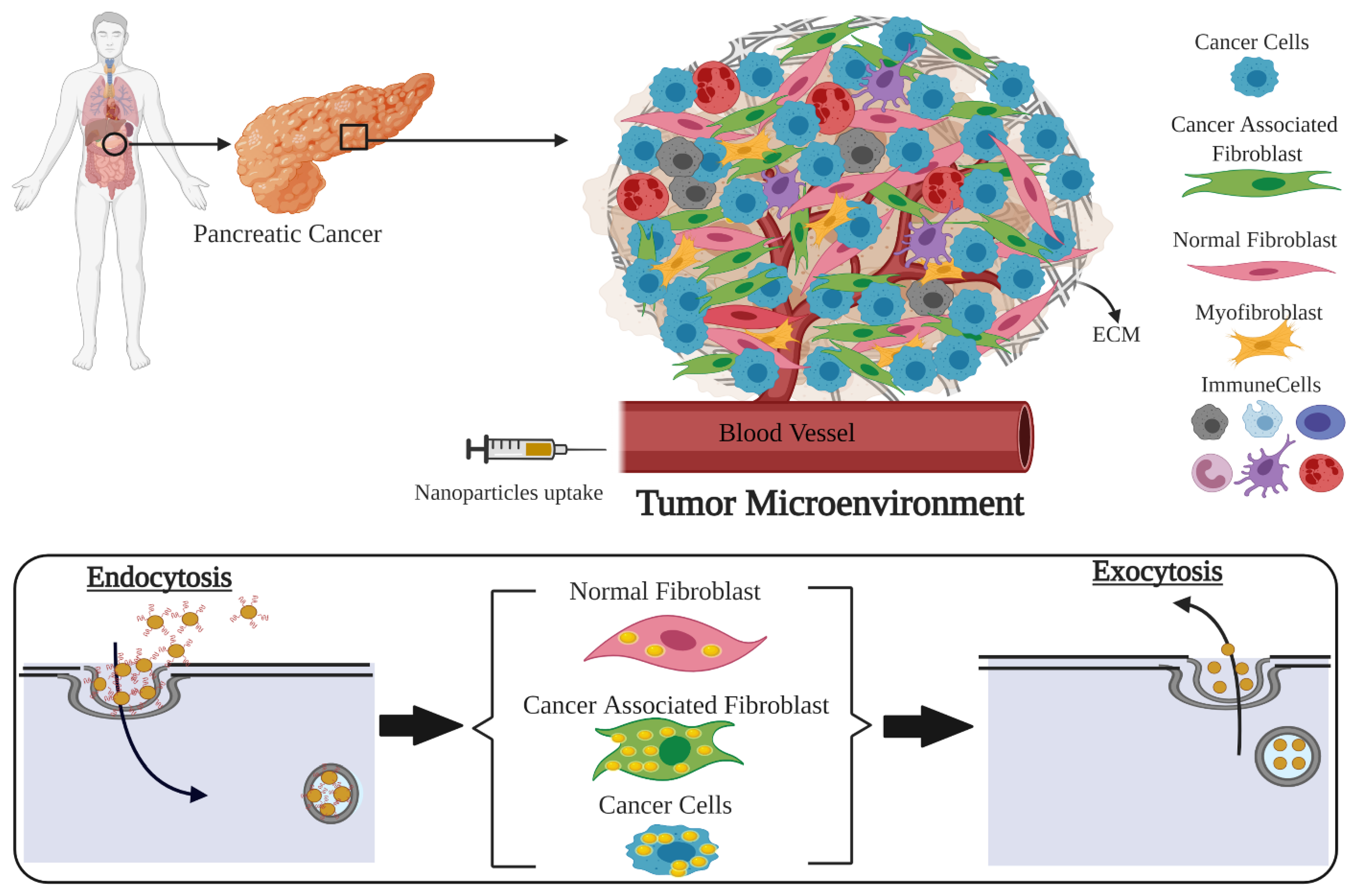
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
- Is there a difference between in vitro uptake of NPs in tumor cells, NFs, and CAFs of pancreatic origin?
- What is the potential of retaining of these NPs within those cells in vitro for efficient delivery of therapeutics?
- Do we see retention of NPs within tumor tissue in vivo for an extended period of time as being successful for the delivery of therapeutics?
2. Materials and Methods
2.1. Gold Nanoparticle Synthesis
2.2. Gold Nanoparticles Functionalization
2.3. Gold Nanoparticles Characterization
2.4. Cell Culture Methodology
2.5. Image Preparation
2.6. Quantification of Cellular Uptake and Retention
2.7. Pancreatic Xenograft Model
2.8. Immunohistochemistry
2.9. Acute and Physical Toxicity Assays
3. Results and Discussion
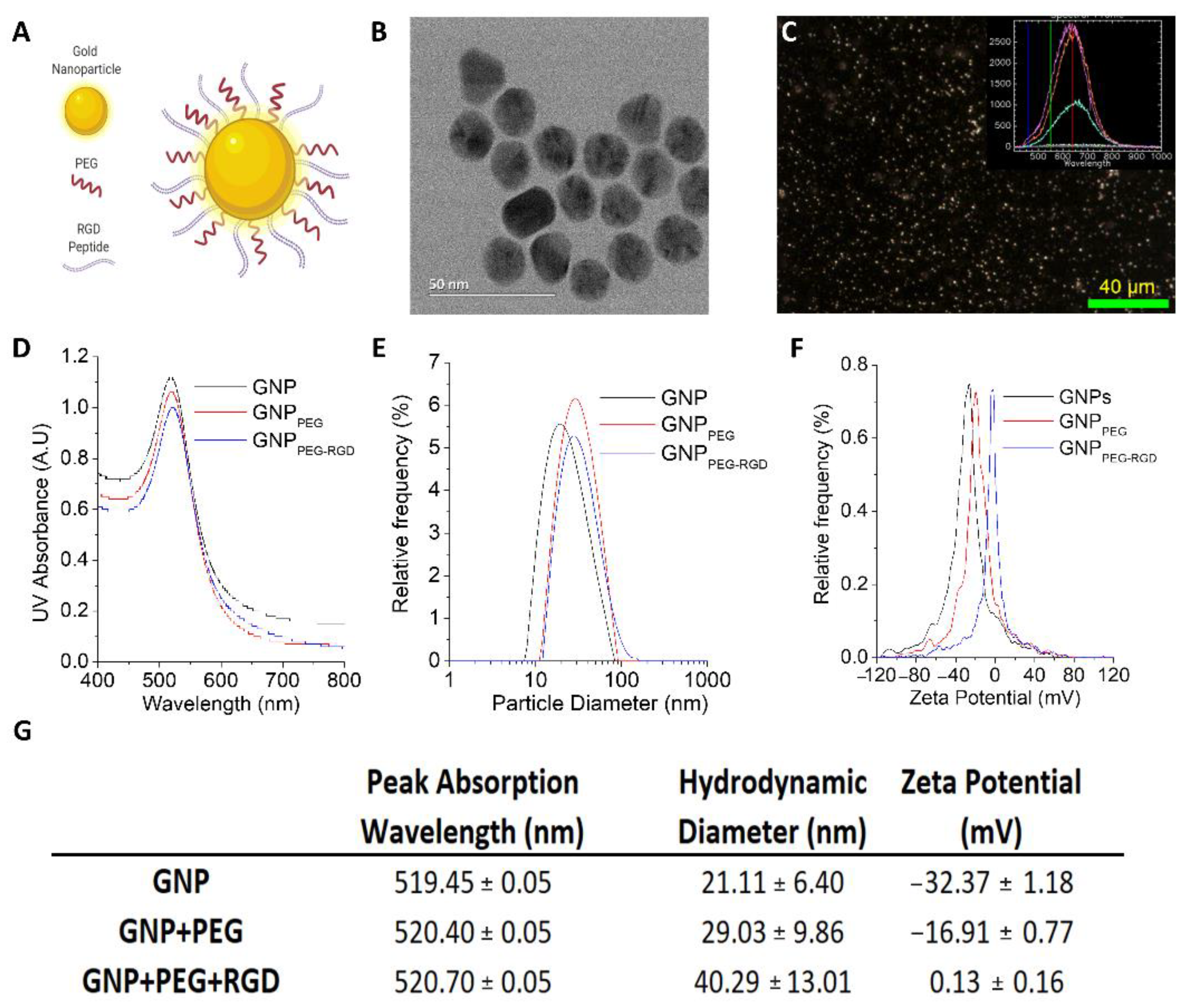
3.1. Characterization of GNP Complexes
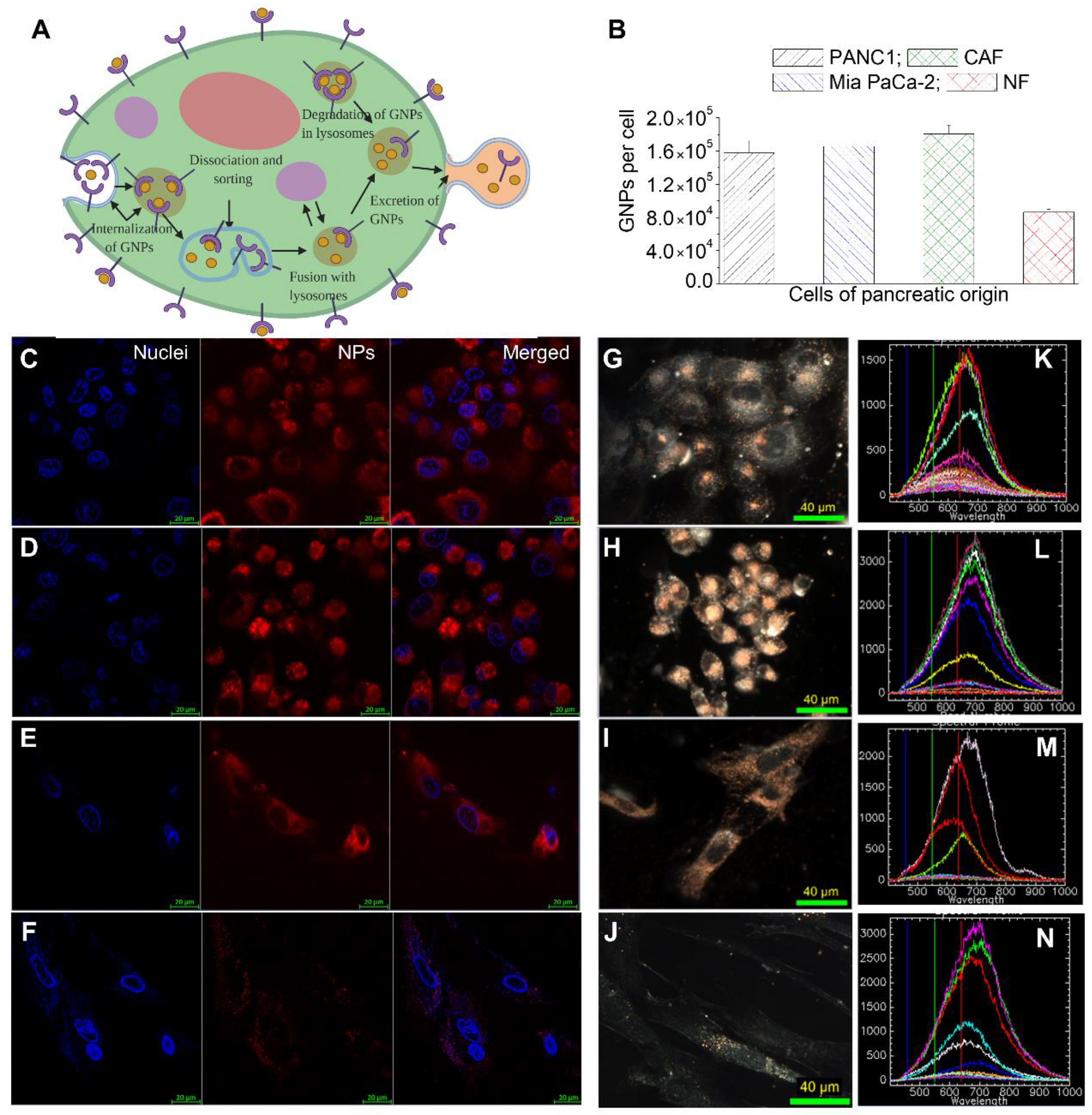
3.2. Cellular Uptake of GNP Complexes in Tumor Cells, Normal Fibroblasts (NFs), and Cancer-Associated Fibroblasts (CAFs) in Monolayer Cell Cultures
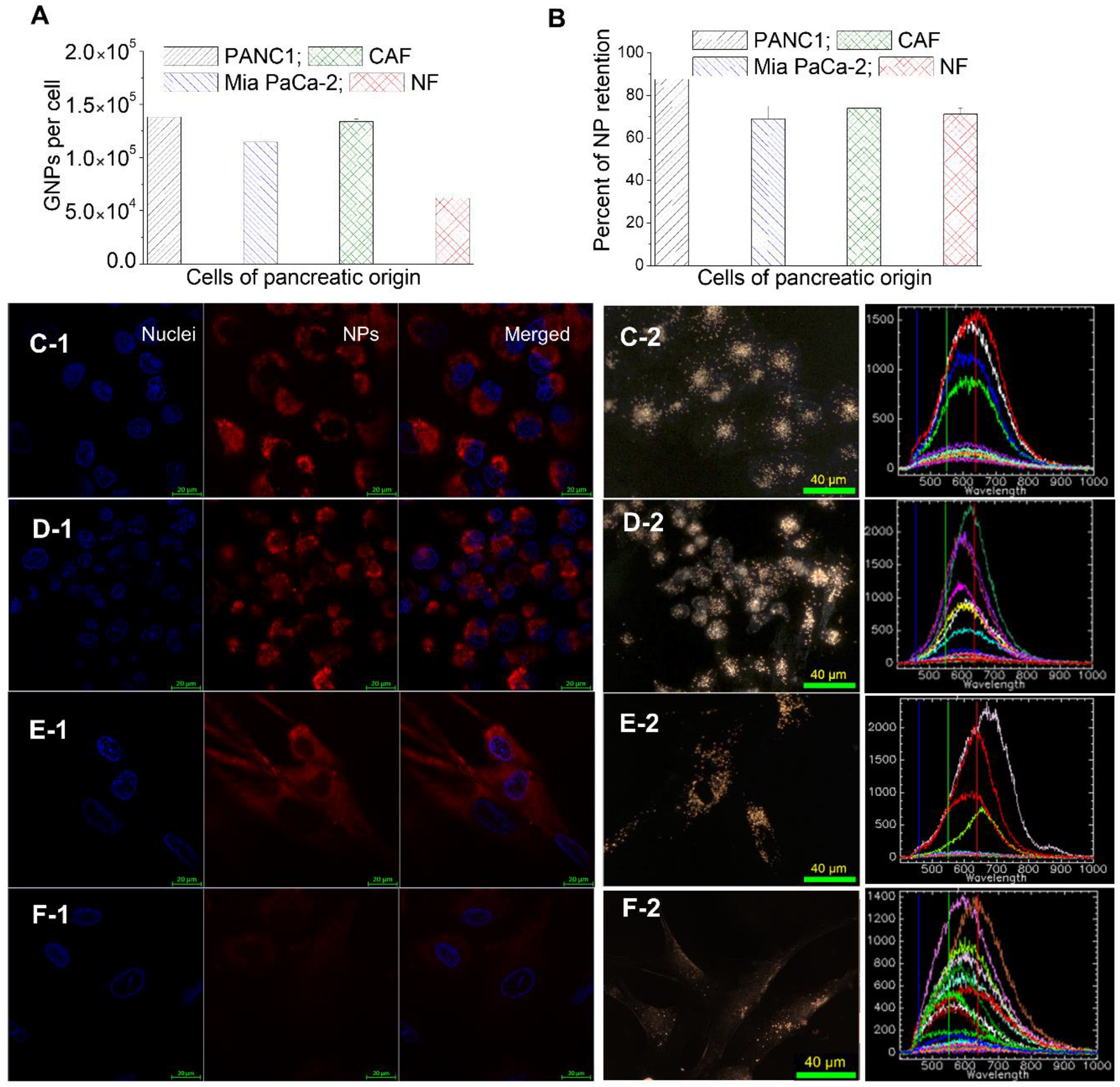
3.3. Retention of GNP Complexes in Tumor Cells, Normal Fibroblasts (NFs), and Cancer-Associated Fibroblasts (CAFs) in Monolayer Cell Cultures
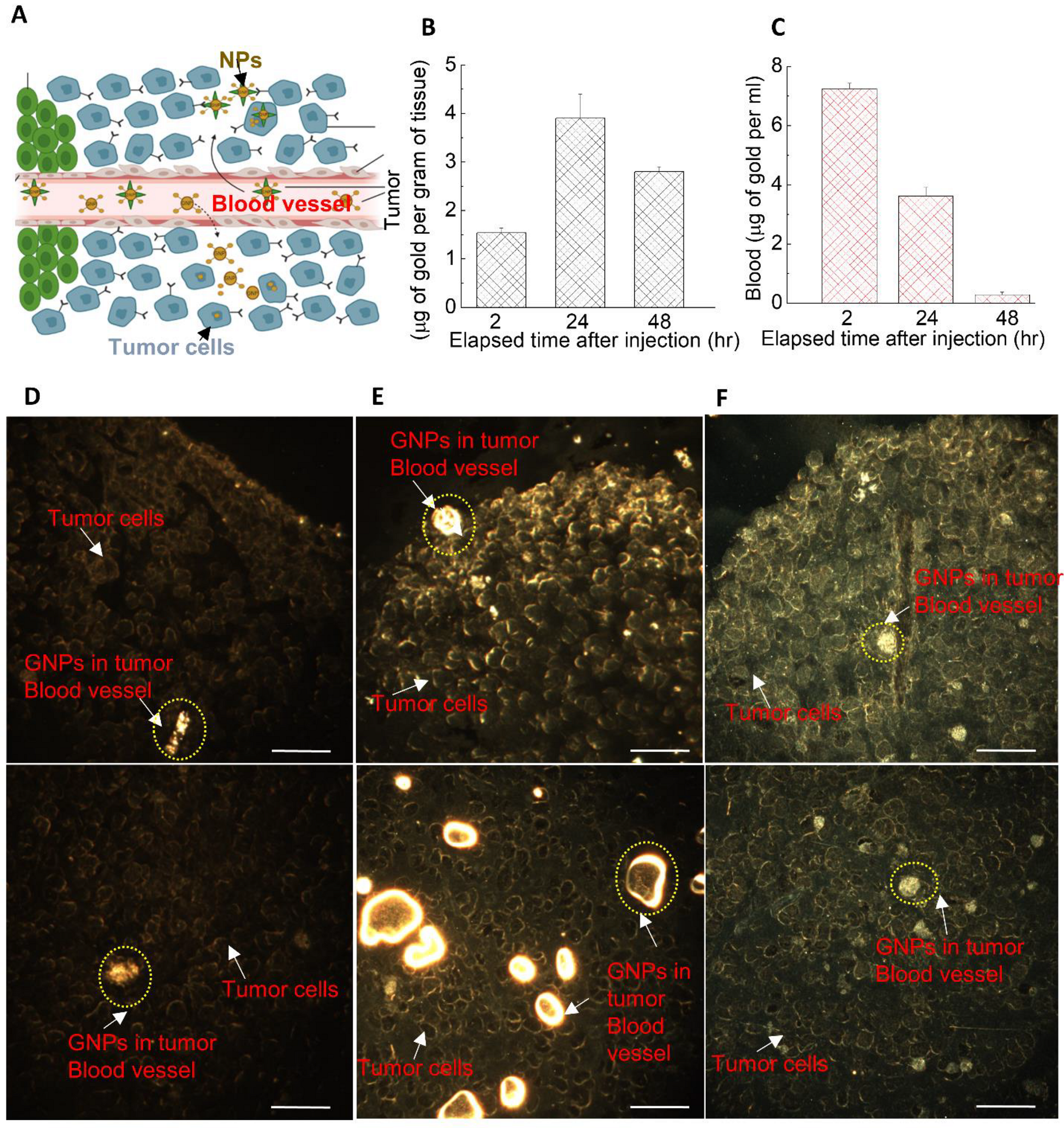
3.4. The Dynamics of GNP Distribution and Retention within Tumor Tissues In Vivo
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, X.; Zhao, G.; Zhao, Y. Combination immunotherapy approaches for pancreatic cancer treatment. Can. J. Gastroenterol. Hepatol. 2018, 2018, 6240467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norton, J.; Foster, D.; Chinta, M.; Titan, A.; Longaker, M. Pancreatic cancer associated fibroblasts (caf): Under-explored target for pancreatic cancer treatment. Cancers 2020, 12, 1347. [Google Scholar] [CrossRef] [PubMed]
- Badiyan, S.N.; Molitoris, J.K.; Chuong, M.D.; Regine, W.F.; Kaiser, A. The role of radiation therapy for pancreatic cancer in the adjuvant and neoadjuvant settings. Surg. Oncol. Clin. N. Am. 2017, 26, 431–453. [Google Scholar] [CrossRef]
- Hall, W.A.; Goodman, K.A. Radiation therapy for pancreatic adenocarcinoma, a treatment option that must be considered in the management of a devastating malignancy. Radiat. Oncol. 2019, 14, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, L.; Schilling, D.; Dobiasch, S.; Raulefs, S.; Santiago Franco, M.; Buschmann, D.; Pfaffl, M.W.; Schmid, T.E.; Combs, S.E. The Emerging Role of miRNAs for the Radiation Treatment of Pancreatic Cancer. Cancers 2020, 12, 3703. [Google Scholar] [CrossRef]
- Prasanna, P.G.S.; Stone, H.B.; Wong, R.S.; Capala, J.; Bernhard, E.J.; Vikram, B.; Coleman, C.N. Normal tissue protection for improving radiotherapy: Where are the gaps? Transl. Cancer Res. 2012, 1, 35–48. [Google Scholar]
- Gao, Y.; Gao, D.; Shen, J.; Wang, Q. A Review of Mesoporous Silica Nanoparticle Delivery Systems in Chemo-Based Combination Cancer Therapies. Front. Chem. 2020, 8, 1086. [Google Scholar] [CrossRef]
- Bromma, K.; Chithrani, D.B. Advances in Gold Nanoparticle-Based Combined Cancer Therapy. Nanomaterials 2020, 10, 1671. [Google Scholar] [CrossRef]
- Schuemann, J.; Bagley, A.F.; Berbeco, R.; Bromma, K.; Butterworth, K.T.; Byrne, H.L.; Chithrani, B.D.; Cho, S.H.; Cook, J.R.; Favaudon, V.; et al. Roadmap for metal nanoparticles in radiation therapy: Current status, translational challenges, and future directions. Phys. Med. Biol. 2020, 65, 21RM02. [Google Scholar] [CrossRef]
- Schuemann, J.; Berbeco, R.; Chithrani, D.B.; Cho, S.H.; Kumar, R.; McMahon, S.J.; Sridhar, S.; Krishnan, S. Roadmap to Clinical Use of Gold Nanoparticles for Radiation Sensitization. Int. J. Radiat. Oncol. 2016, 94, 189–205. [Google Scholar] [CrossRef] [Green Version]
- Jelveh, S.; Chithrani, D.B. Gold Nanostructures as a Platform for Combinational Therapy in Future Cancer Therapeutics. Cancers 2011, 3, 1081–1110. [Google Scholar] [CrossRef]
- Liu, T.; Zhou, L.; Li, D.; Andl, T.; Zhang, Y. Cancer-Associated Fibroblasts Build and Secure the Tumor Microenvironment. Front. Cell Dev. Biol. 2019, 7, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiga, K.; Hara, M.; Nagasaki, T.; Sato, T.; Takahashi, H.; Takeyama, H. Cancer-associated fibroblasts: Their characteristics and their roles in tumor growth. Cancers 2015, 7, 2443–2458. [Google Scholar] [CrossRef] [PubMed]
- Henke, E.; Nandigama, R.; Ergün, S. Extracellular Matrix in the Tumor Microenvironment and Its Impact on Cancer Therapy. Front. Mol. Biosci. 2020, 6, 160. [Google Scholar] [CrossRef] [Green Version]
- Walker, C.; Mojares, E.; Hernández, A.D.R. Role of Extracellular Matrix in Development and Cancer Progression. Int. J. Mol. Sci. 2018, 19, 3028. [Google Scholar] [CrossRef] [Green Version]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [Green Version]
- Desmouliere, A.; Darby, I.A.; Laverdet, B.; Bonté, F. Fibroblasts and myofibroblasts in wound healing. Clin. Cosmet. Investig. Dermatol. 2014, 7, 301–311. [Google Scholar] [CrossRef] [Green Version]
- Albrengues, J.; Bertero, T.; Grasset, E.; Bonan, S.; Maiel, M.; Bourget, I.; Philippe, C.; Serrano, C.H.; Benamar, S.; Croce, O.; et al. Epigenetic switch drives the conversion of fibroblasts into proinvasive cancer-associated fibroblasts. Nat. Commun. 2015, 6, 10204. [Google Scholar] [CrossRef] [Green Version]
- Alkasalias, T.; Moyano-Galceran, L.; Arsenian-Henriksson, M.; Lehti, K. Fibroblasts in the Tumor Microenvironment: Shield or Spear? Int. J. Mol. Sci. 2018, 19, 1532. [Google Scholar] [CrossRef] [Green Version]
- Ortiz-Otero, N.; Clinch, A.B.; Hope, J.; Wang, W.; Reinhart-King, C.A.; King, M.R. Cancer associated fibroblasts confer shear resistance to circulating tumor cells during prostate cancer metastatic progression. Oncotarget 2020, 11, 1037–1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosein, A.N.; Brekken, R.A.; Maitra, A. Pancreatic cancer stroma: An update on therapeutic targeting strategies. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 487–505. [Google Scholar] [CrossRef] [PubMed]
- Alhussan, A.; Bozdoğan, E.; Chithrani, D. Combining Gold Nanoparticles with Other Radiosensitizing Agents for Unlocking the Full Potential of Cancer Radiotherapy. Pharmaceutics 2021, 13, 442. [Google Scholar] [CrossRef]
- Lammel, T.; Mackevica, A.; Johansson, B.R.; Sturve, J. Endocytosis, intracellular fate, accumulation, and agglomeration of titanium dioxide (TiO2) nanoparticles in the rainbow trout liver cell line RTL-W1. Environ. Sci. Pollut. Res. 2019, 26, 15354–15372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dykman, L.A.; Khlebtsov, N.G. Immunological properties of gold nanoparticles. Chem. Sci. 2016, 8, 1719–1735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manson, J.; Kumar, D.; Meenan, B.J.; Dixon, D. Polyethylene glycol functionalized gold nanoparticles: The influence of capping density on stability in various media. Gold Bull. 2011, 44, 99–105. [Google Scholar] [CrossRef]
- Cruje, C.; Yang, C.; Uertz, J.; Van Prooijen, M.; Chithrani, B.D. Optimization of PEG coated nanoscale gold particles for enhanced radiation therapy. RSC Adv. 2015, 5, 101525–101532. [Google Scholar] [CrossRef]
- Spadavecchia, J.; Movia, D.; Moore, C.; Maguire, C.M.; Moustaoui, H.; Casale, S.; Volkov, Y.; Prina-Mello, A. Targeted polyethylene glycol gold nanoparticles for the treatment of pancreatic cancer: From synthesis to proof-of-concept in vitro studies. Int. J. Nanomed. 2016, 11, 791–822. [Google Scholar] [CrossRef] [Green Version]
- Cruje, C.; Chithrani, B.D.; Chithrani, D. Integration of Peptides for Enhanced Uptake of PEGylayed Gold Nanoparticles. J. Nanosci. Nanotechnol. 2015, 15, 2125–2131. [Google Scholar] [CrossRef]
- Wu, P.-H.; Onodera, Y.; Ichikawa, Y.; Rankin, E.B.; Giaccia, A.J.; Watanabe, Y.; Qian, W.; Hashimoto, T.; Shirato, H.; Nam, J.-M. Targeting integrins with RGD-conjugated gold nanoparticles in radiotherapy decreases the invasive activity of breast cancer cells. Int. J. Nanomed. 2017, 12, 5069–5085. [Google Scholar] [CrossRef] [Green Version]
- Biscaglia, F.; Ripani, G.; Rajendran, S.; Benna, C.; Mocellin, S.; Bocchinfuso, G.; Meneghetti, M.; Palleschi, A.; Gobbo, M. Gold Nanoparticle Aggregates Functionalized with Cyclic RGD Peptides for Targeting and Imaging of Colorectal Cancer Cells. ACS Appl. Nano Mater. 2019, 2, 6436–6444. [Google Scholar] [CrossRef]
- Chithrani, B.D.; Ghazani, A.A.; Chan, W.C.W. Determining the size and shape dependence of gold nanoparticle uptake into mammalian cells. Nano Lett. 2006, 6, 662–668. [Google Scholar] [CrossRef]
- Yohan, D.; Cruje, C.; Lu, X.; Chithrani, D.B. Size-Dependent Gold Nanoparticle Interaction at Nano–Micro Interface Using Both Monolayer and Multilayer (Tissue-Like) Cell Models. Nano-Micro Lett. 2016, 8, 44–53. [Google Scholar] [CrossRef] [Green Version]
- Amendola, V.; Meneghetti, M. Size Evaluation of Gold Nanoparticles by UV−vis Spectroscopy. J. Phys. Chem. C 2009, 113, 4277–4285. [Google Scholar] [CrossRef]
- Yang, C.; Neshatian, M.; Van Prooijen, M.; Chithrani, D.B. Cancer Nanotechnology: Enhanced Therapeutic Response Using Peptide-Modified Gold Nanoparticles. J. Nanosci. Nanotechnol. 2014, 14, 4813–4819. [Google Scholar] [CrossRef]
- Gao, H.; Shi, W.; Freund, L.B. From The Cover: Mechanics of receptor-mediated endocytosis. Proc. Natl. Acad. Sci. USA 2005, 102, 9469–9474. [Google Scholar] [CrossRef] [Green Version]
- Stetler-Stevenson, W.G.; Aznavoorian, S.; Liotta, L.A. Tumor cell interactions with the extracellular matrix during invasion and metastasis. Annu. Rev. Cell Biol. 1993, 9, 541–573. [Google Scholar] [CrossRef]
- Wolfe, T.; Chatterjee, D.; Lee, J.; Grant, J.D.; Bhattarai, S.; Tailor, R.; Goodrich, G.; Nicolucci, P.; Krishnan, S. Targeted gold nanoparticles enhance sensitization of prostate tumors to megavoltage radiation therapy in vivo. Nanomed. Nanotechnol. Biol. Med. 2015, 11, 1277–1283. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.D.; Wu, H.Y.; Wu, D.; Wang, Y.Y.; Chang, J.H.; Zhai, Z.B.; Meng, A.M.; Liu, P.X.; Zhang, L.A.; Fan, F.Y. Toxicologic effects of gold nanoparticles in vivo by different administration routes. Int. J. Nanomed. 2010, 5, 771–781. [Google Scholar] [CrossRef] [Green Version]
- Bailly, A.-L.; Correard, F.; Popov, A.; Tselikov, G.; Chaspoul, F.; Appay, R.; Al-Kattan, A.; Kabashin, A.V.; Braguer, D.; Esteve, M.-A. In vivo evaluation of safety, biodistribution and pharmacokinetics of laser-synthesized gold nanoparticles. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Khoo, A.M.; Cho, S.H.; Reynoso, F.J.; Aliru, M.; Aziz, K.; Bodd, M.; Yang, X.; Ahmed, M.F.; Yasar, S.; Manohar, N.; et al. Radiosensitization of prostate cancers in vitro and in vivo to erbium-filtered orthovoltage x-rays using actively targeted gold nanoparticles. Sci. Rep. 2017, 7, 18044. [Google Scholar] [CrossRef] [Green Version]
- Bromma, K.; Banister, A.; Kowalewski, A.; Cicon, L.; Chithrani, D.B. Elucidating the fate of nanoparticles among key cell components of the tumor microenvironment for promoting cancer nanotechnology. Cancer Nanotechnol. 2020, 11, 1–8. [Google Scholar] [CrossRef]
- Bromma, K.; Cicon, L.; Beckham, W.; Chithrani, D.B. Gold nanoparticle mediated radiation response among key cell components of the tumour microenvironment for the advancement of cancer nanotechnology. Sci. Rep. 2020, 10, 1–14. [Google Scholar] [CrossRef]
- Jang, I.; Beningo, K. Integrins, CAFs and Mechanical Forces in the Progression of Cancer. Cancers 2019, 11, 721. [Google Scholar] [CrossRef] [Green Version]
- Dominguez, C.X.; Müller, S.; Keerthivasan, S.; Koeppen, H.; Hung, J.; Gierke, S.; Breart, B.; Foreman, O.; Bainbridge, T.W.; Castiglioni, A.; et al. Single-cell rna sequencing reveals stromal evolution into lrrc15 (+) myofibroblasts as a determinant of patient response to cancer immunotherapy. Cancer Discov. 2020, 10, 232–253. [Google Scholar] [CrossRef] [Green Version]
- Kemi, N.; Eskuri, M.; Herva, A.; Leppänen, J.; Huhta, H.; Helminen, O.; Saarnio, J.; Karttunen, T.J.; Kauppila, J.H. Tumour-stroma ratio and prognosis in gastric adenocarcinoma. Br. J. Cancer 2018, 119, 435–439. [Google Scholar] [CrossRef] [Green Version]
- Graizel, D.; Zlotogorski-Hurvitz, A.; Tsesis, I.; Rosen, E.; Kedem, R.; Vered, M. Oral cancer-associated fibroblasts predict poor survival: Systematic review and meta-analysis. Oral Dis. 2020, 26, 733–744. [Google Scholar] [CrossRef]
- Chithrani, B.D.; Chan, W.C.W. Elucidating the mechanism of cellular uptake and removal of protein-coated gold nanoparticles of different sizes and shapes. Nano Lett. 2007, 7, 1542–1550. [Google Scholar] [CrossRef]
- Chithrani, B.D. Intracellular uptake, transport, and processing of gold nanostructures. Mol. Membr. Biol. 2010, 27, 299–311. [Google Scholar] [CrossRef]
- Yang, C.; Uertz, J.; Yohan, D.; Chithrani, B.D. Peptide modified gold nanoparticles for improved cellular uptake, nuclear transport, and intracellular retention. Nanoscale 2014, 6, 12026–12033. [Google Scholar] [CrossRef]
- Yang, C.; Uertz, J.; Chithrani, D.B. Colloidal gold-mediated delivery of bleomycin for improved outcome in chemotherapy. Nanomaterials 2016, 6, 48. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.A.; Åberg, C.; Salvati, A.; Dawson, K.A. Role of cell cycle on the cellular uptake and dilution of nanoparticles in a cell population. Nat. Nanotechnol. 2011, 7, 62–68. [Google Scholar] [CrossRef]
- Maeda, H.; Fang, J.; Inutsuka, T.; Kitamoto, Y. Vascular permeability enhancement in solid tumor: Various factors, mechanisms involved and its implications. Int. Immunopharmacol. 2003, 3, 319–328. [Google Scholar] [CrossRef]
- Goodrich, G.P.; Bao, L.; Gill-Sharp, K.; Sang, K.L.; Wang, J.; Payne, J.D. Photothermal therapy in a murine colon cancer model using near-infrared absorbing gold nanorods. J. Biomed. Opt. 2010, 15, 018001. [Google Scholar] [CrossRef]
- Hainfeld, J.F.; Dilmanian, F.A.; Slatkin, D.N.; Smilowitz, H.M. Radiotherapy enhancement with gold nanoparticles. J. Pharm. Pharmacol. 2008, 60, 977–985. [Google Scholar] [CrossRef] [Green Version]
- Albertini, B.; Mathieu, V.; Iraci, N.; Van Woensel, M.; Schoubben, A.; Donnadio, A.; Greco, S.M.L.; Ricci, M.; Temperini, A.; Blasi, P.; et al. Tumor targeting by peptide-decorated gold nanoparticles. Mol. Pharm. 2019, 16, 2430–2444. [Google Scholar] [CrossRef] [Green Version]
- Puvanakrishnan, P.; Park, J.; Chatterjee, D.; Krishnan, S.; Tunnell, J.W. In vivo tumor targeting of gold nanoparticles: Effect of particle type and dosing strategy. Int. J. Nanomed. 2012, 7, 1251–1258. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alhussan, A.; Bromma, K.; Bozdoğan, E.P.D.; Metcalfe, A.; Karasinska, J.; Beckham, W.; Alexander, A.S.; Renouf, D.J.; Schaeffer, D.F.; Chithrani, D.B. Investigation of Nano-Bio Interactions within a Pancreatic Tumor Microenvironment for the Advancement of Nanomedicine in Cancer Treatment. Curr. Oncol. 2021, 28, 1962-1979. https://0-doi-org.brum.beds.ac.uk/10.3390/curroncol28030183
Alhussan A, Bromma K, Bozdoğan EPD, Metcalfe A, Karasinska J, Beckham W, Alexander AS, Renouf DJ, Schaeffer DF, Chithrani DB. Investigation of Nano-Bio Interactions within a Pancreatic Tumor Microenvironment for the Advancement of Nanomedicine in Cancer Treatment. Current Oncology. 2021; 28(3):1962-1979. https://0-doi-org.brum.beds.ac.uk/10.3390/curroncol28030183
Chicago/Turabian StyleAlhussan, Abdulaziz, Kyle Bromma, Ece Pinar Demirci Bozdoğan, Andrew Metcalfe, Joanna Karasinska, Wayne Beckham, Abraham S. Alexander, Daniel J. Renouf, David F. Schaeffer, and Devika B. Chithrani. 2021. "Investigation of Nano-Bio Interactions within a Pancreatic Tumor Microenvironment for the Advancement of Nanomedicine in Cancer Treatment" Current Oncology 28, no. 3: 1962-1979. https://0-doi-org.brum.beds.ac.uk/10.3390/curroncol28030183