Treatment Outcomes for Primary Hepatic Angiosarcoma: National Cancer Database Analysis 2004–2014



Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
3. Results
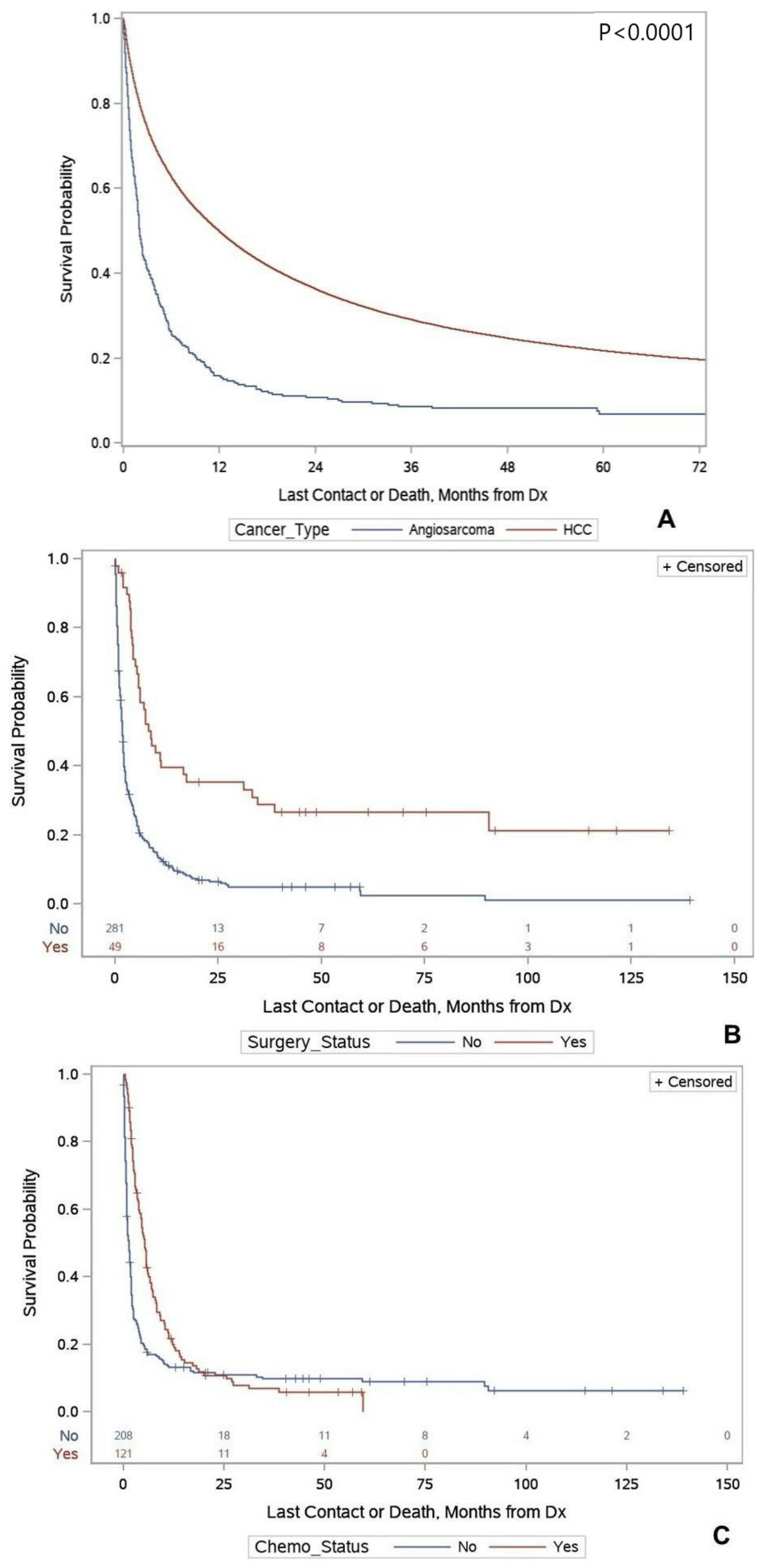
Primary Hepatic Angiosarcoma versus Hepatocellular Carcinoma
4. Discussion
4.1. Primary Hepatic Angiosarcoma
4.2. Primary Hepatic Angiosarcoma versus Hepatocellular Carcinoma
4.3. Limitations of the Study
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zocchetti, C. Liver angiosarcoma in humans: Epidemiologic considerations. Med. Lav. 2001, 92, 39–53. [Google Scholar]
- Elliott, P.; Kleinschmidt, I. Angiosarcoma of the liver in Great Britain in proximity to vinyl chloride sites. Occup. Environ. Med. 1997, 54, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, P.; Bhadana, U.; Singh, R.A.; Ahuja, A. Primary hepatic angiosarcoma. Eur. J. Surg. Oncol. 2015, 41, 1137–1143. [Google Scholar] [CrossRef] [PubMed]
- Block, J.B. Angiosarcoma of the Liver Following Vinyl Chloride Exposure. JAMA 1974, 229, 53–54. [Google Scholar] [CrossRef] [PubMed]
- Gaballah, A.H.; Jensen, C.T.; Palmquist, S.; Pickhardt, P.J.; Duran, A.; Broering, G.; Elsayes, K.M. Angiosarcoma: Clinical and imaging features from head to toe. Br. J. Radiol. 2017, 90, 20170039. [Google Scholar] [CrossRef]
- Pickhardt, P.J.; Kitchin, D.; Lubner, M.G.; Ganeshan, D.M.; Bhalla, S.; Covey, A.M. Primary hepatic angiosarcoma: Multi-institutional comprehensive cancer centre review of multiphasic CT and MR imaging in 35 patients. Eur. Radiol. 2015, 25, 315–322. [Google Scholar] [CrossRef]
- Young, R.J.; Brown, N.J.; Reed, M.W.; Hughes, D.; Woll, P.J. Angiosarcoma. Lancet Oncol. 2010, 11, 983–991. [Google Scholar] [CrossRef]
- Cao, J.; Wang, J.; He, C.; Fang, M. Angiosarcoma: A review of diagnosis and current treatment. Am. J. Cancer Res. 2019, 9, 2303–2313. [Google Scholar]
- Wilson, G.C.; Lluis, N.; Nalesnik, M.A.; Nassar, A.; Serrano, T.; Ramos, E.; Torbenson, M.; Asbun, H.J.; Geller, D.A. Hepatic Angiosarcoma: A Multi-institutional, International Experience with 44 Cases. Ann. Surg. Oncol. 2019, 26, 576–582. [Google Scholar] [CrossRef]
- Zheng, Y.W.; Zhang, X.W.; Zhang, J.L.; Hui, Z.Z.; Du, W.J.; Li, R.M.; Ren, X.B. Primary hepatic angiosarcoma and potential treatment options. J. Gastroenterol. Hepatol. 2014, 29, 906–911. [Google Scholar] [CrossRef] [Green Version]
- Huang, I.H.; Wu, Y.-Y.; Huang, T.-C.; Chang, W.-K.; Chen, J.-H. Statistics and outlook of primary hepatic angiosarcoma based on clinical stage. Oncol. Letters. 2016, 11, 3218–3222. [Google Scholar] [CrossRef] [Green Version]
- Duan, X.F.; Li, Q. Primary hepatic angiosarcoma: A retrospective analysis of 6 cases. J. Dig. Dis. 2012, 13, 381–385. [Google Scholar] [CrossRef]
- Zhu, Y.P.; Chen, Y.M.; Matro, E.; Chen, R.B.; Jiang, Z.N.; Mou, Y.P.; Hu, H.J.; Huang, C.J.; Wang, G.Y. Primary hepatic angiosarcoma: A report of two cases and literature review. World J. Gastroenterol. 2015, 21, 6088–6096. [Google Scholar] [CrossRef]
- Lerro, C.C.; Robbins, A.S.; Phillips, J.L.; Stewart, A.K. Comparison of cases captured in the national cancer data base with those in population-based central cancer registries. Ann. Surg. Oncol. 2013, 20, 1759–1765. [Google Scholar] [CrossRef]
- Kim, H.R.; Rha, S.Y.; Cheon, S.H.; Roh, J.K.; Park, Y.N.; Yoo, N.C. Clinical features and treatment outcomes of advanced stage primary hepatic angiosarcoma. Ann. Oncol. 2009, 20, 780–787. [Google Scholar] [CrossRef]
- Wang, Z.-B.; Yuan, J.; Chen, W.; Wei, L.-X. Transcription factor ERG is a specific and sensitive diagnostic marker for hepatic angiosarcoma. World J. Gastroenterol. 2014, 20, 3672. [Google Scholar] [CrossRef]
- Molina, E.; Hernandez, A. Clinical manifestations of primary hepatic angiosarcoma. Dig. Dis. Sci. 2003, 48, 677–682. [Google Scholar] [CrossRef]
- Groeschl, R.T.; Miura, J.T.; Oshima, K.; Gamblin, T.C.; Turaga, K.K. Does histology predict outcome for malignant vascular tumors of the liver? J. Surg. Oncol. 2014, 109, 483–486. [Google Scholar] [CrossRef]
- Tripke, V.; Heinrich, S.; Huber, T.; Mittler, J.; Hoppe-Lotichius, M.; Straub, B.K.; Lang, H. Surgical therapy of primary hepatic angiosarcoma. BMC Surg. 2019, 19, 5. [Google Scholar] [CrossRef]
- Gutierrez, J.C.; Perez, E.A.; Moffat, F.L.; Livingstone, A.S.; Franceschi, D.; Koniaris, L.G. Should soft tissue sarcomas be treated at high-volume centers? An analysis of 4205 patients. Ann. Surg. 2007, 245, 952–958. [Google Scholar] [CrossRef]
- Martin-Broto, J.; Hindi, N.; Cruz, J.; Martinez-Trufero, J.; Valverde, C.; De Sande, L.M.; Sala, A.; Bellido, L.; De Juan, A.; Rubió-Casadevall, J.; et al. Relevance of Reference Centers in Sarcoma Care and Quality Item Evaluation: Results from the Prospective Registry of the Spanish Group for Research in Sarcoma (GEIS). Oncologist 2019, 24, e338–e346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, A.C.; Ethun, C.G.; Liu, Y.; Lopez-Aguiar, A.G.; Tran, T.B.; Poultsides, G.; Valerie Grignol, J.; Howard, H.; Bedi, M.T.; Gamblin, C.; et al. Studying a Rare Disease Using Multi-Institutional Research Collaborations vs Big Data: Where Lies the Truth? J. Am. Coll. Surg. 2018, 227, 357.e3–366.e3. [Google Scholar] [CrossRef] [PubMed]
- Buehler, D.; Rice, S.R.; Moody, J.S.; Rush, P.; Hafez, G.R.; Attia, S.; Longley, B.J.; Kozak, K.R. Angiosarcoma outcomes and prognostic factors: A 25-year single institution experience. Am. J. Clin. Oncol. 2014, 37, 473–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, S.; Wang, Y.; Wang, Z.; Shao, J.; Ye, Z. Survival predictors of metastatic angiosarcomas: A surveillance, epidemiology, and end results program population-based retrospective study. BMC Cancer 2020, 20, 778. [Google Scholar] [CrossRef]
- Fury, M.G.; Antonescu, C.R.; Van Zee, K.J.; Brennan, M.E.; Maki, R.G. A 14-year retrospective review of angiosarcoma: Clinical characteristics, prognostic factors, and treatment outcomes with surgery and chemotherapy. Cancer J. 2005, 11, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Matthaei, H.; Krieg, A.; Schmelzle, M.; Boelke, E.; Poremba, C.; Rogiers, X.; Knoefel, W.T.; Peiper, M. Long-term survival after surgery for primary hepatic sarcoma in adults. Arch Surg. 2009, 144, 339–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakayama, H.; Masuda, H.; Fukuzawa, M.; Takayama, T.; Hemmi, A. Metastasis of hepatic angiosarcoma to the gastric vein. J. Gastroenterol. 2004, 39, 193–194. [Google Scholar] [CrossRef]
- Timaran, C.H.; Grandas, O.H.; Bell, J.L. Hepatic angiosarcoma: Long-term survival after complete surgical removal. Am. Surg. 2000, 66, 1153–1157. [Google Scholar]
- Özden, I.; Bilge, O.; Erkan, M.; Cevikbaş, U.; Acarlı, K. Five years and 4 months of recurrence-free survival in hepatic angiosarcoma. J. Hepato Biliary Pancreat. Surg. 2003, 10, 250–252. [Google Scholar] [CrossRef]
- Mangla, A.; Yadav, U. Cancer, Leiomyosarcoma; StatPearls: Treasure Island, FL, USA, 2020. [Google Scholar]
- Villanueva, A. Hepatocellular Carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Xie, H.; Hu, M.; Huang, T.; Hu, Y.; Sang, N.; Zhao, Y. Recent progress in treatment of hepatocellular carcinoma. Am. J. Cancer Res. 2020, 10, 2993–3036. [Google Scholar] [PubMed]
- Van der Graaf, W.T.; Blay, J.Y.; Chawla, S.P.; Kim, D.W.; Bui-Nguyen, B.; Casali, P.G.; Schöffski, P.; Aglietta, M.; Staddon, A.P.; Beppu, Y.; et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): A randomised, double-blind, placebo-controlled phase 3 trial. Lancet 2012, 379, 1879–1886. [Google Scholar] [CrossRef]
- Orlando, G.; Adam, R.; Mirza, D.; Soderdahl, G.; Porte, R.J.; Paul, A.; Burroughs, A.K.; Seiler, C.A.; Colledan, M.; Graziadei, I.; et al. Hepatic hemangiosarcoma: An absolute contraindication to liver transplantation—the European Liver Transplant Registry experience. Transplantation 2013, 95, 872–877. [Google Scholar] [CrossRef] [PubMed]
- Konstantinidis, I.T.; Nota, C.; Jutric, Z.; Ituarte, P.; Chow, W.; Chu, P.; Singh, G.; Warner, S.G.; Melstrom, L.G.; Fong, Y. Primary liver sarcomas in the modern era: Resection or transplantation? J. Surg. Oncol. 2018, 117, 886–891. [Google Scholar] [CrossRef]


{kind=link}
| Histologic Type | |||
|---|---|---|---|
| Angiosarcoma | HCC | p | |
| Overall n | 346 | 118,066 | |
| Age, Mean (SD) | 62.9 (13.7) | 62.4 (11.1) | 0.458 |
| Sex, n (%) | |||
| Male | 224 (64.7%) | 90,367 (76.8%) | <0.0001 |
| Female | 122 (35.3%) | 27,266 (23.2%) | |
| Race, n (%) | |||
| White | 290 (85.8%) | 85,200 (7.4%) | <0.0001 |
| Black | 25 (7.4%) | 18,712 (15.9%) | |
| American Indian | 1 (0.3%) | 839 (0.7%) | |
| Asian and Pacific Islander | 22 (6.5%) | 9477 (8.1%) | |
| Ethnicity, n (%) | |||
| Non Spanish | 284 (88.8%) | 97,216 (86.5%) | 0.245 |
| Hispanic | 36 (11.3%) | 15,142 (13.5%) | |
| Metro Status, n (%) | |||
| Metro | 270 (81.1%) | 99,948 (87.3%) | 0.002 |
| Urban | 54 (16.2%) | 12,986 (11.5%) | |
| Rural | 9 (2.7%) | 1470 (1.3%) | |
| Insurance Status, n (%) | |||
| Not Insured | 15 (4.5%) | 7772 (6.8%) | <0.0001 |
| Private Insurance | 146 (43.7%) | 38,481 (33.5%) | |
| Medicaid | 22 (6.6%) | 17,587 (15.3%) | |
| Medicare | 145 (43.4%) | 48,763 (42.5%) | |
| Other Government | 6 (1.8%) | 2120 (1.9%) | |
| Region, n (%) | |||
| Northeast | 63 (19.1%) | 24,451 (21.1%) | <0.0001 |
| Midwest | 104 (31.6%) | 22,811 (19.7%) | |
| South | 98 (29.8%) | 45,337 (39.1%) | |
| West | 64 (19.5%) | 23,382 (20.2%) | |
| Facility Type, n (%) | |||
| Community Cancer Program | 21 (6.4%) | 6335 (5.5%) | 0.141 |
| Comprehensive Community Cancer Program | 104 (31.6%) | 32,083 (27.7%) | |
| Academic/Research Program | 164 (49.8%) | 65,237 (56.3%) | |
| Integrated Network Cancer Program | 40 (12.2%) | 12,326 (10.6%) | |
| Charlson/Deyo Score | |||
| 0 | 229 (66.2%) | 55,031 (46.8%) | <0.0001 |
| 1 | 57 (16.5%) | 31,722 (27.0%) | |
| 2 | 20 (5.8%) | 12,403 (10.5%) | |
| ≥3 | 40 (11.6%) | 18,477 (15.7) | |
| Metastasis | 43 (26.2%) | 5407 (8.6%) | <0.0001 |
| Bone | 24 (13.0%) | 2509 (3.8%) | |
| Lung | 19 (10.3%) | 2997 (4.5%) | |
| Treatment Modalities used | |||
| Surgery, n (%) | 50 (14.6%) | 31,248 (26.7%) | <0.0001 |
| Radiation, n (%) | 12 (3.5%) | 9946 (8.5%) | 0.001 |
| Chemotherapy, n (%) | |||
| No | 218 (64.3%) | 56,023 (55.0%) | 0.086 |
| Yes (single agent) | 82 (24.2%) | 34,755 (34.1%) | |
| Yes (multi-agent) | 39 (11.5%) | 11,161 (11.0) | |
| Alive, n (%) | 34 (9.8%) | 31,158 (26.5%) | <0.0001 |
| Received Surgery | Received Chemotherapy | |||||
|---|---|---|---|---|---|---|
| No | Yes | p | No | Yes | p | |
| Overall n | 293 (85.4%) | 50 (14.6%) | 218 (64.3%) | 121 (42.0%) | ||
| Age, Mean (SD) | 64.2 (13.4) | 55.9 (13.4) | <0.0001 | 65.0 (13.7) | 59.7 (12.6) | <0.0001 |
| Sex, n (%) | ||||||
| Male | 198 (67.6%) | 25 (50%) | 0.016 | 140 (64.2%) | 79 (65.3%) | 0.84 |
| Female | 95 (32.4%) | 25 (40%) | 78 (35.8%) | 42 (34.7%) | ||
| Race, n (%) | ||||||
| White | 245 (83.6%) | 44 (88.0%) | 0.93 | 184 (84.4%) | 100 (82.6%) | 0.85 |
| Black | 21 (7.2%) | 3 (6.0%) | 14 (6.4%) | 11 (9.1%) | ||
| American Indian | 1 (0.3%) | 0 (0%) | 1 (0.5%) | 0 (0%) | ||
| Asian and Pacific Islander | 20 (6.8%) | 2 (4.0%) | 14 (6.4%) | 7 (5.8%) | ||
| Ethnicity, n (%) | ||||||
| Non-Spanish | 243 (89.7%) | 39 (83.0%) | 0.18 | 175 (87.5%) | 102 (90.3%) | 0.46 |
| Hispanic | 28 (10.3%) | 8 (17.0%) | 25 (12.5%) | 11 (9.7%) | ||
| Metro Status, n (%) | ||||||
| Metro | 231 (81.9%) | 36 (75.0%) | 0.51 | 166 (79.1%) | 98 (84.5%) | 0.28 |
| Urban | 44 (15.6%) | 9 (20.8%) | 39 (18.6%) | 14 (12.0%) | ||
| Rural | 7 (2.5%) | 2 (4.2%) | 5 (2.4%) | 4 (3.5%) | ||
| Insurance Status, n (%) | ||||||
| Not Insured | 11 (3.9%) | 3 (6.1%) | 0.099 | 10 (4.7%) | 4 (3.5%) | 0.004 |
| Private Insurance | 118 (41.8%) | 27 (55.1%) | 79 (36.7%) | 64 (56.6%) | ||
| Medicaid | 18 (6.4%) | 4 (8.3%) | 20 (9.3%) | 2 (1.8%) | ||
| Medicare | 131 (46.5%) | 13 (26.5%) | 102 (47.4%) | 41 (36.8%) | ||
| Other Government | 4 (1.4%) | 2 (4.1%) | 4 (1.9%) | 2 (1.8%) | ||
| Region, n (%) | ||||||
| Northeast | 54 (19.3%) | 7 (15.2%) | 0.18 | 39 (18.5%) | 24 (21.2%) | 0.004 |
| Midwest | 89 (31.8%) | 15 (32.6%) | 55 (216.1%) | 47 (41.6%) | ||
| South | 78 (27.9%) | 19 (41.3%) | 66 (31.3%) | 30 (26.6%) | ||
| West | 59 (21.1%) | 5 (10.9%) | 51 (24.2%) | 12 (106%) | ||
| Facility Type, n (%) | ||||||
| Community Cancer Program | 19 (6.8%) | 1 (2.2%) | <0.0001 | 17 (8.1%) | 4 (3.5%) | 0.097 |
| Comprehensive Community Cancer Program | 99 (35.4%) | 4 (8.7%) | 73 (34.6%) | 29 (25.7%) | ||
| Academic/Research Program | 127 (45.4%) | 36 (78.3%) | 98 (46.5%) | 65 (57.5%) | ||
| Integrated Network Cancer Program | 35 (12.5%) | 5 (10.9%) | 23 (10.9%) | 15 (13.3%) | ||
| Charlson/Deyo Score | ||||||
| 0 | 192 (65.5%) | 34 (68.0%) | 0.83 | 131 (60.1%) | 94 (77.7%) | 0.002 |
| 1 | 50 (17.1%) | 7 (14.0%) | 36 (16.5%) | 18 (14.9%) | ||
| 2 | 16 (5.5%) | 4 (8.0%) | 17 (7.8%) | 6 (5.0%) | ||
| ≥3 | 35 (12.0%) | 5 (10%) | 34 (15.6%) | 33 (12.5%) | ||
| Metastasis | 42 (29.0%) | 1 (5.3%) | 0.027 | 16 (22.3%) | 21 (37.5%) | 0.073 |
| Radiation, n (%) | 10 (3.4%) | 2 (4.0%) | 0.84 | 5 (2.3%) | 6 (5.0%) | 0.39 |
| Chemotherapy, n (%) | 109 (45.6%) | 12 (26.1%) | 0.014 | -- | -- | |
| Surgery, n (%) | -- | -- | 34 (20.7%) | 12 (9.9%) | 0.014 | |
| Alive, n (%) | 20 (6.8%) | 13 (26.0%) | <0.001 | 23 (10.6%) | 11 (9.1%) | 0.44 |
| Median Survival | Cox Proportional Hazards | ||||
|---|---|---|---|---|---|
| Median Survival (95% CI) | Univariate HR (95% CI) | p | Multivariable HR *,† (95% CI) | p | |
| Surgery Status | |||||
| Yes | 7.69 (5.46–16.92) | 0.34 (0.24–0.49) | <0.0001 | 0.23 (0.15–0.37) | <0.0001 |
| No | 1.77 (1.48–1.94) | Ref | Ref | ||
| Chemotherapy Status | |||||
| Yes | 5.09 (3.77–5.81) | 0.59 (0.47–0.76) | 0.001 | 0.44 (0.32–0.60) | <0.0001 |
| No | 1.15 (0.85–1.61) | Ref | Ref | ||
| Median Survival (Months) | Cox Proportional Hazards | ||||
|---|---|---|---|---|---|
| Median Survival (95% CI) | Univariate HR (95% CI) | p | Multivariable HR * (95% CI) | p | |
| Cancer Type | |||||
| Angiosarcoma | 1.94 (1.77–2.36) | 2.39 (2.13–2.67) | <0.0001 | 2.41 (2.10–2.77) | <0.0001 |
| HCC | 10.35 (10.22–10.51) | Ref | Ref | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mangla, A.; Cioffi, G.; Barnholtz-Sloan, J.S.; Lee, R.T. Treatment Outcomes for Primary Hepatic Angiosarcoma: National Cancer Database Analysis 2004–2014. Curr. Oncol. 2022, 29, 3637-3646. https://0-doi-org.brum.beds.ac.uk/10.3390/curroncol29050292
Mangla A, Cioffi G, Barnholtz-Sloan JS, Lee RT. Treatment Outcomes for Primary Hepatic Angiosarcoma: National Cancer Database Analysis 2004–2014. Current Oncology. 2022; 29(5):3637-3646. https://0-doi-org.brum.beds.ac.uk/10.3390/curroncol29050292
Chicago/Turabian StyleMangla, Ankit, Gino Cioffi, Jill S. Barnholtz-Sloan, and Richard T. Lee. 2022. "Treatment Outcomes for Primary Hepatic Angiosarcoma: National Cancer Database Analysis 2004–2014" Current Oncology 29, no. 5: 3637-3646. https://0-doi-org.brum.beds.ac.uk/10.3390/curroncol29050292