Catalytic Fast Pyrolysis: A Review

1
Applied Environmental Science and Technology, School of Engineering, University of Alaska Anchorage, 3211 Providence Drive, ENGR 201, Anchorage, AK 99508, USA
2
Agricultural and Forestry Experiment Station, University of Alaska Fairbanks, 1509 S. Georgeson Drive, Palmer, AK 99645, USA
*
Author to whom correspondence should be addressed.
Energies 2013, 6(1), 514-538; https://0-doi-org.brum.beds.ac.uk/10.3390/en6010514
Submission received: 9 November 2012
/
Revised: 7 December 2012
/
Accepted: 21 December 2012
/
Published: 21 January 2013
Abstract
:Catalytic pyrolysis is a promising thermochemical conversion route for lignocellulosic biomass that produces chemicals and fuels compatible with current, petrochemical infrastructure. Catalytic modifications to pyrolysis bio-oils are geared towards the elimination and substitution of oxygen and oxygen-containing functionalities in addition to increasing the hydrogen to carbon ratio of the final products. Recent progress has focused on both hydrodeoxygenation and hydrogenation of bio-oil using a variety of metal catalysts and the production of aromatics from bio-oil using cracking zeolites. Research is currently focused on developing multi-functional catalysts used in situ that benefit from the advantages of both hydrodeoxygenation and zeolite cracking. Development of robust, highly selective catalysts will help achieve the goal of producing drop-in fuels and petrochemical commodities from wood and other lignocellulosic biomass streams. The current paper will examine these developments by means of a review of existing literature.
1. Introduction
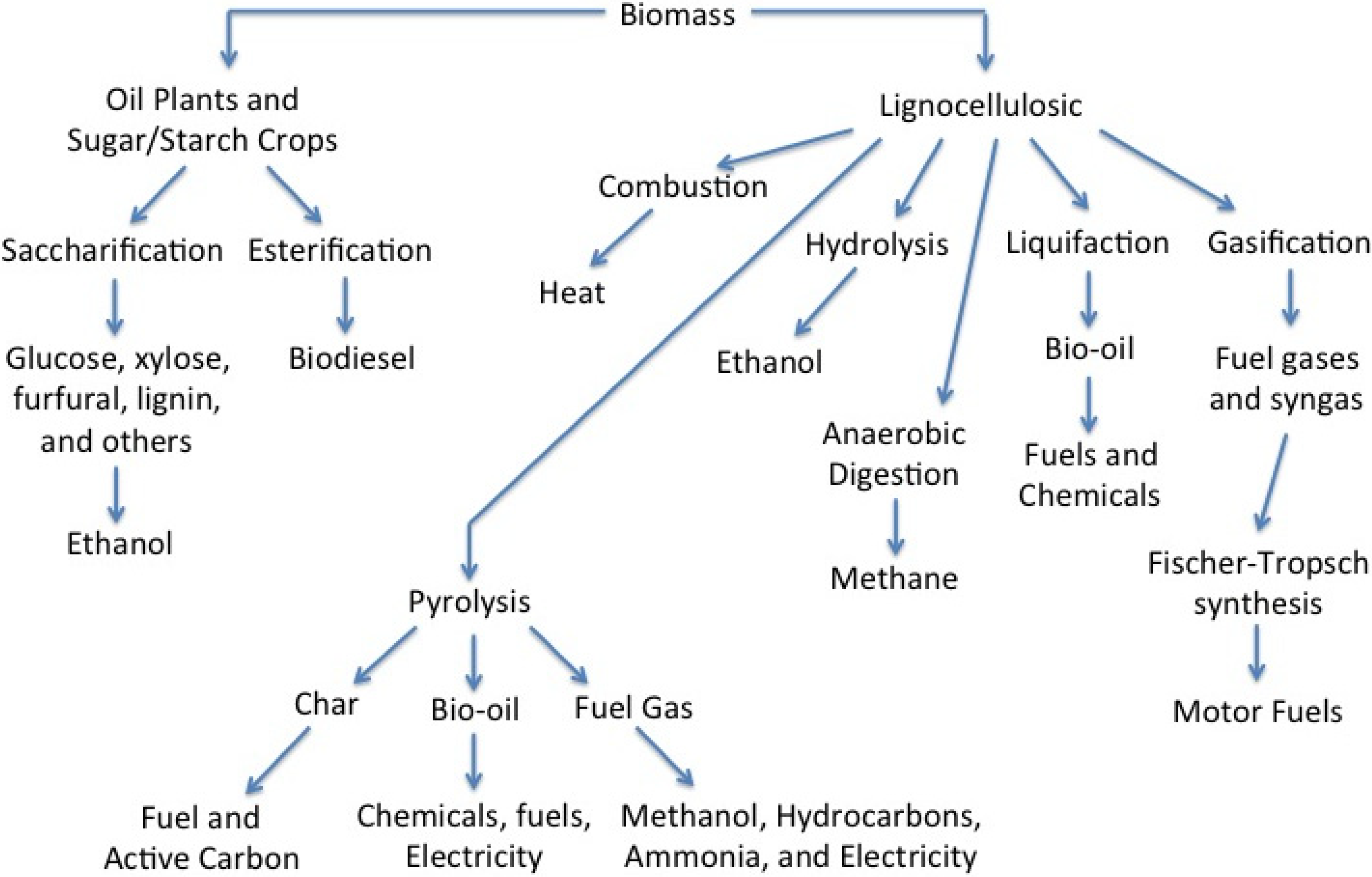
Growing interest in renewable technologies has led to the development of hydroelectric, solar, wave, geothermal, and wind power plants that can potentially displace fossil fuel based energy production [1]. However, existing infrastructures require hydrocarbons for manufacturing goods ranging from plastics and chemicals to fuel oil, lubricating oil, and hydraulic fluids. This has led many to look towards biomass as the only sustainable alternative to fossil fuels capable of yielding petroleum like products [2,3,4,5,6]. As is the case with all potentially disruptive technologies, though, proper environmental mitigation and lifecycle planning must be included when considering the production and use of biofuels [7,8]. Hydrocarbons can be produced from biomass via biological, biochemical, and thermochemical processes as shown in Figure 1 [9].
Figure 1.
Schematic of biomass conversion technologies.

Pyrolysis is touted to be one of the most promising thermochemical technologies with the potential to convert cheap, local, and abundant lignocellulosic biomass such as grasses and trees into a useful form [10,11,12,13], and commercial scale plants, such as Kior in Mississippi, have begun construction and operations. Thermal technologies include direct combustion, gasification, liquefaction, and pyrolysis, all of which have the benefit of using lignocellulosic materials. Only pyrolysis and liquefaction produce a liquid product, and although recent work on liquefaction has shown promising results [14,15,16], it is currently a nascent technology with high capital costs due to the high pressures required. Pyrolysis has the advantage of low capital investment and a liquid final product that can be transported and converted via catalysis to fuels and valuable products such as food flavorings, fertilizers, resins, and other specialty chemicals that are fully compatible with existing petroleum infrastructure. This provides significant economic advantages over ethanol that requires parallel infrastructures. Furthermore, all pyrolysis products can be utilized in the pyrolysis system. Gas can be burned to help dry the incoming biomass and operate the reactor [5,17], and char and ash are promising soil amendments [18] and a potential C sink [19]. Chars can also be used as a fuel [20], and there is potential to expand the resource base to include non-traditional feedstock such as lignin [21], sewage sludge, chicken litter [22], and even salmon waste [23]. The current paper will examine the catalytic processing necessary to transform lignocellulosic biomass into hydrocarbons using pyrolysis platforms.
2. Pyrolysis
Pyrolysis is the thermochemical decomposition of biomass at temperatures between 400 and 650 °C in the absence of O2. The decomposition process releases volatile species, while the solid, non-volatiles are collected as bio-char. A portion of the gas-phase volatiles condense into a black, viscous fluid termed bio-oil [24] that has a variety of synonyms including pyrolysis oil, bio-crude oil, bio-fuel oil, wood liquid, wood oil, liquid smoke, wood distillates, pyroligneous tar, and pyroligneous acid [5]. Pyrolysis methods differ in their residence time, temperature, and heating rate, which in turn greatly affect the percentages of gas, char, and liquid products in a semi-predictable manner, while the resulting, individual chemical species remain hard to predict and quantify [5,10]. Pyrolysis methods can be grouped into two large categories, slow and fast (or flash) pyrolysis.
Slow pyrolysis consists of slow heating rates of 0.1–1 °C/s, a residence time anywhere from hours to minutes, and a temperature range of 400–600 °C. It has been used for centuries to produce methanol and yields approximately equal quantities of char, gas, and liquid [5,10,25].
Fast pyrolysis is a relatively new, promising technology involving a high liquid yield achieved through rapid heating rates of 10 to >1000 °C/s, short residence times of <2 s, temperatures of 400–650 °C, and rapid quenching of the vapors [5,6]. For a more extensive review of reactors, processes, and their corresponding liquid products see Bridgwater et al. [6].
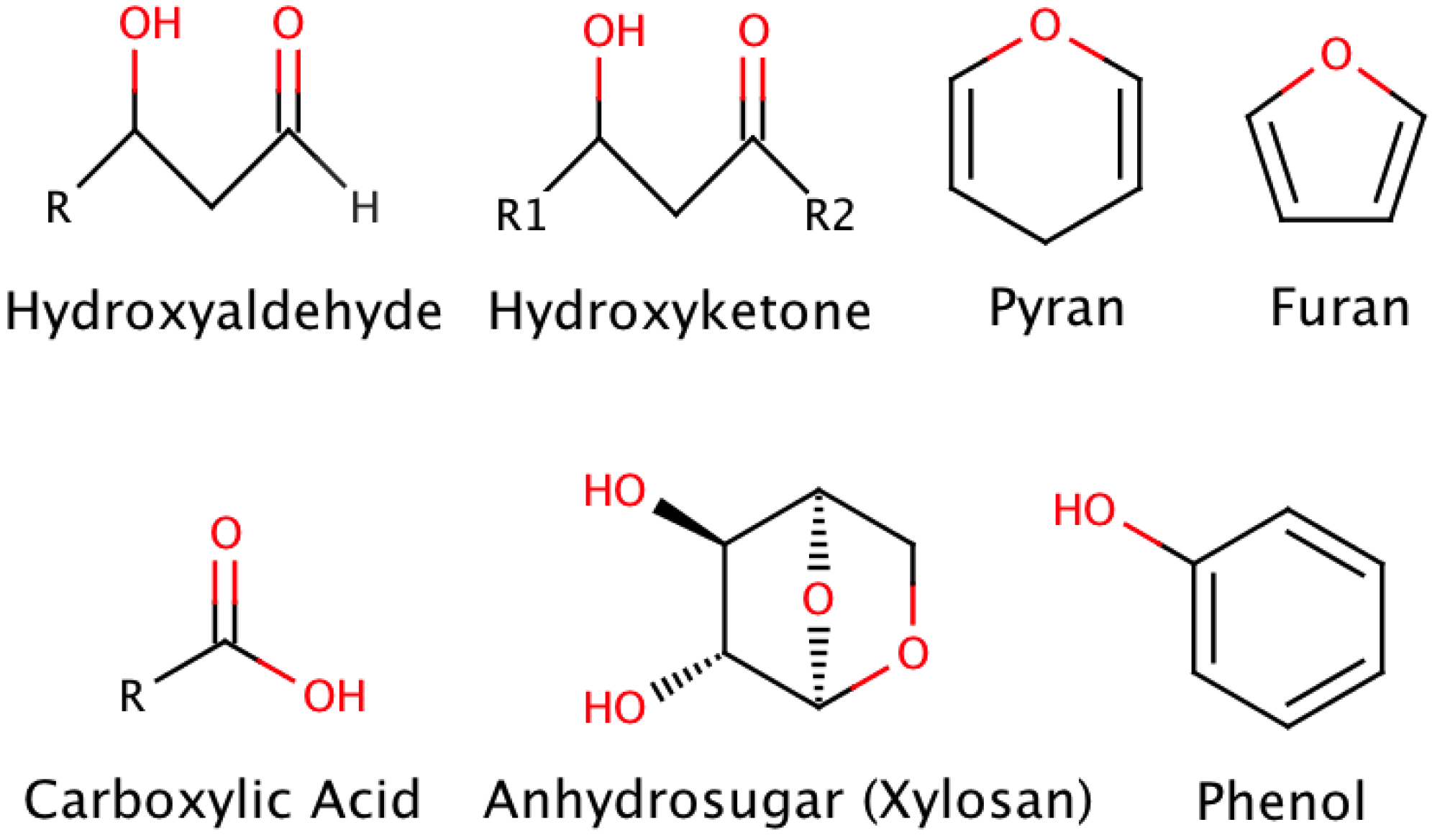
Bio-oil varies according to process conditions and feedstock. Most studies have focused on fast pyrolysis oil that can be generalized as consisting of hydroxyaldehydes, hydroxyketones, carboxylic acids, furan/pyran ring containing compounds, anhydrosugars, phenolic compounds and oligomeric fragments of lignocellulosic polymers with some examples represented in Scheme 1. These products stem from the original biomass composition consisting of cellulose, hemicelluloses, lignin, extractives, lipids, proteins, simple sugars, starches, water, hydrocarbons, ash, and other compounds [5].
Scheme 1.
Representative compounds of bio-oil.

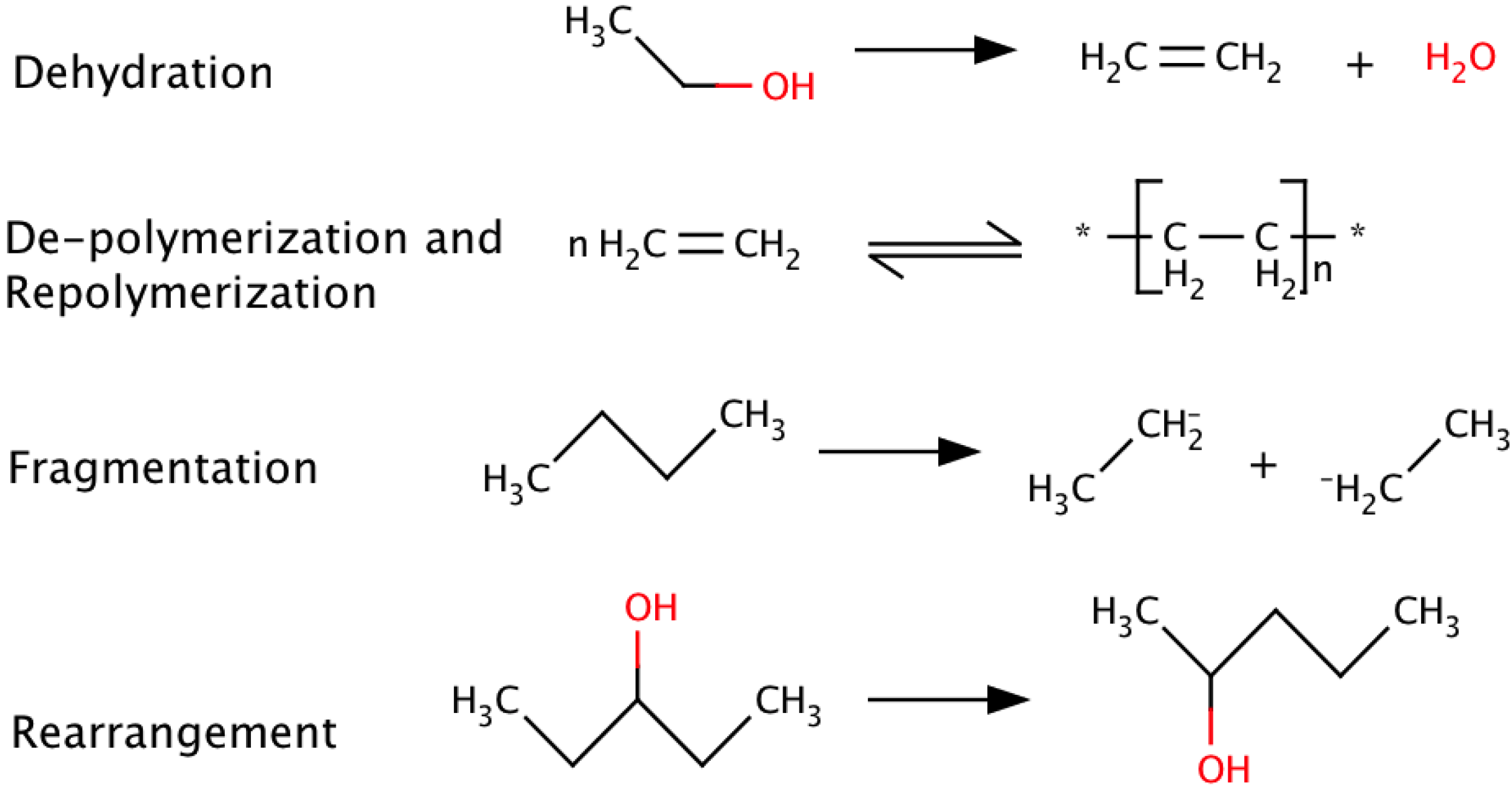
Reactions under pyrolysis conditions are complex and not fully understood due to the range of reaction temperatures and the complex biomass composition, but they can be generally classified as a simultaneous mix of dehydration, depolymerization, re-polymerization, fragmentation, rearrangement, and condensation, as represented by some examples in Scheme 2. These reactions result in a bio-oil containing over 300 individual compounds [22,26,27]. For more extensive reviews of the elemental and chemical composition of biomass and bio-oil see Yanik et al. [28], Branca et al. [29], Lu et al. [13], Valtiner et al. [30], Ralph et al. [31], Evans et al. [32], Marsman et al. [33], and Sipila et al. [34].
Scheme 2.
Representative pyrolysis reactions.

From an applied perspective, bio-oil can be burned directly as a substitute for fuel oil in various static applications such as boilers and furnaces [6]. However, it is not miscible with fossil fuels, but is miscible with water up to 35%–40%. The result is that the liquid gradually “ages” as the polyphenols polymerize, leading to a variable viscosity, ranging from 10 to 10,000 cp. It “ages” faster with exposure to light, O2, or heat above 80 °C, leading to storage issues. The pH is relatively low due to the presence of organic acids, further increasing corrosion and storage issues. There are suspended solids ranging from 0.3% to 3%, which have the potential to negatively affect combustion characteristics by plugging nozzles, and the presence of oligomeric fragments can lead to solid, tar-like deposits on pipes and reactors [35]. Bio-oil characteristics are contrasted to petroleum crude oil in Table 1 [22,36] to highlight areas where bio-oil needs improvements.
To overcome these issues, bio-oil must be modified chemically or ‘upgraded’ by the removal and modification of unwanted compounds, most of which contain oxygen. Many times, the final product is evaluated by its O/C ratio and H/C ratio, with low O/C and high H/C ratios indicating a higher quality liquid product. These ratios are especially important if the bio-oil is to be used as a substitute for demanding petrochemical applications such as transportation fuels that need a clean, homogenous fuel stream [37]. To achieve these results, bio-oil can be re-volatilized in a catalytic environment, generally referred to as catalytic upgrading, or biomass can be pyrolyzed in the presence of catalysts that affect the desired changes before the initial condensation, generally referred to as catalytic fast pyrolysis. Ideally, a single reactor would fulfill all process requirements and produce a quality liquid fuel from the initial, solid biomass [38,39]. Due to its potential in a wide variety of situations, many consider catalytic fast pyrolysis to be the most promising way to improve the final, liquid composition [5,40]. The challenge is to design catalysts that positively affect all of the over 300 volatilized, organic compounds through various cracking and reforming reactions. This is important for biorefineries and will help them become cost competitive with current infrastructure. Improving catalysts’ activity, selectivity, and stability is essential for progress in the field of biomass utilization.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Composition | Bio-oil | Crude Oil |
|---|---|---|
| Water (wt %) | 15–30 | 0.1 |
| pH | 2.8–3.8 | - |
| density (kg/L) | 1.05–1.25 | 0.86 |
| viscosity 50 °C (cP) | 40–100 | 180 |
| HHV (MJ/kg) | 16–19 | 44 |
| C (wt %) | 55–65 | 83-86 |
| O (wt %) | 28–40 | <1 |
| H (wt %) | 5–7 | 11–14 |
| S (wt %) | <0.05 | <4 |
| N (wt %) | <0.4 | <1 |
| Ash (wt %) | <0.2 | 0.1 |
| H/C | 0.9–1.5 | 1.5–2.0 |
| O/C | 0.3–0.5 | ≈0 |
3. Catalytic Upgrading
3.1. Introduction
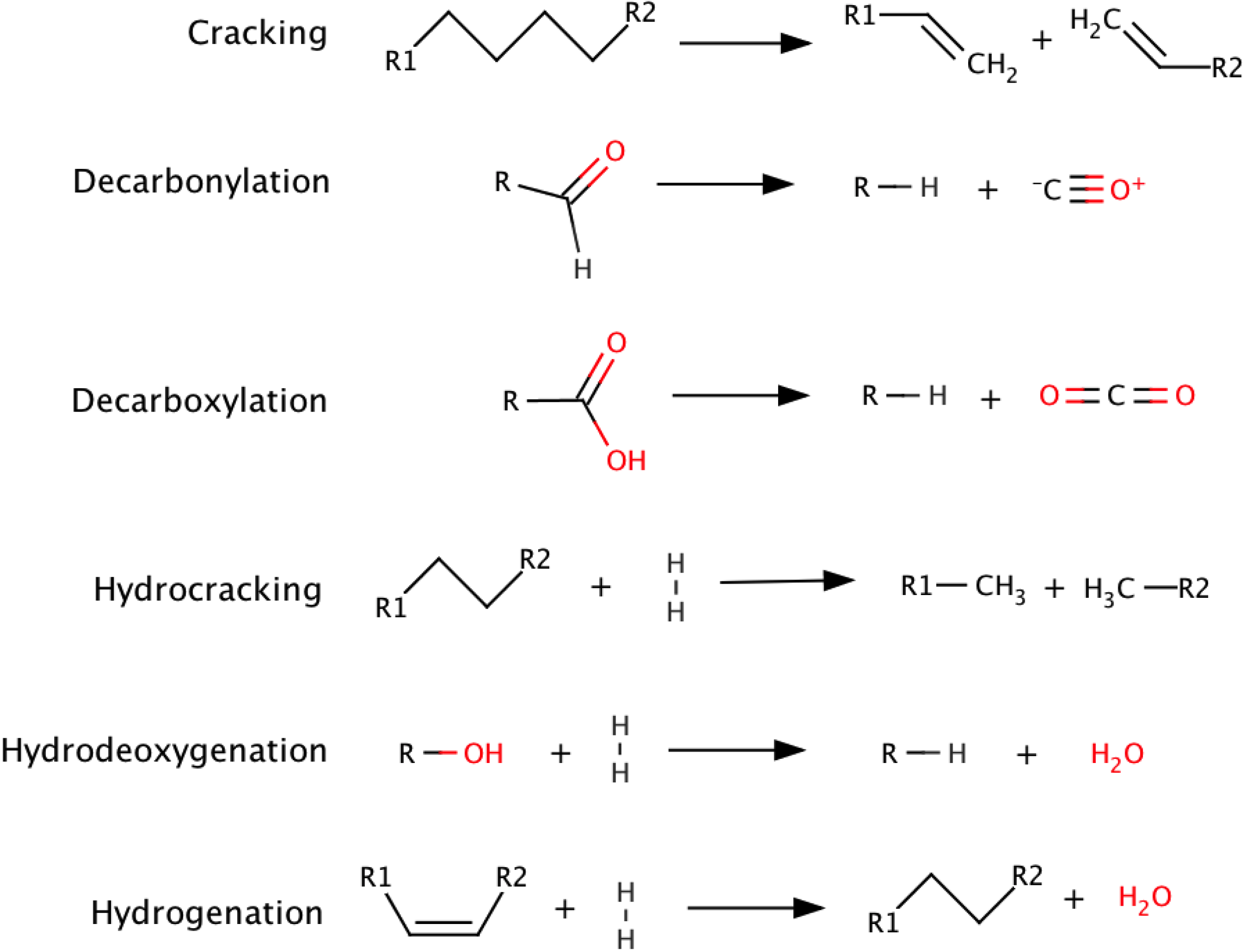
The addition of catalysts to the pyrolysis system enhances reactions that include cracking, decarbonylation, decarboxylation, hydrocracking, hydrodeoxygenation, and hydrogenation as summarized in Scheme 3 [22].
Scheme 3.
Representative catalytic upgrading reactions.

As shown in Scheme 3, catalytic upgrading can enhance the properties of bio-oil by removing oxygenated compounds via H2O and CO2, reducing molecular weight, and altering chemical structures to resemble those of petrochemical products. Interestingly, biomass has within its chemical framework naturally present ions that can have catalytic effects under pyrolysis reaction conditions [41], such as reducing the bio-oil yield in favor of char and gaseous species [42]. This effect was observed with the most abundant inorganic components of biomass, including silica, Na, K, Mg, and Ca [43]. Both the cations and anions present were shown to have an effect on the pyrolysis products [44], while demineralization was shown to increase the yield of anhydrosugars [45]. To study this, biomass was stripped of its natural minerals, and these catalytic elements were then reintroduced individually using ion exchange and sorption. The way in which these ions were reintroduced into the biomass had an affect on the final yields with ion exchange showing a greater catalytic effect than sorbed ions [46].
Catalytic effects of inorganics found in ash have also been studied. Patwardhan et al. [47] studied the pyrolysis of pure cellulose in the presence of switchgrass ash, a representative mix of water-soluble salts of alkali and alkaline earth metals. It was discovered that as little as 0.5% ash resulted in a tripling of the formic acid and quadrupling of glycoaldehyde, whereas the levoglucosan yield was halved. The comparative yields of each chemical species were changed, but the individual species themselves were unaffected. Most of these inorganic salts can be extracted with an acid wash, even though some alkaline earth metals associated with organic macromolecules are resistant to extraction. It is therefore important to take into consideration that pyrolysis is inherently a catalytic process unless pretreated, and when studying the selectivity of additional catalysts, the catalytic effects of various naturally occurring minerals should be evaluated.
In industrial settings, it is required that additional catalysts not found in natural plant biomass are added in order to achieve the desired upgrading effects. However, due to the diverse mixture of species and reactions present, bio-oil cannot currently be upgraded using a single technique. For example, oligomeric fragments cause coking in a steam reforming process but are desirable for catalytic cracking or hydrocracking [48,49].
Overcoming issues like these will require further development of catalysts beyond what has been previously studied [25]. Catalysts should be highly active, selective to particular products, resistant to deactivation, readily recycled, and cheap. Most catalysts used in the upgrading of bio-oil are supported, meaning the metal catalyst is dispersed on a different and cheaper material such as carbon, silica, or alumina. Supported catalysts are cost effective and can generally be used at higher temperatures due to the dispersed metal particles’ resistance to sintering, or melting together. Furthermore, the support itself can act as a catalyst. Different catalysts have different mechanisms for eliminating oxygen and catalyzing other desired reactions as shown in Scheme 2 and Scheme 3. Two main routes for upgrading have dominated the research and development fields, hydrodeoxygenation and zeolite cracking.
3.2. Hydrodeoxygenation
Hydrodeoxygenation (HDO) is a hydrogenolysis process that removes oxygen from a feedstock. Carbon-oxygen bonds are cleaved with hydrogen in the presence of a catalyst producing CO2 and H2O and partially eliminating oxygen from the final product. This type of reaction can be achieved through sulfide/oxide and transition metal catalysts through a combination of chemisorption, proton donation, and desorption. The catalyst reactivity relies on the number and strength of both Lewis acid and Brønsted acid sites on the catalyst/support system. Supports affect both the acidity and reactivity of the catalyst compound. Ideally, they should be stable and have a low affinity for carbon formation and a high affinity for activation of oxy-compounds [22].
HDO produces a quality, energy dense, non-corrosive, naphtha-like product, which can be further upgraded to other chemicals [25]. HDO also has the advantage of easy separation of water and benefits to turbines from the separation of alkali metals from the hydrocarbon product when oxygen levels are reduced to less than 2%. However, it has the major disadvantage of consuming large quantities of high-pressure H2, 600–1000 L/kg bio-oil, thus increasing costs [41]. It is important to note, though, that consumption is also high for some petroleum feedstocks that are difficult to hydrocrack. Just like the petroleum industry in the past, the bio-oil industry needs further technological development. Fortunately, feedstocks for biofuels are not fundamentally more difficult to upgrade than those in the petroleum industry [50]. Furthermore, if HDO is emphasized as opposed to hydrogenation of the aromatic rings, hydrogen consumption can be significantly reduced while raising the final octane rating [51].
Using current technology, hydrogen is generally co-fed into the system at a pressure of 75–300 bar. To a certain extent, high pressure helps increase the solubility of the H2 in the bio-oil and thereby increase hydrogenation via better accessibility to the catalyst. However, even at a low pressure of 5 bar, positive results have been observed [52]. Typical residence times range from 3 to 4 h for batch reactors. Using a Ru/C catalyst at 350 °C and 200 bar in an autoclave setup, final liquid yield was highest for a 4 h reaction time [53]. Temperatures range from 250 to 450 °C. Coke formation generally increases with time and temperature, as does deoxygenation [22]. From 360 to 390 °C and a 5 min to 1 h reaction time, deoxygenation was shown to increase in the presence of a cobalt molybdate catalyst in an autoclave setup [54]. However, for HDO of wood based bio-oil over a Pd/C catalyst in a fixed bed reactor at 140 bar, the degree of deoxygenation plateaued at 340 °C and gave way to cracking. Furthermore, the gas yield increased three-fold and the final liquid yield decreased from 75% to 56% when increasing the temperature from 310 to 360 °C [55]. Clearly catalyst choice and operating conditions impact the final products. A balance must therefore be struck between residence time, temperature, deoxygenation, gas, and coke formation.
Current research focuses on improving catalyst effectiveness and longevity. The petroleum industry has been eliminating sulfur from petrochemicals for years using a similar hydrogenolysis process called hydrodesulfurization (HDS). Therefore, many catalysts have already been extensively studied by the petroleum industry for HDS. Due to the similarities between the two, much of the knowledge about HDO has been extrapolated from previous HDS experience with cobalt/molybdenum and Ni, Co, Mo in oxide forms on silica and alumina supports. It has been noted that the high acidity of Al2O3 supports causes unacceptable coking, and the material has furthermore been shown to sinter at high temperatures in the presence of water and therefore deactivate [56].
Catalytic work has therefore been undertaken on both catalysts and supports. For example, research has shown that neutral carriers such as C generally show lower coke formation than alumina supports [57]. Longevity of the catalyst and therefore low coke formation is important because catalyst functionality including oil yield and resulting H/C ratio is directly affected by the number of recycles, even if O/C ratios seem to be unaffected [53]. Different supports have also been shown to give different final liquid yields and degrees of deoxygenation. For example, Ru/TiO2 and Ru/C yielded more final liquid with lower oxygen content than Ru/Al2O3 in an autoclave setup at 450 °C and 350 bar of H2 [58]. A possible mechanism for HDO of a lignin-derived compound such as guaiacol on a supported metal catalyst is given in Scheme 4 [12,22].
Scheme 4.
Possible mechanism for HDO of guaiacol.

Several alternative approaches to more traditional catalyst development have also been explored. Gutierrez et al. [56] found that multiple noble metals used simultaneously interacted with each other and gave different results than simply the two separate catalyst results combined. HDO of guaiacol was studied using zirconia-supported mono- and bi-metallic noble metal catalysts in a batch reactor at 80 bar. Rh, Pd, Pt, RhPd, RhPt, and PdPt all performed better in the hydrogenation of guaiacol at 100 °C and at 300 °C than the conventional sulfided CoMo/Al2O3 catalyst which deactivated due to carbon deposition and contaminated the product with sulfur. Rh and RhPt completely converted the guaiacol at 100 °C to a variety of products, but deoxygenation only took place at 300 °C. The bimetallic catalysts containing Rh gave better results than the monometallic Pt and Pd catalysts in terms of guaiacol hydrogenation. The PdPt catalyst was less reactive than that of the separate monometallic catalysts. This indicates that catalysts react with each other in a variety of ways when used simultaneously, in addition to reacting with the biomass. These interactions are not fully understood.
Reduction of H2 consumption has been studied using the donation of H2 by a liquid with an easily donated, acidic proton that lowers the H2 pressure required. Xiong et al. [59] studied obtaining hydrogen from formic acid over Ni, Pd, and Ru on various supports in a batch reactor at 40 bar, a temperature of 150–230 °C, and a reaction time of 5–7 h. The catalyst supports did not make a significant difference, but Ni and Ru outperformed Pd in final product quality. Oxygen content was lowered while a reduction in the unsaturated components was observed, along with the conversion of organic acids into esters. All of these reactions occurred without obvious coke formation. In another study, Kunkes et al. [60] described a method of reforming polyol over Pt-Re/C, where reforming refers to C-C cleavage and the corresponding production of H2, CO, and CO2. These products can be used to de-oxygenate the remainder of the feed thus eliminating the need for co-feeding of hydrogen while producing gasoline and diesel type hydrocarbons. Similarly, Pt elements can promote the water-gas shift (WGS) reaction [61]. Pt/Al2O3 has been studied with a model bio-oil in an autoclave at 350 °C under 10 bar of nitrogen. The catalyst showed selectivity for producing H2 in situ, eliminating the need for co-feeding of hydrogen [62]. Clearly, further research on catalysts and corresponding reaction conditions modified to obtain desired products is needed. See Wildschut et al. [58] and Elliott [51] for more in depth work on HDO including product properties and molecular composition of HDO oils. For a comparison of several methods for characterizing HDO oils see Oasmaa et al. [63]. Table 2 [22] provides a summary of the yield, product quality, and operating conditions for several catalysts investigated for HDO. DOD (%) stands for degree of deoxygenation.
| Catalyst | Temp. (°C) | Pressure (bar) | DOD (%) | O/C | H/C | Oil Yield (wt %) | Ref. |
|---|---|---|---|---|---|---|---|
| Co-MoS2/Al2O3 | 350 | 200 | 81 | 0.8 | 1.3 | 26 | [58] |
| Co-MoS2/Al2O4 | 370 | 300 | 100 | 0 | 1.8 | 33 | [64] |
| Ni-MoS2/Al2O3 | 350 | 200 | 74 | 0.1 | 1.5 | 28 | [58] |
| Ni-MoS2/Al2O4 | 400 | 85 | 28 | - | - | 84 | [65] |
| Pd/C | 350 | 200 | 85 | 0.7 | 1.6 | 65 | [58] |
| Pd/C | 340 | 140 | 64 | 0.1 | 1.5 | 48 | [55] |
| Pd/ZrO2 | 300 | 80 | - | 0.1 | 1.3 | - | [56] |
| Pt/Al2O3/SiO2 | 400 | 85 | 45 | - | - | 81 | [65] |
| Pt/ZrO2 | 300 | 80 | - | 0.2 | 1.5 | - | [56] |
| Rh/ZrO2 | 300 | 80 | - | 0 | 1.2 | - | [56] |
| Ru/Al2O3 | 350 | 200 | 78 | 0.4 | 1.2 | 36 | [58] |
| Ru/C | 350–400 | 230 | 73 | 0.1 | 1.5 | 38 | [66] |
| Ru/C | 350 | 200 | 86 | 0.8 | 1.5 | 53 | [58] |
| Ru/TiO2 | 350 | 200 | 77 | 1 | 1.7 | 67 | [58] |
3.3. Catalytic Cracking with Zeolites
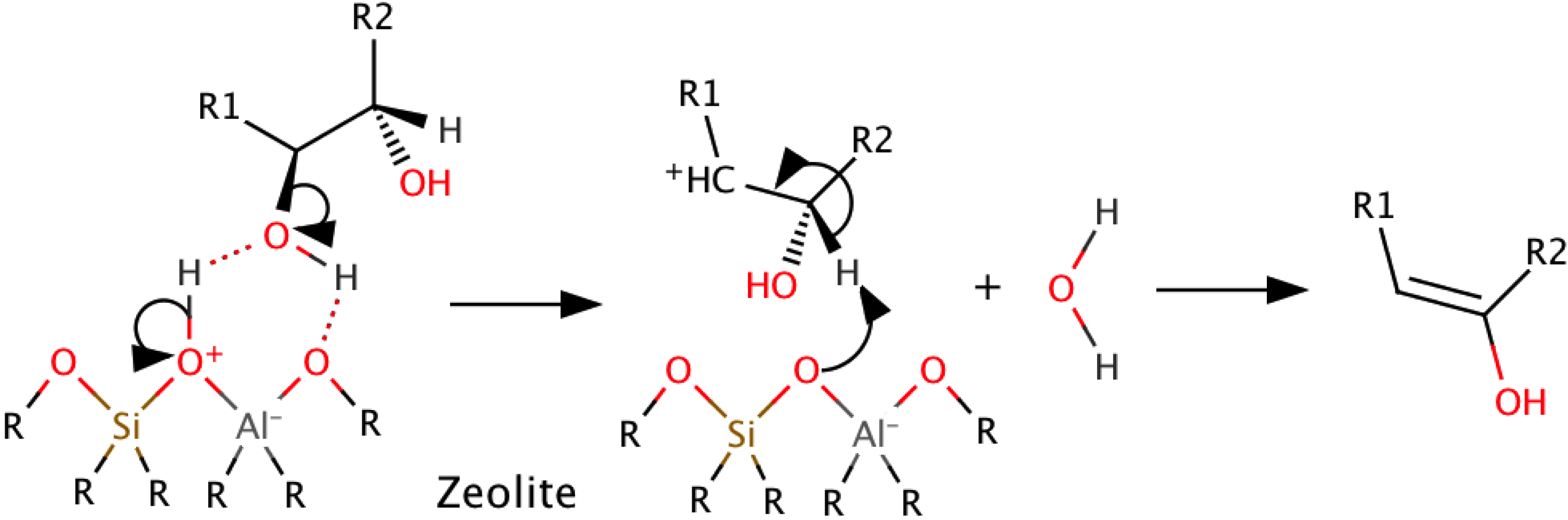
Zeolites are complex, three-dimensional porous structures with varying elemental compositions that exhibit catalytic activity in up to 50% of their volume. Cracking and dehydration are the main reactions seen. Adsorption of the oxy-compound occurs on an acid site. This is followed by either decomposition or bimolecular monomer dehydration, as determined by pore size [22]. As with sulfide/oxide and transition metal catalysts, the acidity of the zeolite affects the reactivity and yields, with high acidity leading to a higher affinity for C and water formation [67]. In HZSM-5, one of the most widely used zeolites, acidity is linked to the Si/Al ratio, with a low ratio indicating high acidity. Pore blockage from polymerization and polycondensation reactions causes deactivation of the catalyst. Zeolites should have correct pore size and acidic sites to promote desired reactions while minimizing carbon formation [22]. A possible mechanism for dehydration of hydroxyl containing compounds (i.e., carbohydrates) on an aluminosilicate zeolite is given in Scheme 5 [22].
Zeolites produce aromatics at atmospheric pressures without H2 requirements. The final product generally has a low heating value. This is due both to its low H/C ratio and high O/C ratio as compared to HDO oils [6,22]. Research is generally conducted at temperatures from 350 to 600 °C. For HZSM-5, yields are in the 15% range with predictions of 23% [41]. Excessive C production and therefore catalyst coking is a problem. In one study, coke deposition at all temperatures led to a decrease in the catalytic activity after only 30 min time on stream [38]. Furthermore, coke has been shown to significantly increase at temperatures above 400 °C [68]. Some coke can be burned off, but irreversible dealumination and loss of acid sites occurs at temperatures as low as 450 °C in the presence of water [69]. Research on the reduction of coking is important, with a variety of approaches showing promise. For example, the recycling of non-condensable gases into a catalytic reactor has the potential to reduce char/coke yields while increasing oil yields [70].
Scheme 5.
Aluminosilicate zeolite dehydration of model compound.

The elemental composition of the fast pyrolysis bio-oil feedstock, as measured by its H/Ceff ratio has been determined to have a large impact on the production of olefins, aromatics, and coke. Experiments have shown that pyrolytic bio-oil feedstocks with a ratio of at least 1.2 or higher perform better in zeolite cracking upgrading [39]. Ten feedstocks were studied over HZSM-5. Yields of olefins and aromatics increased while coke production decreased with increasing H/Ceff ratios. Catalyst life increased as coke yield decreased. This suggests that it may be beneficial to increase the H/Ceff ratio to 1.2 through hydrogenation of the bio-oil feedstock before upgrading with a zeolite catalyst.
Another method of effectively increasing the H/Ceff ratio is to co-pyrolyze a hydrogen donor such as methanol. Horne et al. [71] studied the pyrolysis of wood waste in a dual zone, fluidized bed reactor. Pyrolysis was carried out at 550 °C. Varying amounts of methanol were also injected, and the pyrolysis/methanol gases were passed over a fixed bed of HZSM-5 held at 500 °C. There was an overall increase in hydrocarbon products including alkylated phenols and aromatics. The alkylated products are thought to compete with the production of polyaromatic hydrocarbons (PAHs are carcinogenic compounds and therefore not desirable) and therefore explain the corresponding decrease in 3-, 4-, and 5-ring PAHs. The addition of methanol also increased water formation and decreased CO and CO2 yields, which is in keeping with findings by Chen et al. [72]. However, these observations do not agree with a study by Evans et al. [73] that showed an increase in the CO2/H2O ratio. In all cases, methanol had a synergistic effect on yields, and further research on co-pyrolysis seems justified. Separation of compounds that don’t react well with the particular catalyst being used could also be used in place of increasing the H/Ceff ratio. Gayubo et al. [74] has proposed separating aldehydes and phenols before upgrading over HZSM-5.
Temperature was found to have an impact on product composition with high temperatures producing more light gases. Cheng [38] studied furan conversion to aromatics and olefins using HZSM-5. Products included CO, CO2, allene, C2-C6 olefins, benzene, toluene, styrene, benzofuran, indene, and naphthalene. Varying temperatures favored different final products. At 450 °C, benzofuran and coke formed. At 500–600 °C, aromatics formed. At 650 °C, olefins were produced along with CO and aromatics.
H2O also plays a role in chemical conversions. The conversion of anisole was studied in a quartz tube reactor over HZSM-5 at 400 °C [75]. The presence of controlled quantities of water in the feed was discovered to have a positive effect on catalytic activity. Interestingly, in this study, the deactivation rate of the zeolite was unrelated to the addition of water. Furthermore, H2 was shown to have a positive effect on reducing coke formation, which is affirmed by other studies [76]. This is especially true when a metal function is present [77].
Other zeolites have been studied in comparison to HZSM-5 with mixed results. In one study using a fixed bed micro-reactor operating at 1 bar, 3.6 weight hourly space velocity (WHSV), and 330–410 °C, HZSM-5 was found to yield more hydrocarbons than HY, H-mordenite, silicalite, and other silica-alumina zeolites. HZSM-5 and H-mordenite also showed higher selectivity for aromatics than aliphatics while the other catalysts produced the opposite effect [78]. However, other zeolites do have some advantages. In a similar study, silicalite produced the least coke. Silica-alumina best converted the non-volatile fraction of the bio-oil. HZSM-5 produced the most yield in the gasoline boiling point range while HY and H-mordenite produced less yield and in the kerosene boiling point range [79]. In another study, catalytic pyrolysis of pinewood over zeolites HBeta-25, HY-12, HZSM-5-23, and HMOR-20 was conducted in a fluidized bed reactor at 450 °C [80]. Quartz sand was used for non-catalytic pyrolysis. Oil from each bed was chemically characterized and compared. Overall, ketones and phenols were produced. HZSM-5 produced more ketones and less alcohols and acids than other catalysts. It also produced more liquid, similar in quantity to the quartz bed but containing more water. Mordenite and quartz produced almost no PAHs. One major advantage noted is that the catalysts were regenerated successfully.
There is also the potential to use biomass in a traditional petroleum fluid catalytic cracker (FCC) [81,82]. Corma et al. [83] studied the cracking of glycerol and sorbitol using various FCC catalysts co-fed with vacuum gasoil (a product corresponding to an Arabian Light petroleum product). This work suggests that biomass and petroleum derived oil streams can be co-fed in an industrial, petroleum catalytic reactor producing a desirable final product. Samolada et al. [40] also studied co-feeding of hydrotreated bio-oil in a traditional FCC reactor with promising results. For more extensive information on zeolite upgrading see Lappas et al. [84], Williams et al. [85], Murata et al. [86], and Jae et al. [87]. Table 3 provides a summary of the operating conditions and products for several catalysts that have been investigated for zeolite cracking.
3.4. Catalyst Development
Synthesis of catalysts from renewable materials is an attractive area of active research due to the cost of mining and synthesizing catalysts. Lin et al. [92] studied the successful synthesis of ZSM-5 and ZSM-48 from the ashes of gasified biomass containing 70%–87% amorphous silica. The resulting zeolites showed high silica contents and high crystallinity (>99%). Muradov et al. [93] studied the catalytic effects of duckweed derived biochar in the reforming of CH4-CO2 at 800 °C. Untreated biochar showed low activity and rapidly deactivated. CO2 treated biochar showed higher activity but also deactivated due to carbon deposits from the decomposition of CH4. However, activated carbon has been shown previously in this paper to be an effective support for noble metals, and therefore biomass derived, activated carbon seems promising. Future research in this area is needed, as a self-sustaining system for energy production that includes catalysts would be ideal.
| Catalyst | Temp. (°C) | Feedstock | Catalyst Effects | Ref. |
|---|---|---|---|---|
| HZSM-5 with varying Si/Al2O3 ratios | 500–764 | Kraft Lignin | Decreasing the SiO2/Al2O3 ratio from 200/1 to 25/1 and increasing the catalyst-to-lignin ratio from 1:1 to 20:1 decreased the oxygenates and increased the aromatics. Aromatics yield increased from 500 to 650 °C and then decreased at higher temperatures. Under optimal reaction conditions, the aromatic yields were 2.0% (EHI 0.08) and 5.2% (EHI 0.35). | [88] |
| HZSM-5, Na/ZSM5, HBeta, and HUSY | 650 | Alkaline lignin | H-USY had the largest pore size and lowest Si/Al ratio (7) and had the best liquid yield of 75% and aromatic yield of 40%. | [89] |
| ZSM-5, Al/MCM-41, Al-MSU-F, ZnO, ZrO2, CeO2, Cu2Cr2O5, Criterion-534, alumina-stabilized ceria-MI-575, slate, char and ashes derived from char and biomass | 500 | Cassava rhizome | ZSM-5, Al/MCM-41, Al-MSU-F type, Criterion-534, alumina-stabilized ceria-MI-575, Cu2Cr2O5, and biomass-derived ash were selective to the reduction of most oxygenated lignin derivatives. ZSM-5, Criterion-534, and Al-MSU-F catalysts enhanced the formation of aromatic hydrocarbons and phenols. No single catalyst was found to reduce all carbonyl products but ZSM-5, Criterion-534 and MI-575 could reduce most of the carbonyl products that contained hydroxyl groups. ZSM-5, Criterion-534, Al/MCM-41, Al-MSU-F, copper chromite, char and ashes increased acetic, formic, and lactic acid. MI-575 did not increase acids. | [90] |
| Dolomite | 500–800 | Waste olive husks | Dolomite increased cracking and gas production. | [91] |
| HZSM-5, Al/MCM-41, Al-MSU-F, and alumina-stabilized ceria MI-575, pore sizes 5.5, 31, 15, and NA respectively | 500 | Cassava rhizome | HZSM-5 was the most effective catalyst for the production of aromatic hydrocarbons, phenols, and acetic acid and the reduction of oxygenated lignin-derived compounds and carbonyls containing side chain hydroxyl groups. Only MI-575 showed a decrease in acetic acid yields. MI-575 also showed the most increase in methanol with HZSM-5 a close second. | [81] |
Catalysts can either be mixed with the biomass prior to pyrolysis or separated as to only interact with the gaseous reactants. It was discovered that separation of the catalyst and biomass was more effective for the conversion to desirable products [82]. This study involved a catalyst containing Fe and Cr in their oxide forms, chromite (FeCr2O4) in the combined active phase. It showed promising results in terms of limited water production. Chromite produces hydrogen from water and carbon monoxide in the WGS reaction. The catalyst therefore consumes water produced from deoxygenation through the WGS reaction while producing large quantities of hydrogen. Chromite also showed selectivity in converting the heavy phenolic compounds to phenol and light phenolics.
3.4.1. Multistage Catalysis
Some work has been conducted on combining the benefits of HDO and cracking in one process using multiple stages. Early work by Gagnon et al. [94] showed that a catalytic pretreatment consisting of mild hydrogenation of wood-derived, vacuum pyrolysis oils at 80–140 °C and 41–103 bar over a ruthenium catalyst prevented coking and polymerization in the following HDO runs at a standard temperature of 325 °C and 172 bar. Subsequent studies showed that a non-catalytic, hydrothermal pretreatment followed by catalytic pyrolysis showed an increase in final liquid yield [4]. In combination, these studies lead to the conclusion that a pretreatment step can have positive effects on subsequent HDO runs and should be considered an option in future research.
Vispute et al. [11] combined pretreatment, HDO, and catalytic cracking. The first step involved hydrogenation at 125 °C over a Ru/C catalyst, with no coking or reactor plugging observed. The second stage involved further hydrogenation at 325 °C over a Pt/C catalyst. Finally, olefins and aromatics such as benzene, toluene, xylene, ethylene, propylene, and butylene were produced over a HZSM-5 zeolite catalyst, the first five of which are important building blocks for the chemical industry. Coke yield was very low due to the previous hydrogenation steps. As the H/Ceff ratio increases, the thermal stability of the bio-oil improves, reducing coke formation. Furthermore, an increase in the hydrogen content promotes H2O production and therefore a higher yield of hydrocarbons in the products, as less carbon is lost to CO and CO2 during deoxygenation. Finally, using this reactor configuration allows the catalysts to be regenerated in the fluidized bed reactor of the zeolite upgrading stage. Optimization of each step is required and will depend on the cost of hydrogen and therefore the optimal amount of hydrogenation. Wang et al. [95] studied a similar process with the order of each stage reversed. The final product from HDO of catalytic pyrolysis derived bio-oil and HDO of untreated bio-oil were compared. The former showed higher degrees of deoxygenation. These studies all demonstrate that a deoxygenated feedstock significantly improves downstream processing, regardless of the method of deoxygenation in the initial stage. It is therefore critical to find the optimal combination of stages and reactions that will lead to the highest quality product at the lowest possible cost.
3.4.2. Multifunctional Catalysts
Research on the use of cracking and HDO catalysts used simultaneously or combined to form one multifunctional catalyst has shown promising results. Specifically, zeolites doped with metal catalysts have drawn attention, both for the potential product streams generated and because this line of research sheds light on the mechanisms of cracking and reforming which are still not well understood. Ideally, doped zeolites can be integrated into the pyrolysis equipment, catalyzing the production of the desired products in situ during the initial quenching, thus creating a one-step solids-to-liquids unit which benefits both the quality of the liquid yield and the economic potential of commercial applications [96]. Some research on in situ catalytic upgrading has been undertaken including a study using molecular beam mass spectrometry, a quartz micro reactor, and a HZSM-5 catalyst [73]. Techniques such as this allow real time analysis and rapid screening of a variety of catalysts, operating conditions, and biomass streams at minimal expense.
French et al. [37] studied catalytic pyrolysis of biomass in a tubular quartz micro reactor using a molecular-beam mass spectrometer. Approximately 10 mg of biomass covered with 50–100 mg of catalyst was pyrolyzed at 400–600 °C. Aspen wood, Avicel PH-105 cellulose, and straw lignin were tested in combination with 10 commercial zeolites including HZSM-5, and 22 laboratory-prepared ZSM-5 catalysts with Co, Fe, Ni, Ce, Ga, Cu, and Na substituted for the aluminum and hydrogen components. Four laboratory X and Y zeolites and four different silica and alumina materials were also tested. Ni, Co, Fe, and Ga showed the highest yield of hydrocarbons at 16%. Unfortunately, semi-continuous operation caused partial deactivation of the ZSM-5 catalyst after only 4 min. A similar study was conducted on the cracking of naphtha over Fe/HZSM-5 [97]. Yields of light olefins increased with temperature and a moderate WHSV maximized light olefins as expected. However, the interaction of zeolite and transition metal caused increased olefins yields only up to 6% Fe loading, with further loading catalyzing the production of aromatics. Results such as these clearly indicate the need for further catalyst development. It is also important to note that the way in which a catalyst such as Fe/HZSM-5 is prepared can also have an effect on its reactivity [98]. For more information on fast pyrolysis and upgrading see Butler et al. [99].
While catalytic development work continues to occur, there has only been scattered research on multifunctional and novel catalysts. The research spans a wide range of catalyst combinations, temperatures, and feedstocks in an effort to develop fundamental knowledge on the catalytic effects, processes, and conditions. The resulting body of literature, summarized on Table 4, with regards to multifuncional catalysts is built using information from studies using agronomic, algae and woody biomass feedstocks, as well as fundamental studies utilizing model compounds, including those present in biomass, and upgraded bio-oil products. Provided in Table 4 is a summary of the current research on novel and multifunctional catalysts showing operating conditions and resulting products.
Table 4.
Summary of results from current work on HDO, zeolite, multifunctional, and novel catalysts.
| Catalyst | Temp. (°C) | Feedstock | Catalyst Effects | Ref. |
|---|---|---|---|---|
| Pt/HZSM-5 and HZSM-5 | 400–500 | Canola Oil | Pt/HZSM-5 increased isomerization and hydrogenation, increased gas yields, increased C4 iso/n-hydrocarbon ratio, and lowered organic liquid product (OLP) yield. Steam decreased the OLP yield. | [100] |
| Pretreatment with Na2CO3 | 300–450 | Chlorella algae | Na2CO3 lowered the initial degradation temperature. Catalyst also increased gas yield and decreased liquid yield. Resulting bio-oil had higher heating value, more aromatics, and lower acidity. | [101] |
| ZnCl2 impregnated in biomass | 250–500 | Corn cob, fir wood, bagasse, and rice husk | Enhanced charring and dehydration and promoted production of furfural (FF) and acetic acid (AA). Corn cob gave most FF (8%) at 340 °C with 15% ZnCl2 and a yield of 4% AA compared to a non-catalytic yield of FF 0.49%. | [102] |
| MgO at 5%, 10%, 15%, and 20% of raw material | 550 | Cotton seed | Increasing the amount of catalyst decreased the oil yield and increased the gas and char yields. MgO increased the oil quality by reducing oxygen levels from 9.56% to 4.90% and converting almost all of the long chain alkanes and alkenes to lower molecular weight hydrocarbons in the diesel range. | [103] |
| Boric Oxide mixed with biomass | 400 | Empty palm oil fruit bunch and oil palm fronds | Promoted deoxygenation, eliminated 50%–80% of the hydroxyl and methoxy groups, increased both water and char yields, and decreased gas yields. | [104] |
| Al/MCM-41, Al/MCM-48, HZSM-5, Meso-MFI, Pt/ HZSM-5 (0.5%), Pt/Meso-MFI (0.5%) | 450 | Miscanthus | Al/MCM-41, Al/MCM-48, and Meso-MFI produced more phenolics and reduced more oxygenates than HZSM-5. HZSM-5 and Meso-MFI produced aromatics due to their acidic sites. Meso-MFI zeolite, which has both mesopores and high acidity, performed the best overall. Pt enhanced deoxygenation and aromatization in both cases. | [105] |
| Meso-MFI and Pt/Meso-MFI (0.5%, ion exchanged) | 500 | Waste rice husk | Meso-MFI reduced oxygenates by 38%. Pt/Meso-MFI reduced oxygenates by 49%. Both converted heavy phenols to light phenols and aromatics. | [106] |
| Pt/Hbeta, Pt/SiO2, Hbeta | 400 | Anisole | Pt/Hbeta catalyzed both methyl transfer and hydrodeoxygenation at significantly higher rates than the monofunctional catalysts. Formed benzene, toluene, and xylenes with lower hydrogen consumption and a significant reduction in carbon losses. The rate of deactivation and coke deposition were moderately reduced. | [107] |
| Ga/HZSM-5 | 400–550 | Benzaldehyde | Ga/HZSM-5 catalyzed decarbonylation, producing benzene and CO in the absence of H2. In the presence of H2, it catalyzed toluene production. Addition of water increased benzene and reduced toluene. | [108] |
| Zn/HZSM-5 (0.5 and 1.5%) | 300–500 | Furfural | 1.5% Zn/H-ZSM-5 produced slightly more aromatics (~5%) than 0.5% Zn/HZSM-5. Zn/HZSM-5 catalysts yielded more aromatics and olefins and less furan and coke than HZSM-5. | [109] |
| Ce/HZSM-5 | 600 | Glucose | Increased oxygenated compounds and CO while reducing coke. | [110] |
| Hybrid Pt/HZSM-5 (mixture) and Pt/HZSM-5 | 350–450 | Pyrolysis gasoline | Hybrid Pt/HZSM-5 catalyst showed lower metal-support interaction but a higher catalytic activity. Pt/HZSM-5 increased C2+n-alkanes and decreased methane and hydrogen requirements. | [111] |
| Pd/HY, Pt/HY, Ir/HY, Ni/HY | 350–450 | Pyrolysis gasoline | Ir/HY showed better metal dispersion, acidity, hydrogen adsorption, and metal surface exposure than Pt/HY or Pd/HY. Ni/HY catalyzed less hydrogenation than the other three. Hydrogen pressure helped stabilize the catalysts. | [112] |
| (10%) Pd/C, (30%) Pd/C, Pd(OH)2/C, Pd(OAc)2, Pd-PEPPSI-iPr and Pd/Lindlar with Nafion SAC-13 used in every run. | 300 | Various lignin types and lignin model compounds | Various phenols such as guaiacol, pyrocatechol, and resorcinol were formed from lignin. Model compounds were hydrodeoxygenated, demethylated, and demethoxylated. Percentage yields were better than many other HDO techniques. Activity of catalysts was in the following order; Pd(OAc)2 < Pd-PEPPSI-IPr < Pd(OH)2/C < 10% Pd/C < Pd-Lindlar. | [113] |
| Ni/HZSM-5 (1%) | 450 | Bio-oil from Pinus insignis with 60% methanol | 90% conversion of the bio-oil in the feed with a selectivity for aromatics of 0.4 (benzene, toluene, xylenes (BTX) selectivity of 0.25). Rapid coke deposition was observed. | [114] |
| Ni/Al2O3, Ni/CeO2, and Ni/Al2O3-CeO2 with varying percentages of nickel. | 800 | Cellulose | Initial degradation at lower temp. All reduced tar. 30% Ni/CeO2 catalyst yielded least amount of tar and least CO. 30% Ni/Al2O3 produced maximum amount of H2 (43.5 vol % at 800 °C, 15 min residence time). | [115] |
| Ga/HZSM-5 | 600 | Furan with pinewood sawdust | Depending on preparation, Ga/HZSM-5 increased the rate of aromatics production. Ga seemed to increase the rate of decarbonylation and olefin aromatization, whereas HZSM-5 catalyzed other reactions such as oligomerization. 41% of the energy in the wood was converted into usable products. | [116] |
| NiCl2, HZSM-5, Ni/ZSM-5 | 700 | Kraft Lignin | HZSM-5 almost completely decomposed the aliphatic C-O bonds and carbonyl groups and eliminated 80% of the methoxy groups. It showed more deoxygenation than Ni/ZSM-5. NiCl2 reduced liquid yield while increasing the molecular weight and increasing the gas yield. It produced more aromatic carbons and less aliphatic carbons. | [117] |
| Al/MCM-41, Cu-Al/MCM-41, Fe-Al/MCM-41, Zn-Al/MCM-41 | 500 | Lignocel from beech wood and Miscanthus | Lignocel yielded more hydrocarbons and Miscanthus more phenols. All catalysts produced more phenols. A low Si/Al ratio increased product yields and improved final composition. Fe and Cu containing catalysts produced the most phenols. The presence of Al/MCM-41 reduced oxygenated compounds. Cu/MCM-41 promoted the largest increase of H2 in the gas yield. | [118] |
| 31 catalysts mixed with biomass. Included ZnO, CuO, Fe2O3. | 500 | Pine sawdust | A significant decrease in non-volatile fraction and slight decrease in bio-oil yield were obtained with ZnO (reduced the proportion of heavy fraction in the bio-oil with a limited decrease in its yield), CuO (exhibited the highest yields in semi-volatile compounds), Fe2O3, and mixed oxide catalysts containing Cu and Co. | [119] |
| HFer-20, Fe/HFer-20, HY-12, Fe/HY-12, HBeta-25, Fe/HBeta-25 | 400–450 | Pine wood | Iron modified zeolites increased coke and methyl substituted phenols, decreased methoxy substituted phenols, and didn't affect the CO to CO2 ratio. Beta zeolite was the most active in deoxygenation. All zeolites increased levoglucosan. | [120] |
| K2CO3 or Ca(OH)2 mixed with biomass | 700 | Pine wood | K2CO3 was more active producing no saccharides, aldehydes, or alcohols and substantially reducing the formation of acids, furans, and guaiacols. The yields of alkanes and phenols were increased. Ca(OH)2 reduced char, increased liquid, and increased alcohols, opposing the results from K2CO3. | [121] |
| MgO, CaO, TiO2, Fe2O3, NiO, and ZnO | 600 | Poplar Wood | ZnO showed no activity. CaO reduced heavy products including phenols and anhydrosugars and increased formation of cyclopentanones, hydrocarbons, and light products including acetaldehyde, 2-butanone, and methanol. CaO also reduced acids. Other catalysts were not as effective. Fe2O3 produced PAHs. | [13] |
| Ni/C mixed with biomass | 350 | Pubescens | Produced bio-oil with high content of phenols but low contents of acetic acid, furfural, and water. | [122] |
4. Conclusions
The world currently consumes large quantities of non-renewable hydrocarbons at an ever-increasing rate. The reserves of fossil fuels are limited, and there are many other problems associated with their consumption. Biomass has the potential to be a renewable source of hydrocarbons, which could replace a significant percentage of the world’s fossil fuels in a variety of applications. Currently, several biological and chemical routes utilize biomass as a source of chemicals and fuels. However, thermochemical routes can use a wider variety of feedstock, allowing them to utilize non-food based, lignocellulosic material. Specifically, pyrolysis is touted to be an affective process to convert solid biomass to a liquid product in high yields. However, the composition of bio-oil is such that it cannot currently be used in the existing fossil fuel infrastructure for demanding applications, such as transportation fuels. Catalytic upgrading is touted to be a promising method for converting bio-oil into higher quality fuels and chemicals. However, many challenges must still be addressed.
The inherent complexity of bio-oil makes finding a single, comprehensive upgrading method difficult. This is due both to the lack of understanding in relation to specific compounds and the difficulty in controlling multiple reactions occurring all at once. The oil is difficult to transport, combust, store, or upgrade. Better catalysts could help solve these problems, but most catalytic materials are currently expensive and easily deactivated. Furthermore, HDO requires large quantities of H2 at high pressures and zeolite cracking leads to rapid catalyst deactivation and low quality fuel. Finally, scaling any of these technologies up to the size of a biorefinery will prove challenging. Therefore, future research in several areas is required to move the field forward.
Understanding the reaction mechanisms and complex compounds of pyrolysis and bio-oil will certainly progress technological development. Elucidation of the effects of both indigenous catalysts and other added catalysts is also critical for the future of the biofuels and biochemicals industry, along with the effects of biomass impurities on catalyst activity. Both hydrodeoxygenation and zeolite cracking have been studied under a variety of different reaction and catalytic conditions. However, more research on both methods is paramount with a specific focus on combining the best of both into one catalytic fast pyrolysis unit, either single or multi stage. Efforts to reduce pressure and temperature should be undertaken. Research should also include novel approaches to both methods such as co-pyrolyzation with a hydrogen donor. Rapid screening using a micro reactor as described in this paper could be very effective. Work on multifunctional catalysts such as HZSM-5 doped with noble metal catalysts is also a promising area for further study, along with multiple catalysts used simultaneously. Finally, optimization of industrial scale facilities should be undertaken. This review has summarized the current work in this field in the hopes of providing a base of knowledge for further multifunctional catalyst work.
Acknowledgements
The authors would like to acknowledge U.S.D.A. NIFA McIntire-Stennis (ARK 10-10) and the USDA Special Grant, Wood Utilization Research Grant (2010-02721) for financial support.
References
- Demirbas, A. Global renewable energy resources. Energy Sources Part A Recovery Util. Environ. Eff. 2006, 28, 779–792. [Google Scholar] [CrossRef]
- Huber, G.W.; Iborra, S.; Corma, A. Synthesis of transportation fuels from biomass: Chemistry, catalysts, and engineering. Chem. Rev. 2006, 106, 4044–4098. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Ruiz, J.C.; Dumesic, J.A. Catalytic routes for the conversion of biomass into liquid hydrocarbon transportation fuels. Energy Environ. Sci. 2011, 4, 83–99. [Google Scholar] [CrossRef]
- Stephanidis, S.; Nitsos, C.; Kalogiannis, K.; Iliopoulou, E.F.; Lappas, A.A.; Triantafyllidis, K.S. Catalytic upgrading of lignocellulosic biomass pyrolysis vapours: Effect of hydrothermal pre-treatment of biomass. Catal. Today 2011, 167, 37–45. [Google Scholar] [CrossRef]
- Balat, M.; Balat, M.; Kirtay, E.; Balat, H. Main routes for the thermo-conversion of biomass into fuels and chemicals. Part 1: Pyrolysis systems. Energy Convers. Manag. 2009, 50, 3147–3157. [Google Scholar] [CrossRef]
- Bridgwater, A.; Peacocke, G. Fast pyrolysis processes for biomass. Renew. Sustain. Energy Rev. 2000, 4, 1–73. [Google Scholar] [CrossRef]
- Trimble, J.L.; Vanhook, R.I.; Folger, A.G. Biomass for energy: The environmental-issues. Biomass 1984, 6, 3–13. [Google Scholar] [CrossRef]
- Committee on Economic and Environmental Impacts of Increasing Biofuels Production. Renewable Fuel Standard: Potential Economic and Environmental Effects of U.S. Biofuel Policy; National Academies Press: Washington, DC, USA, 2011. [Google Scholar]
- Demirbas, F.M. Biorefineries for biofuel upgrading: A critical review. Appl. Energy 2009, 86, S151–S161. [Google Scholar] [CrossRef]
- Mohan, D.; Pittman, C.U., Jr.; Steele, P.H. Pyrolysis of wood/biomass for bio-oil: A critical review. Energy Fuels 2006, 20, 848–889. [Google Scholar] [CrossRef]
- Vispute, T.P.; Zhang, H.; Sanna, A.; Xiao, R.; Huber, G.W. Renewable chemical commodity feedstocks from integrated catalytic processing of pyrolysis oils. Science 2010, 330, 1222–1227. [Google Scholar] [CrossRef] [PubMed]
- Pandey, M.P.; Kim, C.S. Lignin depolymerization and conversion: A review of thermochemical methods. Chem. Eng. Technol. 2011, 34, 29–41. [Google Scholar] [CrossRef]
- Lu, Q.; Zhang, Z.-F.; Dong, C.-Q.; Zhu, X.-F. Catalytic upgrading of biomass fast pyrolysis vapors with nano metal oxides: An analytical Py-GC/MS study. Energies 2010, 3, 1805–1820. [Google Scholar] [CrossRef]
- Soria, A.J.; McDonald, A.G.; Shook, S.R. Wood solubilization and depolymerization using supercritical methanol. Part 1: Process optimization and analysis of methanol insoluble components (bio-char). Holzforschung 2008, 62, 402–408. [Google Scholar]
- Soria, J.A. Unlocking hydrocarbons from biomass. Agroborealis 2010, 41, 39–40. [Google Scholar]
- Soria, A.J.; McDonald, A.G.; He, B.B. Wood solubilization and depolymerization by supercritical methanol. Part 2: Analysis of methanol soluble compounds. Holzforschung 2008, 62, 409–416. [Google Scholar]
- Speight, J. Synthetic Fuels Handbook: Properties, Process and Performance; McGraw-Hill Professional: New York, NY, USA, 2008. [Google Scholar]
- Lehmann, J.; Rillig, M.C.; Thies, J.; Masiello, C.A.; Hockaday, W.C.; Crowley, D. Biochar effects on soil biota: A review. Soil Biol. Biochem. 2011, 43, 1812–1836. [Google Scholar] [CrossRef]
- Roberts, K.G.; Gloy, B.A.; Joseph, S.; Scott, N.R.; Lehmann, J. Life cycle assessment of biochar systems: Estimating the energetic, economic, and climate change potential. Environ. Sci. Technol. 2010, 44, 827–833. [Google Scholar] [CrossRef] [PubMed]
- Ozcimen, D.; Ersoy-Mericboyu, A. Characterization of biochar and bio-oil samples obtained from carbonization of various biomass materials. Renew. Energy 2010, 35, 1319–1324. [Google Scholar] [CrossRef]
- Nowakowski, D.J.; Bridgwater, A.V.; Elliott, D.C.; Meier, D.; de Wild, P. Lignin fast pyrolysis: Results from an international collaboration. J. Anal. Appl. Pyrolysis 2010, 88, 53–72. [Google Scholar] [CrossRef]
- Mortensen, P.M.; Grunwaldt, J.D.; Jensen, P.A.; Knudsen, K.G.; Jensen, A.D. A review of catalytic upgrading of bio-oil to engine fuels. Appl. Catal. A Gen. 2011, 407, 1–19. [Google Scholar] [CrossRef]
- Freitas, S.R.; Soria, J.A. Development of a gasification system for utilizing fish processing waste and coastal small diameter wood in rural areas. Energy Fuels 2011, 25, 2292–2300. [Google Scholar] [CrossRef]
- Yaman, S. Pyrolysis of biomass to produce fuels and chemical feedstocks. Energy Conver. Manag. 2004, 45, 651–671. [Google Scholar] [CrossRef]
- Bulushev, D.A.; Ross, J.R.H. Catalysis for conversion of biomass to fuels via pyrolysis and gasification: A review. Catal. Today 2011, 171, 1–13. [Google Scholar] [CrossRef]
- Faix, O.; Meier, D.; Fortmann, I. Thermal-degradation products of wood: A collection of electron-impact (EI) mass-spectra of monomeric lignin derived products. Holz als Roh- und Werkstoff 1990, 48, 351–354. [Google Scholar] [CrossRef]
- Faix, O.; Fortmann, I.; Bremer, J.; Meier, D. Thermal degradation products of wood. Eur. J. Wood Wood Prod. 1991, 49, 213–219. [Google Scholar] [CrossRef]
- Yanik, J.; Kommayer, C.; Saglam, M.; Yueksel, M. Fast pyrolysis of agricultural wastes: Characterization of pyrolysis products. Fuel Process. Technol. 2007, 88, 942–947. [Google Scholar] [CrossRef]
- Branca, C.; Giudicianni, P.; Di Blasi, C. GC/MS characterization of liquids generated from low-temperature pyrolysis of wood. Ind. Eng. Chem. Res. 2003, 42, 3190–3202. [Google Scholar] [CrossRef]
- Valtiner, S.M.; Bonn, G.K.; Huck, C.W. Characterization of different types of hay by solid-phase micro-extraction gas chromatography/mass spectrometry and multivariate data analysis. Phytochem. Anal. 2008, 19, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Ralph, J.; Hatfield, R.D. Pyrolysis-GC-MS characterization of forage materials. J. Agric. Food Chem. 1991, 39, 1426–1437. [Google Scholar] [CrossRef]
- Evans, R.J.; Milne, T.A. Molecular characterization of the pyrolysis of biomass. Energy Fuels 1987, 1, 123–137. [Google Scholar] [CrossRef]
- Marsman, J.H.; Wildschut, J.; Mahfud, F.; Heeres, H.J. Identification of components in fast pyrolysis oil and upgraded products by comprehensive two-dimensional gas chromatography and flame ionization detection. J. Chromatogr. A 2007, 1150, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Sipila, K.; Kuoppala, E.; Fagernas, L.; Oasmaa, A. Characterization of biomass-based flash pyrolysis oils. Biomass Bioenergy 1998, 14, 103–113. [Google Scholar] [CrossRef]
- Oasmaa, A.; Czernik, S. Fuel oil quality of biomass pyrolysis oils—State of the art for the end user. Energy Fuels 1999, 13, 914–921. [Google Scholar] [CrossRef]
- Zhang, Q.; Chang, J.; Wang, T.; Xu, Y. Review of biomass pyrolysis oil properties and upgrading research. Energy Conver. Manag. 2007, 48, 87–92. [Google Scholar] [CrossRef]
- French, R.; Czernik, S. Catalytic pyrolysis of biomass for biofuels production. Fuel Process. Technol. 2010, 91, 25–32. [Google Scholar] [CrossRef]
- Cheng, Y.T.; Huber, G.W. Chemistry of furan conversion into aromatics and olefins over HZSM-5: A model biomass conversion reaction. ACS Catal. 2011, 1, 611–628. [Google Scholar] [CrossRef]
- Zhang, H.; Cheng, Y.T.; Vispute, T.P.; Xiao, R.; Huber, G.W. Catalytic conversion of biomass-derived feedstocks into olefins and aromatics with ZSM-5: The hydrogen to carbon effective ratio. Energy Environ. Sci. 2011, 4, 2297–2307. [Google Scholar] [CrossRef]
- Samolada, M.C.; Baldauf, W.; Vasalos, I.A. Production of a bio-gasoline by upgrading biomass flash pyrolysis liquids via hydrogen processing and catalytic cracking. Fuel 1998, 77, 1667–1675. [Google Scholar] [CrossRef]
- Bridgwater, A.V. Production of high grade fuels and chemicals from catalytic pyrolysis of biomass. Catal. Today 1996, 29, 285–295. [Google Scholar] [CrossRef]
- Sekiguchi, Y.; Shafizadeh, F. The effect of inorganic additives on the formation, composition, and combustion of cellulosic char. J. Appl. Polym. Sci. 1984, 29, 1267–1286. [Google Scholar] [CrossRef]
- Raveendran, K.; Ganesh, A.; Khilar, K.C. Influence of mineral matter on biomass pyrolysis characteristics. Fuel 1995, 74, 1812–1822. [Google Scholar] [CrossRef]
- Muller-Hagedorn, M.; Bockhorn, H.; Krebs, L.; Muller, U. A comparative kinetic study on the pyrolysis of three different wood species. J. Anal. Appl. Pyrolysis 2003, 68–69, 231–249. [Google Scholar] [CrossRef]
- Scott, D.S.; Piskorz, J.; Radlein, D. Liquid products from the continuous flash pyrolysis of biomass. Ind. Eng. Chem. Process Des. Dev. 1985, 24, 581–588. [Google Scholar] [CrossRef]
- Richards, G.N.; Zheng, G.C. Influence of metal-ions and of salts on products from pyrolysis of wood: Applications to thermochemical processing of newsprint and biomass. J. Anal. Appl. Pyrolysis 1991, 21, 133–146. [Google Scholar] [CrossRef]
- Patwardhan, P.R.; Satrio, J.A.; Brown, R.C.; Shanks, B.H. Influence of inorganic salts on the primary pyrolysis products of cellulose. Bioresour. Technol. 2010, 101, 4646–4655. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.B.; Valkenburg, C.; Walton, C.W.; Elliott, D.C.; Holladay, J.E.; Stevens, D.J.; Kinchin, C.; Czernik, S. Production of Gasoline and Diesel From Biomass via Fast Pyrolysis, Hydrotreating and Hydrocracking: A Design Case; Pacific Northwest National Laboratory: Richland, WA, USA, 2009. [Google Scholar]
- Marquevich, M.; Czernik, S.; Chornet, E.; Montane, D. Hydrogen from biomass: Steam reforming of model compounds of fast-pyrolysis oil. Energy Fuels 1999, 13, 1160–1166. [Google Scholar] [CrossRef]
- Grange, P.; Laurent, E.; Maggi, R.; Centeno, A.; Delmon, B. Hydrotreatment of pyrolysis oils from biomass: Reactivity of the various categories of oxygenated compounds and preliminary techno-economical study. Catal. Today 1996, 29, 297–301. [Google Scholar] [CrossRef]
- Elliott, D.C. Historical developments in hydroprocessing bio-oils. Energy Fuels 2007, 21. [Google Scholar] [CrossRef]
- Thangalazhy-Gopakumar, S.; Adhikari, S.; Gupta, R.B.; Tu, M.; Taylor, S. Production of hydrocarbon fuels from biomass using catalytic pyrolysis under helium and hydrogen environments. Bioresour. Technol. 2011, 102, 6742–6749. [Google Scholar] [CrossRef] [PubMed]
- Wildschut, J.; Melian-Cabrera, I.; Heeres, H.J. Catalyst studies on the hydrotreatment of fast pyrolysis oil. Appl. Catal. B Environ. 2010, 99, 298–306. [Google Scholar] [CrossRef]
- Su-Ping, Z.; Yong-Jie, Y.; Zhengwei, R.; Tingchen, L. Study of hydrodeoxygenation of bio-oil from the fast pyrolysis of biomass. Energy Sources 2003, 25, 57–65. [Google Scholar] [CrossRef]
- Elliott, D.C.; Hart, T.R.; Neuenschwander, G.G.; Rotness, L.J.; Zacher, A.H. Catalytic hydroprocessing of biomass fast pyrolysis bio-oil to produce hydrocarbon products. Environ. Prog. Sustain. Energy 2009, 28, 441–449. [Google Scholar] [CrossRef]
- Gutierrez, A.; Kaila, R.K.; Honkela, M.L.; Slioor, R.; Krause, A.O.I. Hydrodeoxygenation of guaiacol on noble metal catalysts. Catal. Today 2009, 147, 239–246. [Google Scholar] [CrossRef]
- Damartzis, T.; Zabaniotou, A. Thermochemical conversion of biomass to second generation biofuels through integrated process design- a review. Renew. Sustain. Energy Rev. 2011, 15, 366–378. [Google Scholar] [CrossRef]
- Wildschut, J.; Mahfud, F.H.; Venderbosch, R.H.; Heeres, H.J. Hydrotreatment of fast pyrolysis oil using heterogeneous noble-metal catalysts. Ind. Eng. Chem. Res. 2009, 48, 10324–10334. [Google Scholar] [CrossRef]
- Xiong, W.-M.; Fu, Y.; Zeng, F.-X.; Guo, Q.-X. An in situ reduction approach for bio-oil hydroprocessing. Fuel Process. Technol. 2011, 92, 1599–1605. [Google Scholar] [CrossRef]
- Kunkes, E.L.; Simonetti, D.A.; West, R.M.; Serrano-Ruiz, J.C.; Gartner, C.A.; Dumesic, J.A. Catalytic conversion of biomass to monofunctional hydrocarbons and targeted liquid-fuel classes. Science 2008, 322, 417–421. [Google Scholar] [CrossRef] [PubMed]
- Panagiotopoulou, P.; Kondarides, D.I. A comparative study of the water-gas shift activity of Pt catalysts supported on single (MOx) and composite (MOx/Al2O3, MOx/TiO2) metal oxide carriers. Catal. Today 2007, 127, 319–329. [Google Scholar] [CrossRef]
- Fisk, C.A.; Morgan, T.; Ji, Y.; Crocker, M.; Crofcheck, C.; Lewis, S.A. Bio-oil upgrading over platinum catalysts using in situ generated hydrogen. Appl. Catal. A Gen. 2009, 358, 150–156. [Google Scholar] [CrossRef]
- Oasmaa, A.; Kuoppala, E.; Ardiyanti, A.; Venderbosch, R.H.; Heeres, H.J. Characterization of hydrotreated fast pyrolysis liquids. Energy Fuels 2010, 24, 5264–5272. [Google Scholar] [CrossRef]
- Baldauf, W.; Balfanz, U.; Rupp, M. Upgrading of flash pyrolysis oil and utilization in refineries. Biomass Bioenergy 1994, 7, 237–244. [Google Scholar] [CrossRef]
- Sheu, Y.H.E.; Anthony, R.G.; Soltes, E.J. Kinetic-studies of upgrading pine pyrolytic oil by hydrotreatment. Fuel Process. Technol. 1988, 19, 31–50. [Google Scholar] [CrossRef]
- Venderbosch, R.H.; Ardiyanti, A.R.; Wildschut, J.; Oasmaa, A.; Heeresb, H.J. Stabilization of biomass-derived pyrolysis oils. J. Chem. Technol. Biotechnol. 2010, 85, 674–686. [Google Scholar] [CrossRef]
- Aho, A.; Kumar, N.; Eranen, K.; Salmi, T.; Hupa, M.; Murzin, D.Y. Catalytic pyrolysis of biomass in a fluidized bed reactor: Influence of the acidity of H-beta zeolite. Process Saf. Environ. Prot. 2007, 85, 473–480. [Google Scholar] [CrossRef]
- Gayubo, A.G.; Aguayo, A.T.; Atutxa, A.; Aguado, R.; Bilbao, J. Transformation of oxygenate components of biomass pyrolysis oil on a HZSM-5 zeolite. I. Alcohols and phenols. Ind. Eng. Chem.Res. 2004, 43, 2610–2618. [Google Scholar] [CrossRef]
- Gayubo, A.G.; Aguayo, A.T.; Atutxa, A.; Prieto, R.; Bilbao, J. Deactivation of a HZSM-5 zeolite catalyst in the transformation of the aqueous fraction of biomass pyrolysis oil into hydrocarbons. Energy Fuels 2004, 18, 1640–1647. [Google Scholar] [CrossRef]
- Mante, O.D.; Agblevor, F.A.; Oyama, S.T.; McClung, R. The influence of recycling non-condensable gases in the fractional catalytic pyrolysis of biomass. Bioresour. Technol. 2012, 111, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Horne, P.A.; Nugranad, N.; Williams, P.T. Catalytic coprocessing of biomass-derived pyrolysis vapors and methanol. J. Anal. Appl. Pyrolysis 1995, 34, 87–108. [Google Scholar] [CrossRef]
- Chen, N.Y.; Walsh, D.E.; Koenig, L.R. Fluidized-bed upgrading of wood pyrolysis liquids and related-compounds. In Pyrolysis Oils from Biomass; Soltes, J., Milne, T.A., Eds.; American Chemical Society: Washington, DC, USA, 1988; pp. 277–289. [Google Scholar]
- Evans, R.J.; Milne, T. Molecular-beam, mass-spectrometric studies of wood vapor and model compounds over an HZSM-5 catalyst. In Pyrolysis Oils from Biomass; Soltes, J., Milne, T.A., Eds.; American Chemical Society: Washington, DC, USA, 1988; pp. 311–327. [Google Scholar]
- Gayubo, A.G.; Aguayo, A.T.; Atutxa, A.; Aguado, R.; Olazar, M.; Bilbao, J. Transformation of oxygenate components of biomass pyrolysis oil on a HZSM-5 zeolite. II. Aldehydes, ketones, and acids. Ind. Eng. Chem. Res. 2004, 43, 2619–2626. [Google Scholar] [CrossRef]
- Zhu, X.; Mallinson, R.G.; Resasco, D.E. Role of transalkylation reactions in the conversion of anisole over HZSM-5. Appl. Catal. A Gen. 2010, 379, 172–181. [Google Scholar] [CrossRef]
- Peralta, M.A.; Sooknoi, T.; Danuthai, T.; Resasco, D.E. Deoxygenation of benzaldehyde over CsNax zeolites. J.Mol. Catal. A Chem. 2009, 312, 78–86. [Google Scholar] [CrossRef]
- Santikunaporn, M.; Herrera, J.E.; Jongpatiwut, S.; Resasco, D.E.; Alvarez, W.E.; Sughrue, E.L. Ring opening of decalin and tetralin on HY and Pt/HY zeolite catalysts. J. Catal. 2004, 228, 100–113. [Google Scholar] [CrossRef]
- Adjaye, J.D.; Bakhshi, N.N. Production of hydrocarbons by catalytic upgrading of a fast pyrolysis bio-oil. Part 1. Conversion over various catalysts. Fuel Process. Technol. 1995, 45, 161–183. [Google Scholar] [CrossRef]
- Adjaye, J.D.; Bakhshi, N.N. Upgrading of a wood-derived oil over various catalysts. Biomass Bioenergy 1994, 7, 201–211. [Google Scholar] [CrossRef]
- Aho, A.; Kumar, N.; Eranen, K.; Salmi, T.; Hupa, M.; Murzin, D.Y. Catalytic pyrolysis of woody biomass in a fluidized bed reactor: Influence of the zeolite structure. Fuel 2008, 87, 2493–2501. [Google Scholar] [CrossRef]
- Pattiya, A.; Titiloye, J.O.; Bridgwater, A.V. Fast pyrolysis of cassava rhizome in the presence of catalysts. J. Anal. Appl. Pyrolysis 2008, 81, 72–79. [Google Scholar] [CrossRef]
- Samolada, M.C.; Papafotica, A.; Vasalos, I.A. Catalyst evaluation for catalytic biomass pyrolysis. Energy Fuels 2000, 14, 1161–1167. [Google Scholar] [CrossRef]
- Corma, A.; Huber, G.W.; Sauvanaud, L.; O’Connor, P. Processing biomass-derived oxygenates in the oil refinery: Catalytic cracking (FCC) reaction pathways and role of catalyst. J. Catal. 2007, 247, 307–327. [Google Scholar] [CrossRef]
- Lappas, A.A.; Samolada, M.C.; Iatridis, D.K.; Voutetakis, S.S.; Vasalos, I.A. Biomass pyrolysis in a circulating fluid bed reactor for the production of fuels and chemicals. Fuel 2002, 81, 2087–2095. [Google Scholar] [CrossRef]
- Williams, P.T.; Nugranad, N. Comparison of products from the pyrolysis and catalytic pyrolysis of rice husks. Energy 2000, 25, 493–513. [Google Scholar] [CrossRef]
- Murata, K.; Liu, Y.; Inaba, M.; Takahara, I. Catalytic fast pyrolysis of jatropha wastes. J. Anal. Appl. Pyrolysis 2012, 94, 75–82. [Google Scholar] [CrossRef]
- Jae, J.; Tompsett, G.A.; Lin, Y.-C.; Carlson, T.R.; Shen, J.; Zhang, T.; Yang, B.; Wyman, C.E.; Conner, W.C.; Huber, G.W. Depolymerization of lignocellulosic biomass to fuel precursors: Maximizing carbon efficiency by combining hydrolysis with pyrolysis. Energy Environ. Sci. 2010, 3, 295–303. [Google Scholar] [CrossRef]
- Li, X.; Su, L.; Wang, Y.; Yu, Y.; Wang, C.; Li, X.; Wang, Z. Catalytic fast pyrolysis of Kraft lignin with HZSM-5 zeolite for producing aromatic hydrocarbons. Front. Environ. Sci. Eng. 2012, 6, 295–303. [Google Scholar] [CrossRef]
- Ma, Z.; Troussard, E.; van Bokhoven, J.A. Controlling the selectivity to chemicals from lignin via catalytic fast pyrolysis. Appl. Catal. A Gen. 2012, 423–424, 130–136. [Google Scholar] [CrossRef]
- Pattiya, A.; Titiloye, J.O.; Bridgwater, A.V. Evaluation of catalytic pyrolysis of cassava rhizome by principal component analysis. Fuel 2010, 89, 244–253. [Google Scholar] [CrossRef]
- Encinar, J.M.; Gonzalez, J.F.; Martinez, G.; Roman, S. Catalytic pyrolysis of exhausted olive oil waste. J. Anal. Appl. Pyrolysis 2009, 85, 197–203. [Google Scholar] [CrossRef]
- Kuen-Song, L.; Wang, H.P.; Chang, N.B.; Jou, C.; Hsiao, M. Synthesis of ZSM-type zeolites from biowaste gasification ashes. Energy Sources 2003, 25, 565–576. [Google Scholar] [CrossRef]
- Muradov, N.; Fidalgo, B.; Gujar, A.C.; Garceau, N.; T-Raissi, A. Production and characterization of lemna minor bio-char and its catalytic application for biogas reforming. Biomass Bioenergy 2012, 42, 123–131. [Google Scholar] [CrossRef]
- Gagnon, J.; Kaliaguine, S. Catalytic hydrotreatment of vacuum pyrolysis oils from wood. Ind. Eng. Chem. Res. 1988, 27, 1783–1788. [Google Scholar] [CrossRef]
- Wang, Y.; He, T.; Liu, K.; Wu, J.; Fang, Y. From biomass to advanced bio-fuel by catalytic pyrolysis/hydro-processing: Hydrodeoxygenation of bio-oil derived from biomass catalytic pyrolysis. Bioresour. Technol. 2012, 108, 280–284. [Google Scholar] [CrossRef] [PubMed]
- Li, H.Y.; Yan, Y.J.; Ren, Z.W. Online upgrading of organic vapors from the fast pyrolysis of biomass. J. Fuel Chem. Technol. 2008, 36, 666–671. [Google Scholar] [CrossRef]
- Alyani, M.; Towfighi, J.; Sadrameli, S.M. Effect of process variables on product yield distribution in thermal catalytic cracking of naphtha to light olefins over Fe/HZSM-5. Korean J. Chem. Eng. 2011, 28, 1351–1358. [Google Scholar] [CrossRef]
- Song, H.; Tan, B.; Ozkan, U.S. Novel synthesis techniques for preparation of Co/CeO2 as ethanol steam reforming catalysts. Catal. Lett. 2009, 132, 422–429. [Google Scholar] [CrossRef]
- Butler, E.; Devlin, G.; Meier, D.; McDonnell, K. A review of recent laboratory research and commercial developments in fast pyrolysis and upgrading. Renew. Sustain. Energy Rev. 2011, 15, 4171–4186. [Google Scholar] [CrossRef]
- Katikaneni, S.P.R.; Adjaye, J.D.; Bakhshi, N.N. Conversion of canola oil to various hydrocarbons over Pt/HZSM-5 bifunctional catalyst. Can. J. Chem. Eng. 1997, 75, 391–401. [Google Scholar] [CrossRef]
- Babich, I.V.; van der Hulst, M.; Lefferts, L.; Moulijn, J.A.; O'Connor, P.; Seshan, K. Catalytic pyrolysis of microalgae to high-quality liquid bio-fuels. Biomass Bioenergy 2011, 35, 3199–3207. [Google Scholar] [CrossRef]
- Lu, Q.; Wang, Z.; Dong, C.-Q.; Zhang, Z.-F.; Zhang, Y.; Yang, Y.-P.; Zhu, X.-F. Selective fast pyrolysis of biomass impregnated with ZnCl2: Furfural production together with acetic acid and activated carbon as by-products. J. Anal. Appl. Pyrolysis 2011, 91, 273–279. [Google Scholar] [CrossRef]
- Putun, E. Catalytic pyrolysis of biomass: Effects of pyrolysis temperature, sweeping gas flow rate and MgO catalyst. Energy 2010, 35, 2761–2766. [Google Scholar] [CrossRef]
- Lim, X.Y.; Andresen, J.M. Pyro-catalytic deoxgenated bio-oil from palm oil empty fruit bunch and fronds with boric oxide in a fixed-bed reactor. Fuel Process. Technol. 2011, 92, 1796–1804. [Google Scholar] [CrossRef]
- Park, H.J.; Park, K.-H.; Jeon, J.-K.; Kim, J.; Ryoo, R.; Jeong, K.-E.; Park, S.H.; Park, Y.-K. Production of phenolics and aromatics by pyrolysis of miscanthus. Fuel 2012, 97, 379–384. [Google Scholar] [CrossRef]
- Jeon, M.-J.; Kim, S.-S.; Jeon, J.-K.; Park, S.H.; Kim, J.M.; Sohn, J.M.; Lee, S.-H.; Park, Y.-K. Catalytic pyrolysis of waste rice husk over mesoporous materials. Nanoscale Res. Lett. 2012, 7, 18:1–18:5. [Google Scholar] [CrossRef]
- Zhu, X.; Lobban, L.L.; Mallinson, R.G.; Resasco, D.E. Bifunctional transalkylation and hydrodeoxygenation of anisole over a Pt/Hbeta catalyst. J. Catal. 2011, 281, 21–29. [Google Scholar] [CrossRef]
- Ausavasukhi, A.; Sooknoi, T.; Resasco, D.E. Catalytic deoxygenation of benzaldehyde over gallium-modified ZSM-5 zeolite. J. Catal. 2009, 268, 68–78. [Google Scholar] [CrossRef]
- Fanchiang, W.-L.; Lin, Y.-C. Catalytic fast pyrolysis of furfural over HZSM-5 and Zn/HZSM-5 catalysts. Appl. Catal. A Gen. 2012, 419–420, 102–110. [Google Scholar] [CrossRef]
- Neumann, G.T.; Hicks, J.C. Effects of cerium and aluminum in cerium-containing hierarchical HZSM-5 catalysts for biomass upgrading. Top. Catal. 2012, 55, 196–208. [Google Scholar] [CrossRef]
- Castano, P.; Gutierrez, A.; Villanueva, I.; Pawelec, B.; Bilbao, J.; Arandes, J.M. Effect of the support acidity on the aromatic ring-opening of pyrolysis gasoline over Pt/HZSM-5 catalysts. Catal. Today 2009, 143, 115–119. [Google Scholar] [CrossRef]
- Castano, P.; Pawelec, B.; Fierro, J.L.G.; Arandes, J.M.; Bilbao, J. Aromatics reduction of pyrolysis gasoline (pygas) over HY-supported transition metal catalysts. Appl. Catal. A Gen. 2006, 315, 101–113. [Google Scholar] [CrossRef]
- Liguori, L.; Barth, T. Palladium-nafion sac-13 catalysed depolymerisation of lignin to phenols in formic acid and water. J. Anal. Appl. Pyrolysis 2011, 92, 477–484. [Google Scholar] [CrossRef]
- Valle, B.; Gayubo, A.G.; Aguayo, A.T.; Olazar, M.; Bilbao, J. Selective production of aromatics by crude bio-oil valorization with a nickel-modified HZSM-5 zeolite catalyst. Energy Fuels 2010, 24, 2060–2070. [Google Scholar] [CrossRef]
- Chattopadhyay, J.; Son, J.E.; Pak, D. Preparation and characterizations of Ni-alumina, Ni-ceria and Ni-alumina/ceria catalysts and their performance in biomass pyrolysis. Korean J. Chem. Eng. 2011, 28, 1677–1683. [Google Scholar] [CrossRef]
- Cheng, Y.-T.; Jae, J.; Shi, J.; Fan, W.; Huber, G.W. Production of renewable aromatic compounds by catalytic fast pyrolysis of lignocellulosic biomass with bifunctional Ga/ZSM-5 catalysts. Angew. Chem. Int. Ed. 2012, 124, 1416–1419. [Google Scholar] [CrossRef]
- Ben, H.; Ragauskas, A.J. Pyrolysis of Kraft lignin with additives. Energy Fuels 2011, 25, 4662–4668. [Google Scholar] [CrossRef]
- Antonakou, E.; Lappas, A.; Nilsen, M.H.; Bouzga, A.; Stocker, M. Evaluation of various types of Al-MCM-41 materials as catalysts in biomass pyrolysis for the production of bio-fuels and chemicals. Fuel 2006, 85, 2202–2212. [Google Scholar] [CrossRef]
- Torri, C.; Reinikainen, M.; Lindfors, C.; Fabbri, D.; Oasmaa, A.; Kuoppala, E. Investigation on catalytic pyrolysis of pine sawdust: Catalyst screening by Py-GC-MIP-AED. J. Anal. Appl. Pyrolysis 2010, 88, 7–13. [Google Scholar] [CrossRef]
- Aho, A.; Kumar, N.; Lashkul, A.V.; Eranen, K.; Ziolek, M.; Decyk, P.; Salmi, T.; Holmbom, B.; Hupa, M.; Murzin, D.Y. Catalytic upgrading of woody biomass derived pyrolysis vapours over iron modified zeolites in a dual-fluidized bed reactor. Fuel 2010, 89, 1992–2000. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, F.; Cao, J.; Wang, J. Pyrolysis of pine wood in a slowly heating fixed-bed reactor: Potassium carbonate versus calcium hydroxide as a catalyst. Fuel Process. Technol. 2010, 91, 942–950. [Google Scholar] [CrossRef]
- Luo, J.; Li, J.; Shen, D.; He, L.; Tong, D.; Hu, C. Catalytic pyrolysis of pubescens to phenols over Ni/C catalyst. Sci. China Chem. 2010, 53, 1487–1491. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Dickerson, T.; Soria, J. Catalytic Fast Pyrolysis: A Review. Energies 2013, 6, 514-538. https://0-doi-org.brum.beds.ac.uk/10.3390/en6010514
AMA Style
Dickerson T, Soria J. Catalytic Fast Pyrolysis: A Review. Energies. 2013; 6(1):514-538. https://0-doi-org.brum.beds.ac.uk/10.3390/en6010514
Chicago/Turabian StyleDickerson, Theodore, and Juan Soria. 2013. "Catalytic Fast Pyrolysis: A Review" Energies 6, no. 1: 514-538. https://0-doi-org.brum.beds.ac.uk/10.3390/en6010514