
Novel Triarylamine-Based Hole Transport Materials: Synthesis, Characterization and Computational Investigation

 , and
, and 
Abstract
:
1. Introduction
2. Experimental
2.1. General
2.2. Computational Methods
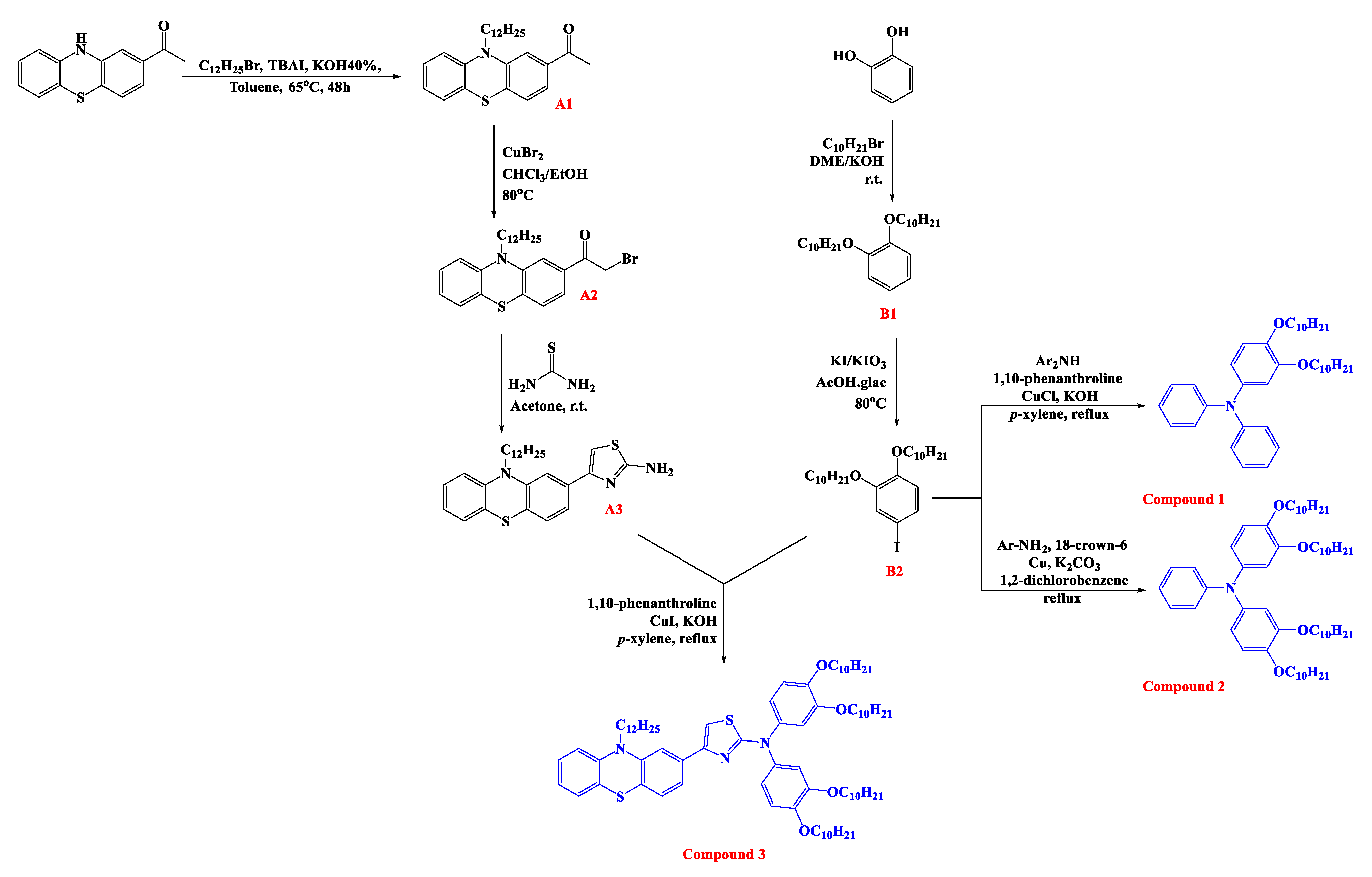
2.3. Synthesis
2.3.1. 1,2-Bis(decyloxy)benzene (B1)
2.3.2. 1,2-Bis(decyloxy)-4-iodobenzene (B2)
2.3.3. 3,4-Bis(decyloxy)-N,N-diphenylaniline (Compound 1)
2.3.4. N-(3,4-Bis(decyloxy)phenyl)-3,4-bis(decyloxy)-N-phenylaniline (Compound 2)
2.3.5. 1-(10-Dodecyl-10H-phenothiazin-2-yl)ethan-1-one (A1)
2.3.6. 2-Bromo-1-(10-dodecyl-10H-phenothiazin-2-yl)ethan-1-one (A2)
2.3.7. 5-(10-Dodecyl-10H-phenothiazin-2-yl)thiazol-2-amine (A3)
2.3.8. N,N-Bis(3,4-bis(decyloxy)phenyl)-5-(10-dodecyl-10H-phenothiazin-2-yl)thiazol-2-amine (Compound 3)
3. Result and Discussion
3.1. Synthesis
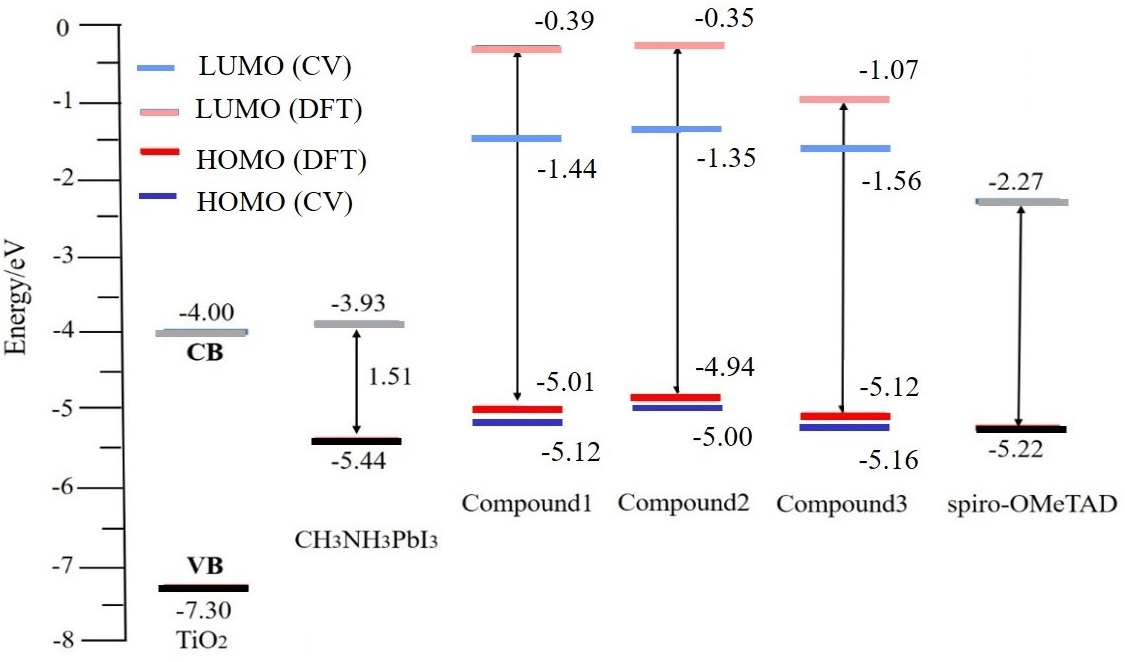
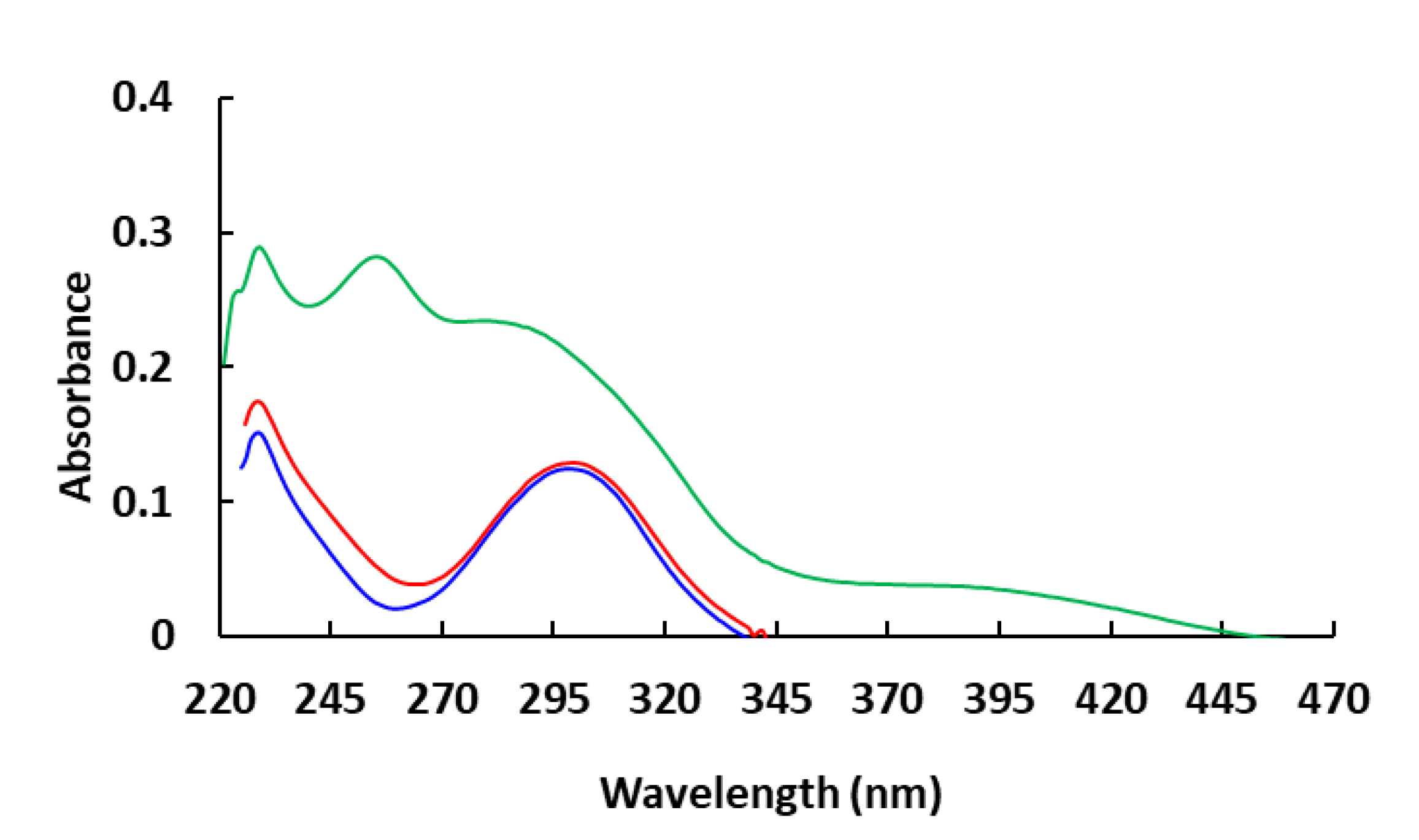
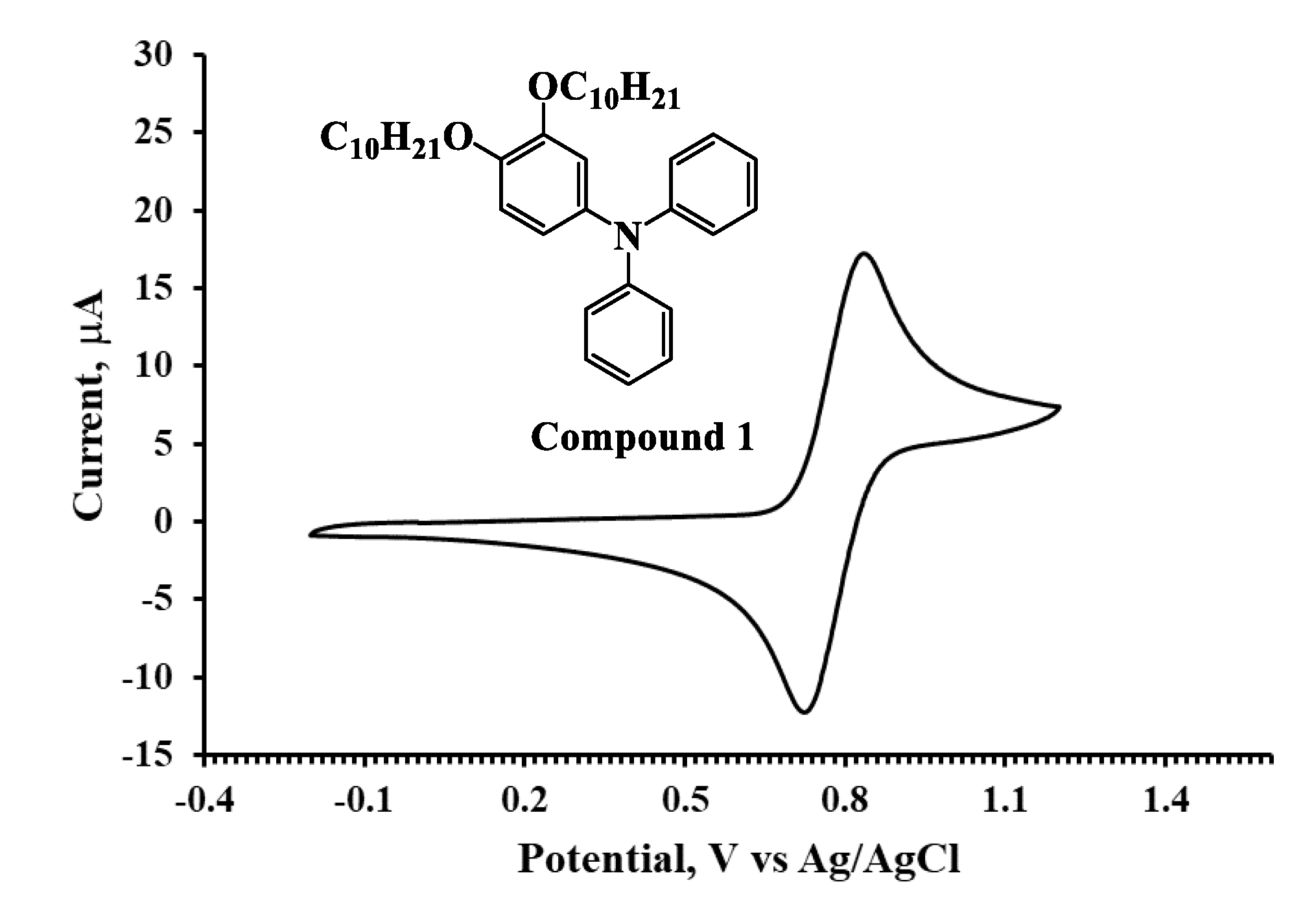
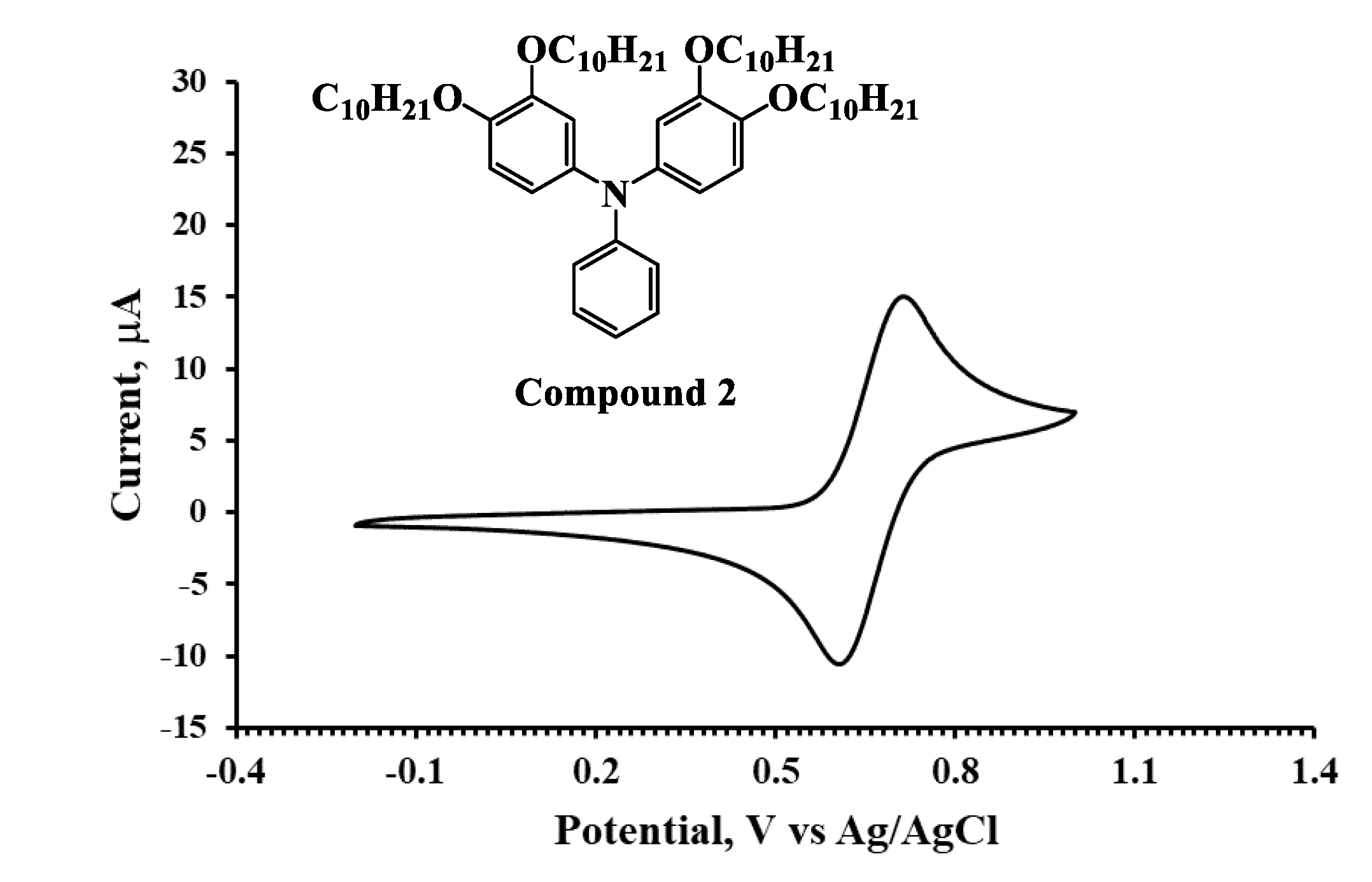
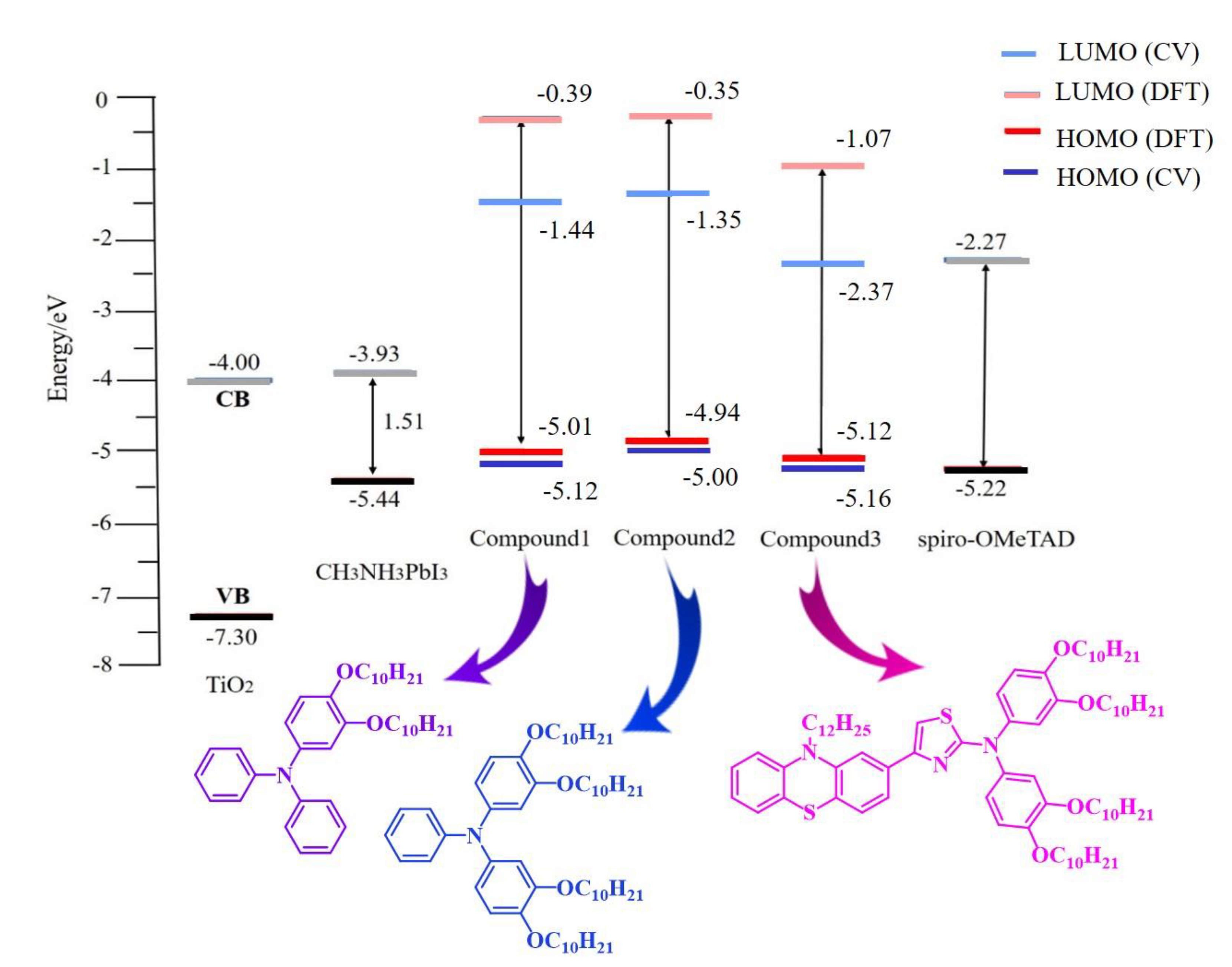
3.2. UV–Vis Absorption and Electrochemical Property
3.3. Computational Investigation
3.3.1. Ground-State Geometries and Frontier Molecular Orbitals
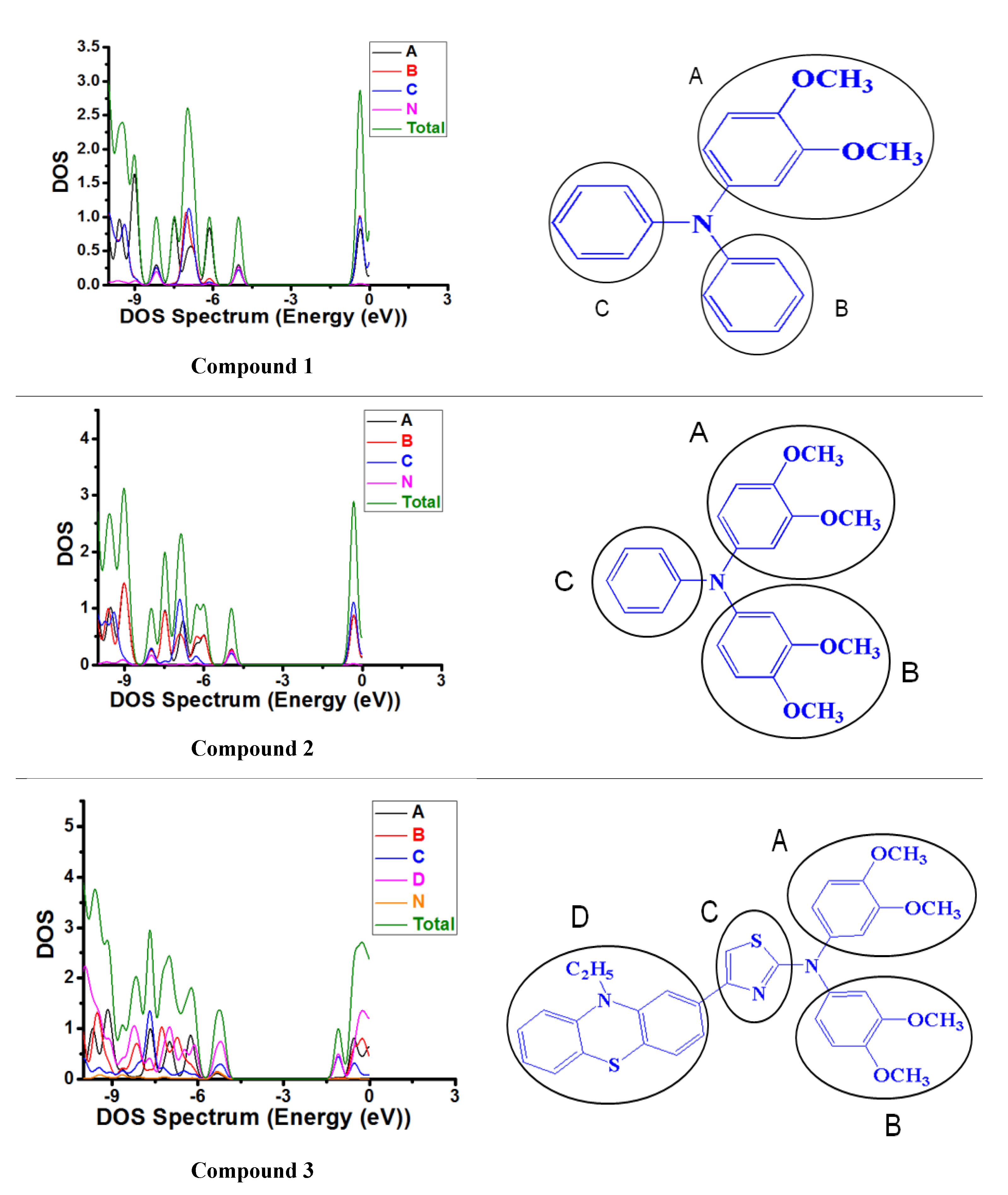
3.3.2. Density of States (DOS) and Frontier Molecular Orbitals
3.3.3. Ionization Potentials (IP), Electron Affinities (EA) and Absolute Hardness
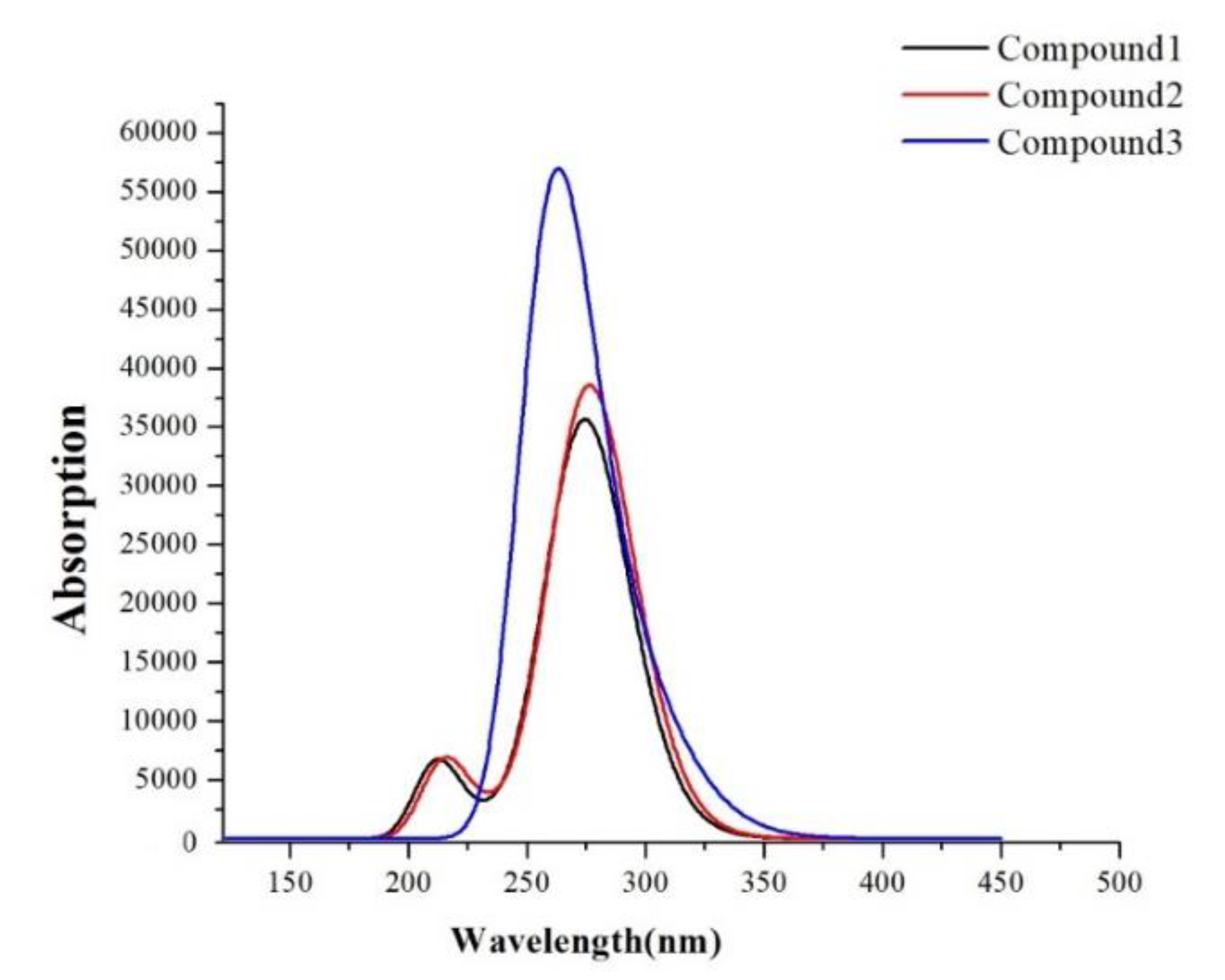
3.3.4. Absorption Spectra
3.3.5. Fluorescence Lifetime
3.3.6. Charge Transport and Hole Mobility
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nhari, L.M.; El-Shishtawy, R.M.; Asiri, A.M. Recent Progress in Organic Hole Transport Materials for Energy Applications. Dye. Pigment. 2021, 109465. [Google Scholar] [CrossRef]
- Dhingra, P.; Singh, P.; Rana, P.J.S.; Garg, A.; Kar, P. Hole-Transporting Materials for Perovskite-Sensitized Solar Cells. Energy Technol. 2016, 4, 891–938. [Google Scholar] [CrossRef]
- Shirota, Y. Organic materials for electronic and optoelectronic devices. J. Mater. Chem. 2000, 10, 1–25. [Google Scholar] [CrossRef]
- Nishimura, H.; Okada, I.; Tanabe, T.; Nakamura, T.; Murdey, R.; Wakamiya, A. Additive-free, Cost-Effective Hole-Transporting Materials for Perovskite Solar Cells Based on Vinyl Triarylamines. ACS Appl. Mater. Interfaces 2020, 12, 32994–33003. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Sun, L. Recent Progress on Hole-Transporting Materials for Emerging Organometal Halide Perovskite Solar Cells. Adv. Energy Mater. 2015, 5, 1500213. [Google Scholar] [CrossRef]
- Sheibani, E.; Yang, L.; Zhang, J. Recent Advances in Organic Hole Transporting Materials for Perovskite Solar Cells. Sol. RRL 2020, 4, 2000461. [Google Scholar] [CrossRef]
- Shariatinia, Z. Recent progress in development of diverse kinds of hole transport materials for the perovskite solar cells: A review. Renew. Sustain. Energy Rev. 2020, 119, 109608. [Google Scholar] [CrossRef]
- Suranagi, S.R.; Singh, R.; Kim, M. Enhancing the power conversion of the perovskite solar cells via structural tuning of BTT(DPP)3-based low bandgap hole transporting material. Dye. Pigment. 2019, 163, 525–532. [Google Scholar] [CrossRef]
- Gao, W.-J.; Yu, H.-J.; Chen, J.; Xiao, J.; Fang, J.-K.; Jia, X.-R.; Peng, C.-F.; Shao, G.; Kuang, D.-B. Simple hole-transporting materials containing twin-carbazole moiety and unconjugated flexible linker for efficient and stable perovskite solar cells. Chem. Eng. J. 2021, 405, 126434. [Google Scholar] [CrossRef]
- Guo, J.; Zhang, Y.; Cai, W.; Zhang, Z.; He, R.; Shen, W.; Li, M. Effect of double bond conjugation on hole mobility of thio-phene-based hole transport materials in perovskite solar cells. Mater. Chem. Phys. 2020, 240, 122058. [Google Scholar] [CrossRef]
- Li, M.; Ma, S.; Mateen, M.; Liu, X.; Ding, Y.; Gao, J.; Yang, Y.; Zhang, X.; Wu, Y.; Dai, S. Facile donor (D)-π-D triphenyla-mine-based hole transporting materials with different π-linker for perovskite solar cells. Sol. Energy 2020, 195, 618–625. [Google Scholar] [CrossRef]
- Azmi, R.; Nam, S.Y.; Sinaga, S.; Akbar, Z.A.; Lee, C.-L.; Yoon, S.C.; Jung, I.H.; Jang, S.-Y. High-performance dopant-free conjugated small molecule-based hole-transport materials for perovskite solar cells. Nano Energy 2018, 44, 191–198. [Google Scholar] [CrossRef]
- Li, M.; Wu, J.; Wang, G.; Wu, B.; Sun, Z.; Xue, S.; Qiao, Q.; Liang, M. The donor-dependent methoxy effects on the perfor-mance of hole-transporting materials for perovskite solar cells. J. Energy Chem. 2020, 47, 10–17. [Google Scholar] [CrossRef]
- Ashassi-Sorkhabi, H.; Salehi-Abar, P. Evaluation of the performance of stilbene-based hole transport materials with an em-phasis on their configuration for use in perovskite solar cells. Sol. Energy 2019, 188, 951–957. [Google Scholar] [CrossRef]
- Ashassi-Sorkhabi, H.; Salehi-Abar, P.; Asghari, E.; Kazempour, A. Structural effect on the thermodynamic and electrochem-ical properties of pyrene-based hole transport materials. J. Mol. Liq. 2019, 285, 338–346. [Google Scholar] [CrossRef]
- Dong, Y.; Zhu, H.; Cao, X.; Han, Y.-P.; Zhang, H.-Y.; Yang, Q.; Zhang, Y.; Zhao, J.; Yin, G.; Wang, S. Simple 9,10-dihydrophenanthrene based hole-transporting materials for efficient perovskite solar cells. Chem. Eng. J. 2020, 402, 126298. [Google Scholar] [CrossRef]
- Ashassi-Sorkhabi, H.; Salehi-Abar, P. Design of two novel hole transport materials via replacing the core of spiro-OMeTAD with tetrathiafulvalene and tetraazafulvalene for application in perovskite solar cells. Sol. Energy 2018, 173, 132–138. [Google Scholar] [CrossRef]
- Kula, S.; Pająk, A.; Szlapa-Kula, A.; Mieszczanin, A.; Gnida, P.; Lipiński, M.; Schab-Balcerzak, E. 9,9′-Bifluorenylidene de-rivatives as novel hole-transporting materials for potential photovoltaic applications. Dye. Pigment. 2020, 174, 108031. [Google Scholar] [CrossRef]
- Dalkılıç, Z.; Lee, C.B.; Choi, H.; Nar, I.; Yavuz, N.K.; Burat, A.K. Tetra and octa substituted Zn(II) and Cu(II) phthalocyanines: Synthesis, characterization and investigation as hole-transporting materials for inverted type-perovskite solar cells. J. Organomet. Chem. 2020, 922, 121419. [Google Scholar] [CrossRef]
- Guo, J.; Sun, M.; Meng, X.; Zhu, H.; Ma, C.; Hu, S.; Shen, J.; Wang, Q.; Gao, J. Impact of peripheral groups on novel asym-metric phthalocyanine-based hole-transporting materials for perovskite solar cells. Dye. Pigment. 2020, 177, 108301. [Google Scholar] [CrossRef]
- Yin, X.; Wang, C.; Zhao, D.; Shrestha, N.; Grice, C.R.; Guan, L.; Song, Z.; Chen, C.; Li, C.; Chi, G.; et al. Binary hole transport materials blending to linearly tune HOMO level for high efficiency and stable perovskite solar cells. Nano Energy 2018, 51, 680–687. [Google Scholar] [CrossRef]
- Luo, D.; Jin, R. Theoretical characterisation and design of D–π–A star-shaped molecules with triphenylamine as core and diketopyrrolopyrroles as arms for organic solar cells. Mol. Phys. 2018, 117, 1825–1832. [Google Scholar] [CrossRef]
- Wang, Q.; Zeng, Z.; Li, Y.; Chen, X. Efficient strategies for improving the performance of EDOT derivatives and TPA deriva-tives-based hole transport materials for perovskite solar cells. Sol. Energy 2020, 208, 10–19. [Google Scholar] [CrossRef]
- Qiu, M.; Pei, W.; Lu, Q.; Li, Z.; Li, Y.; Liang, J. DFT Characteristics of Charge Transport in DBTP-Based Hole Transport Ma-terials. Appl. Sci. 2019, 9, 2244. [Google Scholar] [CrossRef] [Green Version]
- Wazzan, N.; El-Shishtawy, R.M.; Irfan, A. DFT and TD–DFT calculations of the electronic structures and photophysical properties of newly designed pyrene-core arylamine derivatives as hole-transporting materials for perovskite solar cells. Theor. Chem. Acc. 2017, 137, 9. [Google Scholar] [CrossRef]
- El-Shishtawy, R.M. Functional dyes, and some hi-tech applications. Int. J. Photoenergy 2009, 2009, 434897. [Google Scholar] [CrossRef]
- Calió, L.; Kazim, S.; Grätzel, M.; Ahmad, S. Hole-transport materials for perovskite solar cells. Angew. Chem. Int. Ed. 2016, 55, 14522–14545. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Men-nucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, 864–871. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Huang, S.; Zai, J.; Ma, D.; Hu, Z.; He, Q.; Wu, M.; Chen, D.; Chen, Z.; Qian, X. Improving the catalytic performance of Ni 3 S 4 -PtCo heteronanorods via Mott-Schottky effect toward the reduction of iodine couples in dye-sensitized solar cells. Electrochim. Acta 2017, 241, 89–97. [Google Scholar] [CrossRef]
- Adamo, C.; Jacquemin, D. The calculations of excited-state properties with Time-Dependent Density Functional Theory. Chem. Soc. Rev. 2013, 42, 845–856. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef] [Green Version]
- O’Boyle, N.M.; Tenderholt, A.L.; Langner, K.M. cclib: A library for package-independent computational chemistry algorithms. J. Comput. Chem. 2008, 29, 839–845. [Google Scholar] [CrossRef]
- Chattaraj, P.K.; Sarkar, U.; Roy, D.R. Electrophilicity index. Chem. Rev. 2006, 107, 2065–2091. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, L.; Mao, L.; Zhang, J. Carbazole-substituted NP-based derivative as hole transporting material for highly efficient perovskite solar cells. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2020, 228, 117808. [Google Scholar] [CrossRef]
- Ren, P.; Sun, C.; Shi, Y.; Song, P.; Yang, Y.; Li, Y. Global performance evaluation of solar cells using two models: From charge-transfer and recombination mechanisms to photoelectric properties. J. Mater. Chem. C 2019, 7, 1934–1947. [Google Scholar] [CrossRef]
- Köse, M.E.; Mitchell, W.J.; Kopidakis, N.; Chang, C.H.; Shaheen, S.E.; Kim, K.; Rumbles, G. Theoretical studies on conju-gated phenyl-cored thiophene dendrimers for photovoltaic applications. J. Am. Chem. Soc. 2007, 129, 14257–14270. [Google Scholar] [CrossRef]
- Zou, L.Y.; Ren, A.M.; Feng, J.K.; Liu, Y.L.; Ran, X.Q.; Sun, C.C. Theoretical study on photophysical properties of multi-functional electroluminescent molecules with different π-conjugated bridges. J. Phys. Chem. A 2008, 112, 12172–12178. [Google Scholar] [CrossRef]
- Valeev, E.F.; Coropceanu, V.; Filho, D.A.D.S.; Salman, A.S.; Brédas, J.-L. Effect of Electronic Polarization on Charge-Transport Parameters in Molecular Organic Semiconductors. J. Am. Chem. Soc. 2006, 128, 9882–9886. [Google Scholar] [CrossRef]
- Marcus, R.A. Chemical and Electrochemical Electron-Transfer Theory. Annu. Rev. Phys. Chem. 1964, 15, 155–196. [Google Scholar] [CrossRef]
- Bisquert, J. Interpretation of electron diffusion coefficient in organic and inorganic semiconductors with broad distributions of states. Phys. Chem. Chem. Phys. 2008, 10, 3175–3194. [Google Scholar] [CrossRef]
- El-Shishtawy, R.; Borbone, F.; Al-Amshany, Z.M.; Tuzi, A.; Barsella, A.; Asiri, A.M.; Roviello, A. Thiazole azo dyes with lateral donor branch: Synthesis, structure and second order NLO properties. Dye. Pigment. 2013, 96, 45–51. [Google Scholar] [CrossRef]
- Hsieh, C.-M.; Yu, Y.-L.; Chen, C.-P.; Chuang, S.-C. Effects of the additives n-propylammonium or n-butylammonium iodide on the performance of perovskite solar cells. RSC Adv. 2017, 7, 55986–55992. [Google Scholar] [CrossRef]
- El-Shishtawy, R.M.; Decoppet, J.-D.; Al-Zahrani, F.A.; Cao, Y.; Khan, S.B.; Al-Ghamdi, M.S.; Alhogbi, B.G.; Asiri, A.M.; Zakeeruddin, S.M.; Grätzel, M. Influence of redox electrolyte on the device performance of phenothiazine based dye sensi-tized solar cells. New J. Chem. 2018, 42, 9045–9050. [Google Scholar] [CrossRef]
- Xiao, Y.; Han, G.; Chang, Y.; Zhou, H.; Li, M.; Li, Y. An all-solid-state perovskite-sensitized solar cell based on the dual func-tion polyaniline as the sensitizer and p-type hole-transporting material. J. Power Sources 2014, 267, 1–8. [Google Scholar] [CrossRef]
- Oliveira, E.F.; Lavarda, F.C. [Unesp] Effect of the length of alkyl side chains in the electronic structure of conjugated polymers. Mater. Res. 2014, 17, 1369–1374. [Google Scholar] [CrossRef] [Green Version]
- Jin, R.; Wang, K. Rational Design of Diketopyrrolopyrrole-Based Small Moleculesas Donating Materials for Organic Solar Cells. Int. J. Mol. Sci. 2015, 16, 20326–20343. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | Λ a (nm) | Egb (eV) | Eoxc (V) | Eox (Ferrocene) c (V) | HOMO d (eV) | LUMO e (eV) |
|---|---|---|---|---|---|---|
| Compound 1 | 337 | 3.68 | 0.72 | 0.40 | −5.12 | −1.44 |
| Compound 2 | 340 | 3.65 | 0.60 | 0.40 | −5.00 | −1.35 |
| Compound 3 | 445 | 2.79 | 0.76 | 0.40 | −5.16 | −2.37 |
| Molecules | HOMO (eV) | LUMO (eV) | ΔL-H (eV) |
|---|---|---|---|
| Compound 1 | −5.007438148 | −0.387762165 | 4.619675983 |
| Compound 2 | −4.938049129 | −0.353203712 | 4.584845416 |
| Compound 3 | −5.125263423 | −1.071039917 | 4.054223506 |
| Molecules | Compound 1 | Compound 2 | Compound 3 |
|---|---|---|---|
| IP | 4.826496076 | 4.759131584 | 4.920280097 |
| EA | 0.524809559 | 0.515998514 | 1.29218136 |
| η | 2.150843259 | 2.121566535 | 1.814049369 |
| Molecules | State | Energy | λ | f | CI |
|---|---|---|---|---|---|
| Compound 1 | S1 | 4.4652 | 277.67 | 0.2767 | H-L/0.66564 |
| S2 | 4.5111 | 274.84 | 0.3910 | H-L + 1/0.66581 | |
| S3 | 4.5623 | 271.76 | 0.2050 | H-L + 2/0.65691 | |
| S4 | 4.9924 | 248.34 | 0.0315 | H-L + 4/0.48494 | |
| S5 | 5.0173 | 247.11 | 0.0321 | H-L + 3/0.53146 | |
| S6 | 5.8306 | 212.64 | 0.1661 | H-L + 2/0.38636 | |
| Compound 2 | S1 | 4.4577 | 278.14 | 0.3412 | H-L/0.64282 |
| S2 | 4.4771 | 276.93 | 0.4976 | H-L + 1/0.65286 | |
| S3 | 4.5514 | 272.41 | 0.1049 | H-L + 2/0.65475 | |
| S4 | 4.9754 | 249.19 | 0.0358 | H-L + 3/0.44687 | |
| S5 | 4.9942 | 248.26 | 0.0271 | H-L + 4/0.42356 | |
| S6 | 5.7443 | 215.84 | 0.1691 | H − 1-L + 2/0.50837 | |
| Compound 3 | S1 | 4.0959 | 302.71 | 0.1619 | H-L/0.57662 |
| S2 | 4.3452 | 285.33 | 0.1596 | H − 1-L/0.48943 | |
| S3 | 4.5086 | 274.99 | 0.0480 | H-L + 3/0.39172 | |
| S4 | 4.6656 | 265.74 | 0.1675 | H-L + 1/0.43774 | |
| S5 | 4.7392 | 261.61 | 0.8023 | H − 1-L + 2/0.36926 | |
| S6 | 4.7891 | 258.89 | 0.3343 | H-L + 5/0.42187 |
| Molecules | State | Energy | λ | f | CI | τ1 |
|---|---|---|---|---|---|---|
| Compound 1 | S1 | 3.8032 | 326.00 | 0.2075 | H-L/0.69026 | 7.6785 |
| Compound 2 | S1 | 3.8016 | 326.14 | 0.2227 | H-L/0.68822 | 7.1604 |
| Compound 3 | S1 | 2.9393 | 421.82 | 0.0355 | H-L/0.64191 | 75.141 |
| Molecules | Compound 1 | Compound 2 | Compound 3 |
|---|---|---|---|
| λh | 0.483489079 | 0.495456644 | 0.435409291 |
| λe | 0.319682013 | 0.375718408 | 0.531043686 |
| Molecules | Compound 1 | Compound 2 | Compound 3 |
|---|---|---|---|
| r | 5.36846 | 4.87962 | 4.90008 |
| Vh | 0.00217691 | 0.06802845 | 0.024898413 |
| Ve | 0.001496626 | 0.030204632 | 0.02938829 |
| kh (×1011) | 0.0106386 | 9.14143 | 2.33463 |
| ke (×1011) | 0.0301456 | 6.58764 | 1.16805 |
| µh | 0.000059301 | 0.042098 | 0.010842 |
| µe | 0.00016803 | 0.030337 | 0.0054243 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nhari, L.M.; El-Shishtawy, R.M.; Lu, Q.; Li, Y.; Asiri, A.M. Novel Triarylamine-Based Hole Transport Materials: Synthesis, Characterization and Computational Investigation. Materials 2021, 14, 3128. https://0-doi-org.brum.beds.ac.uk/10.3390/ma14113128
Nhari LM, El-Shishtawy RM, Lu Q, Li Y, Asiri AM. Novel Triarylamine-Based Hole Transport Materials: Synthesis, Characterization and Computational Investigation. Materials. 2021; 14(11):3128. https://0-doi-org.brum.beds.ac.uk/10.3390/ma14113128
Chicago/Turabian StyleNhari, Laila M., Reda M. El-Shishtawy, Qiuchen Lu, Yuanzuo Li, and Abdullah M. Asiri. 2021. "Novel Triarylamine-Based Hole Transport Materials: Synthesis, Characterization and Computational Investigation" Materials 14, no. 11: 3128. https://0-doi-org.brum.beds.ac.uk/10.3390/ma14113128