
Modified Mesoporous Carbon Material (Pb-N-CMK-3) Obtained by a Hard-Templating Route, Dicyandiamide Impregnation and Electrochemical Lead Particles Deposition as an Electrode Material for the U(VI) Ultratrace Determination

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Apparatus
2.2. Reagents and Solutions
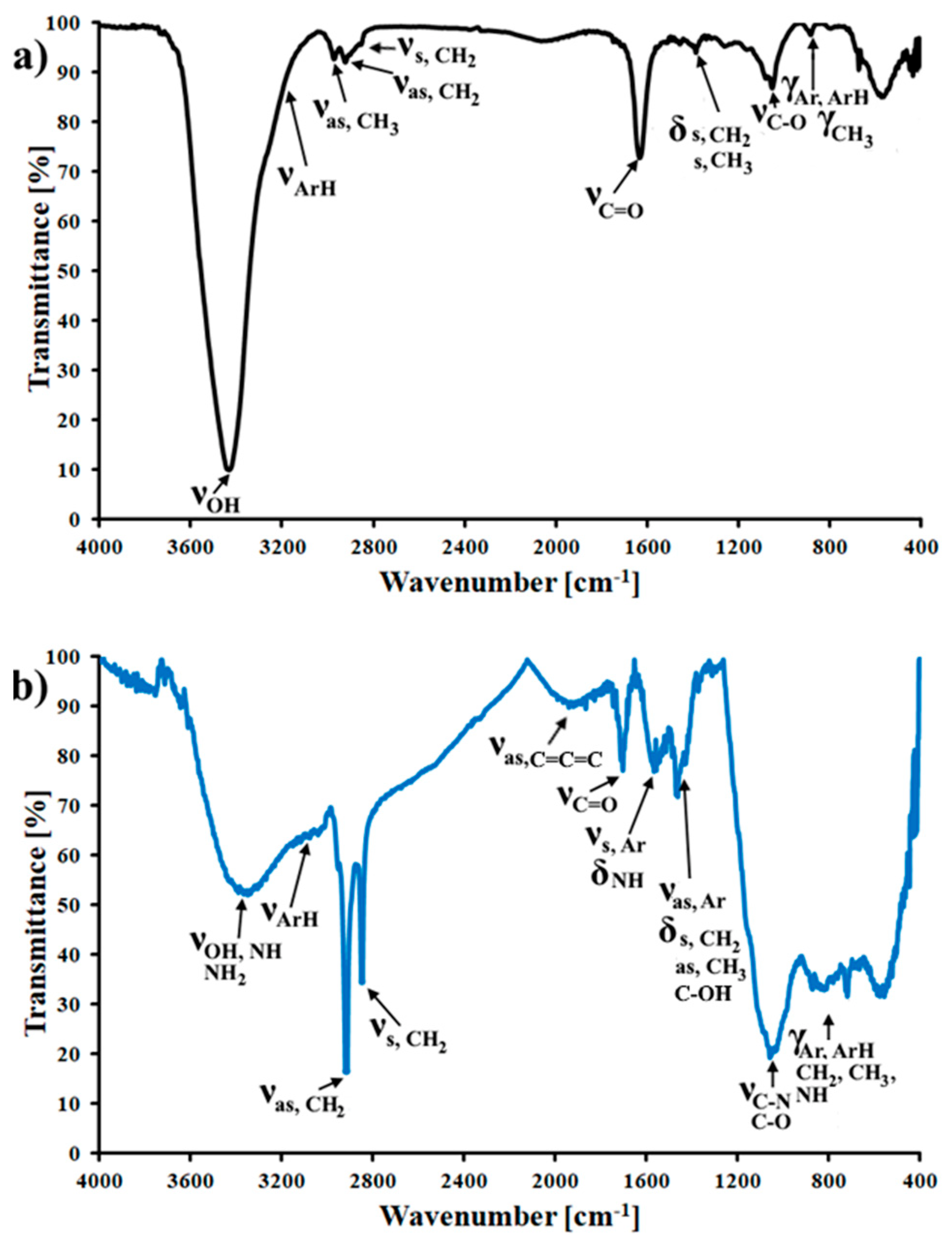
2.3. Synthesis of N-CMK-3
2.4. Preparation of Pb-N-CMK-3/GCE and U(VI) Analysis
3. Results and Discussion
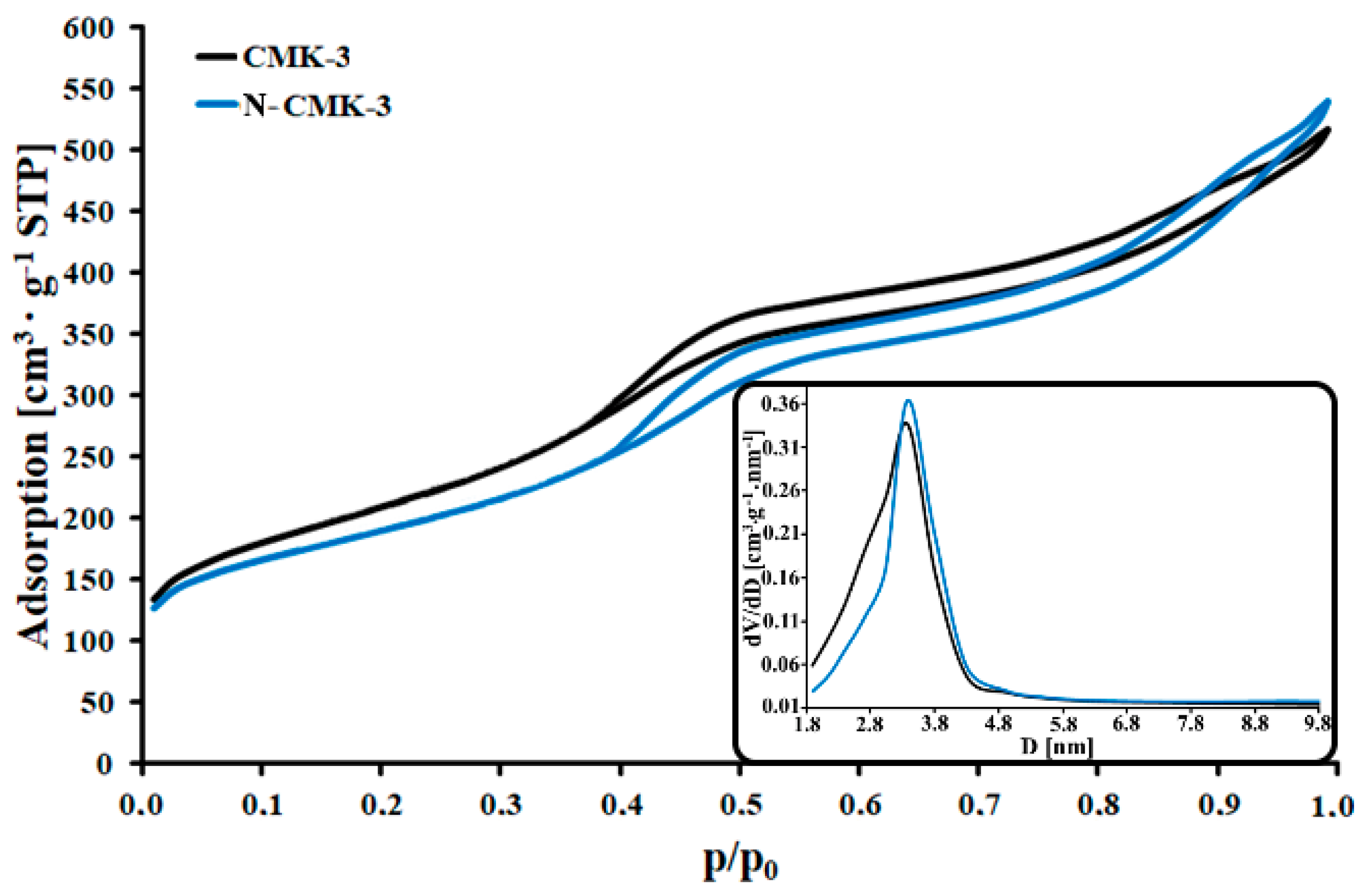
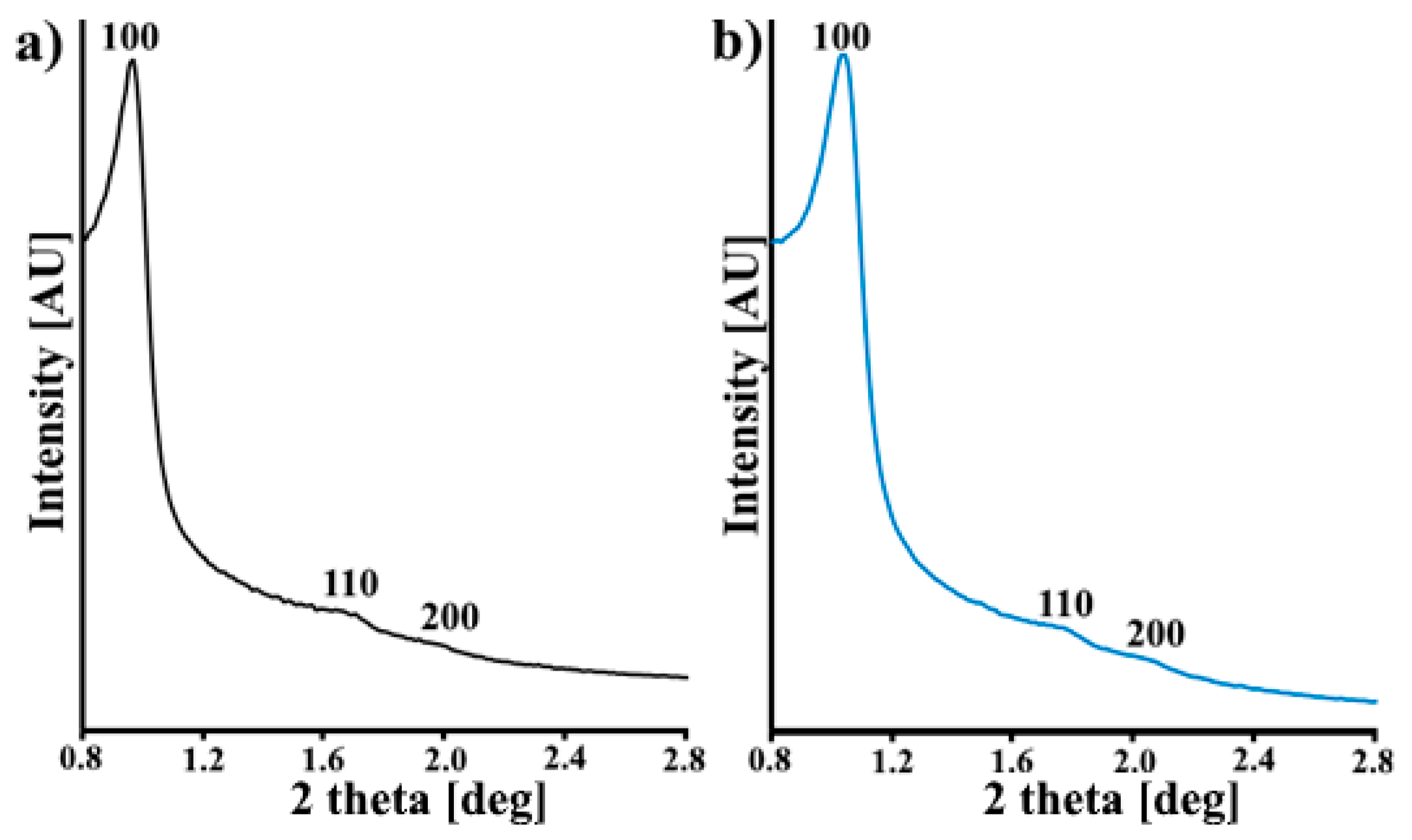
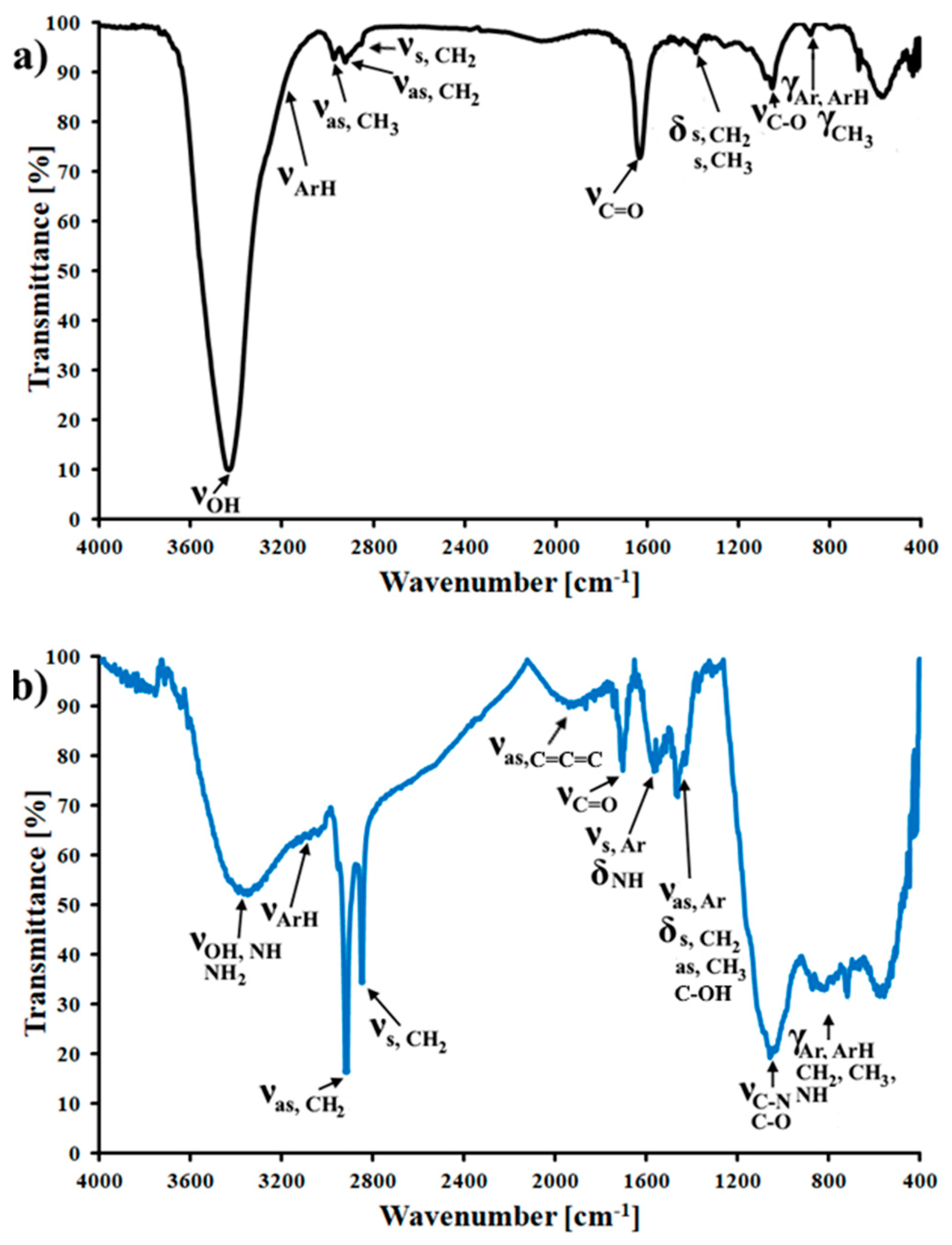
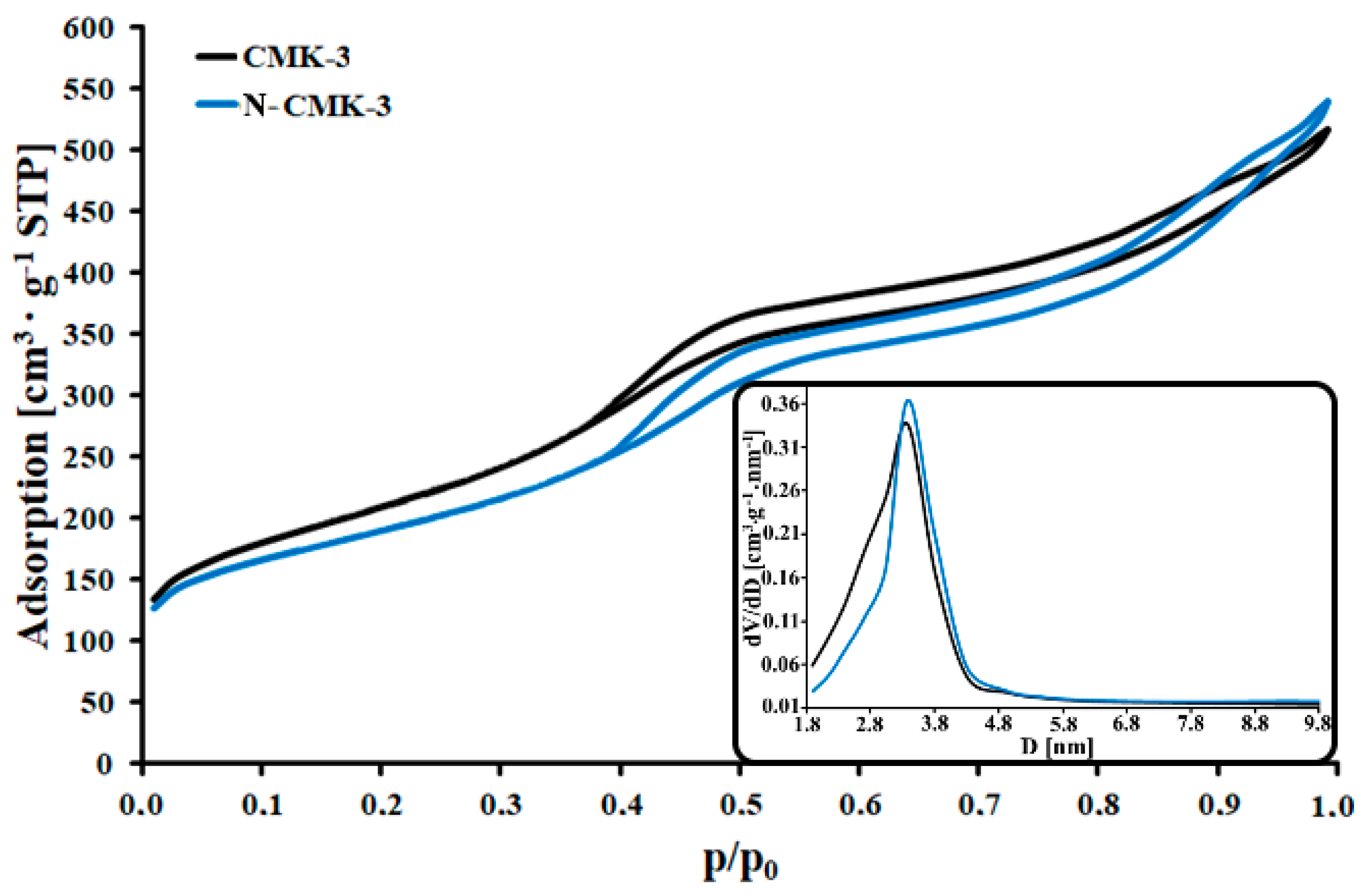
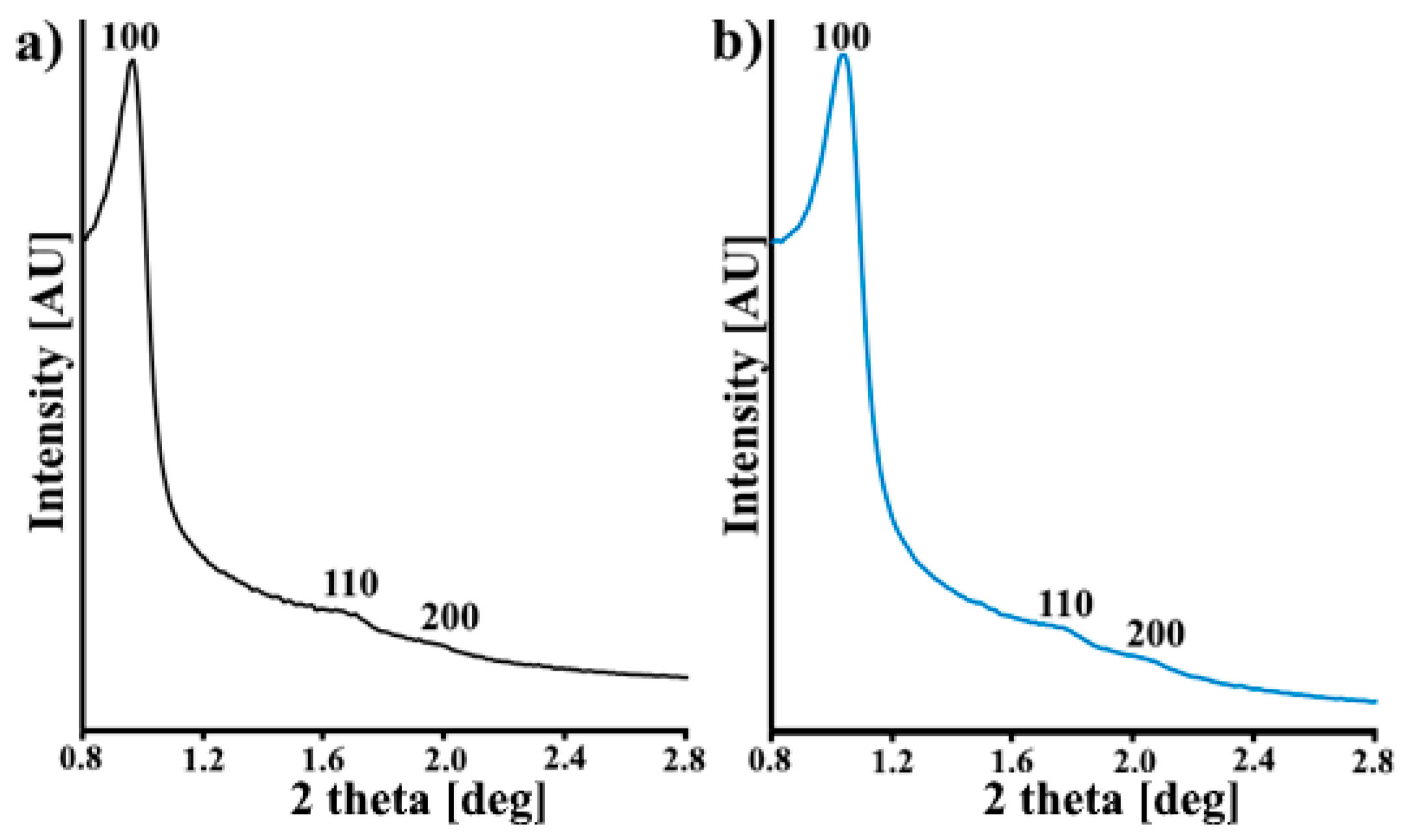
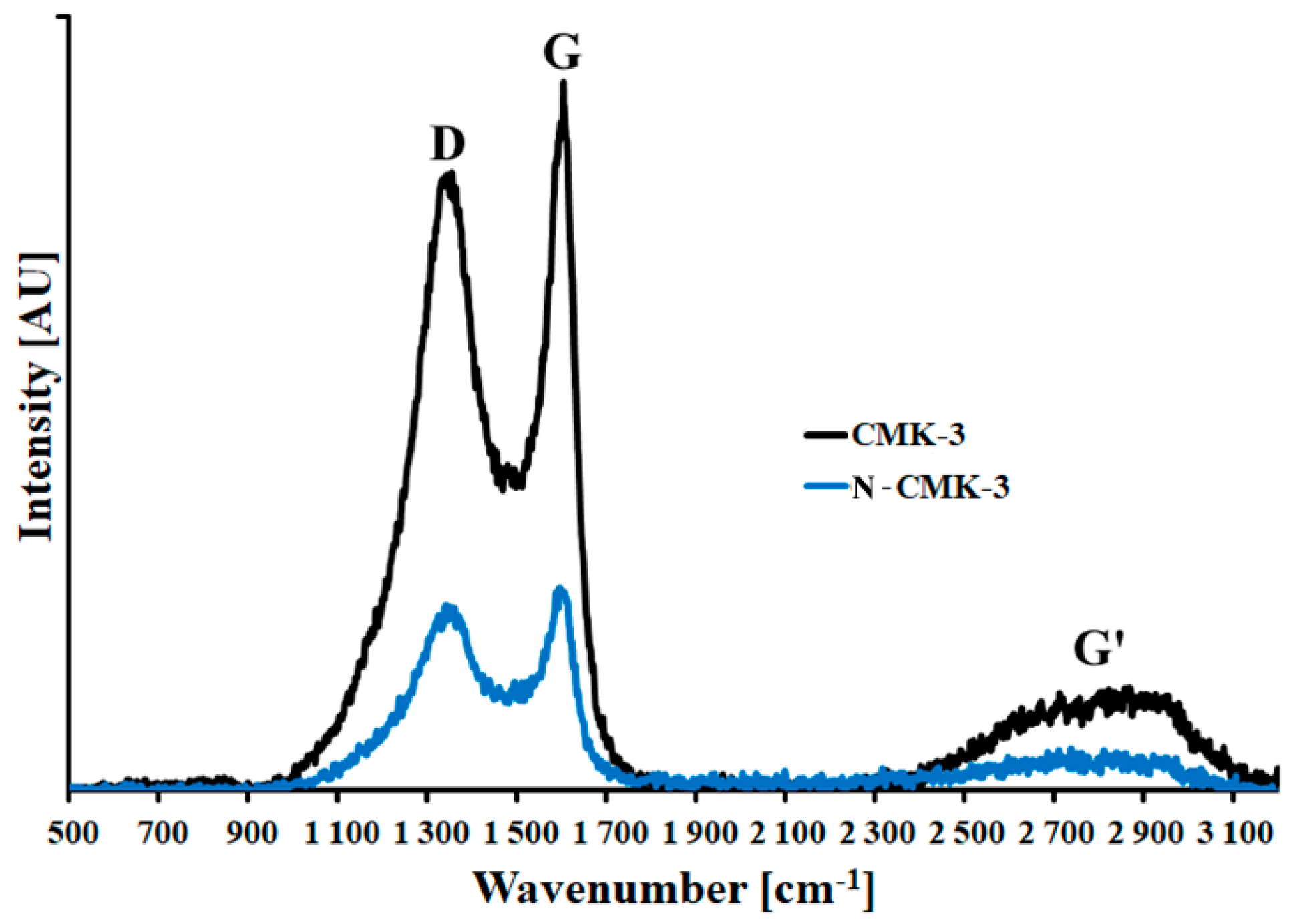
3.1. Characterization of the CMK-3 and N-CMK-3
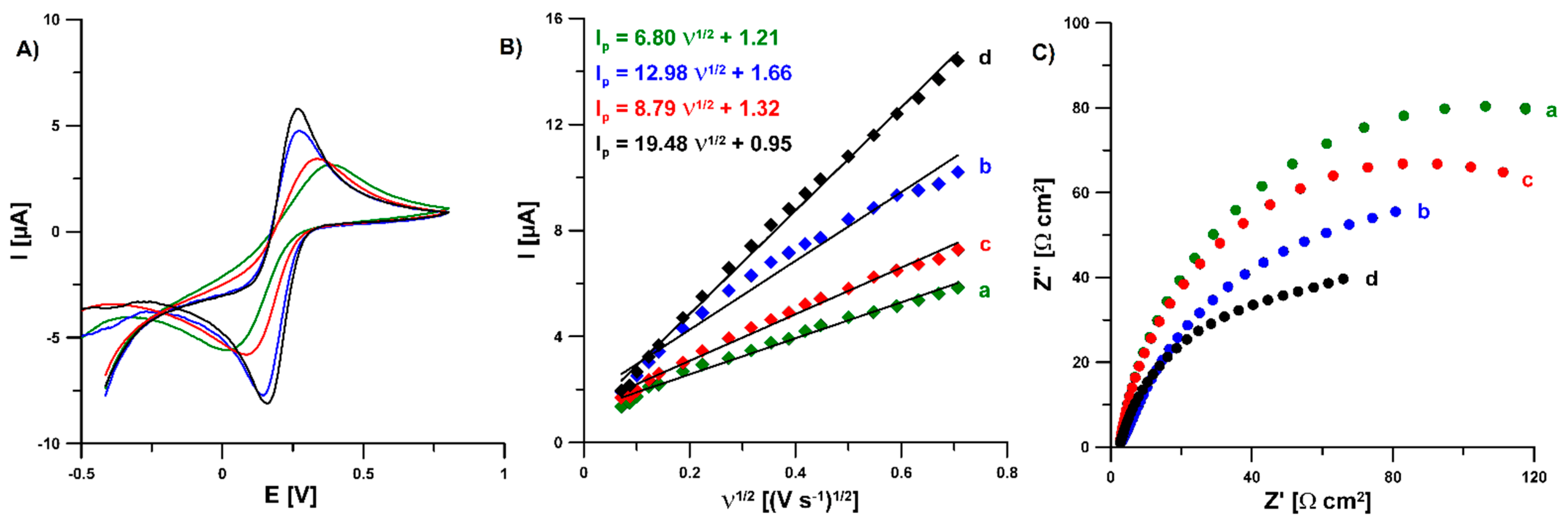
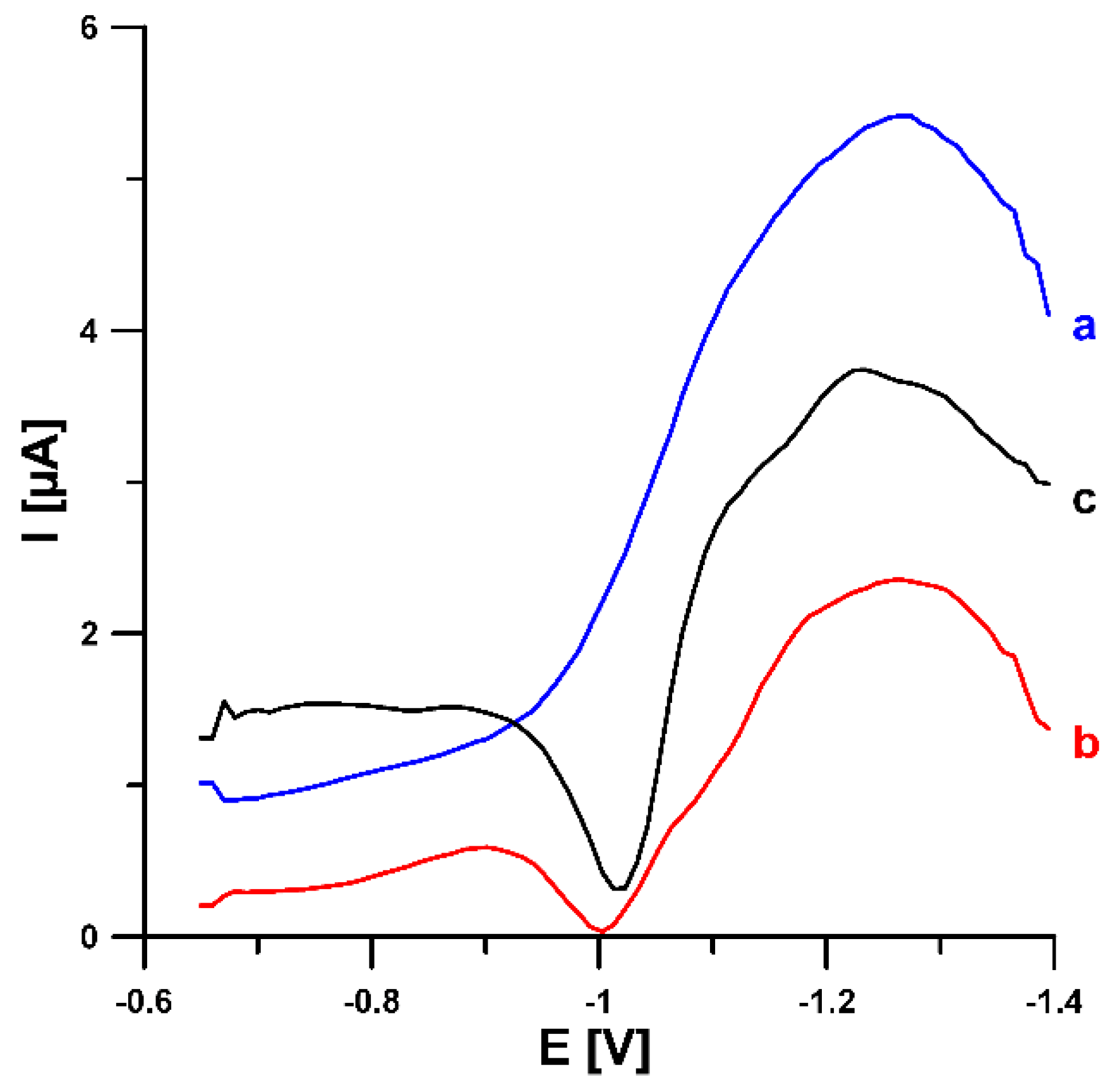
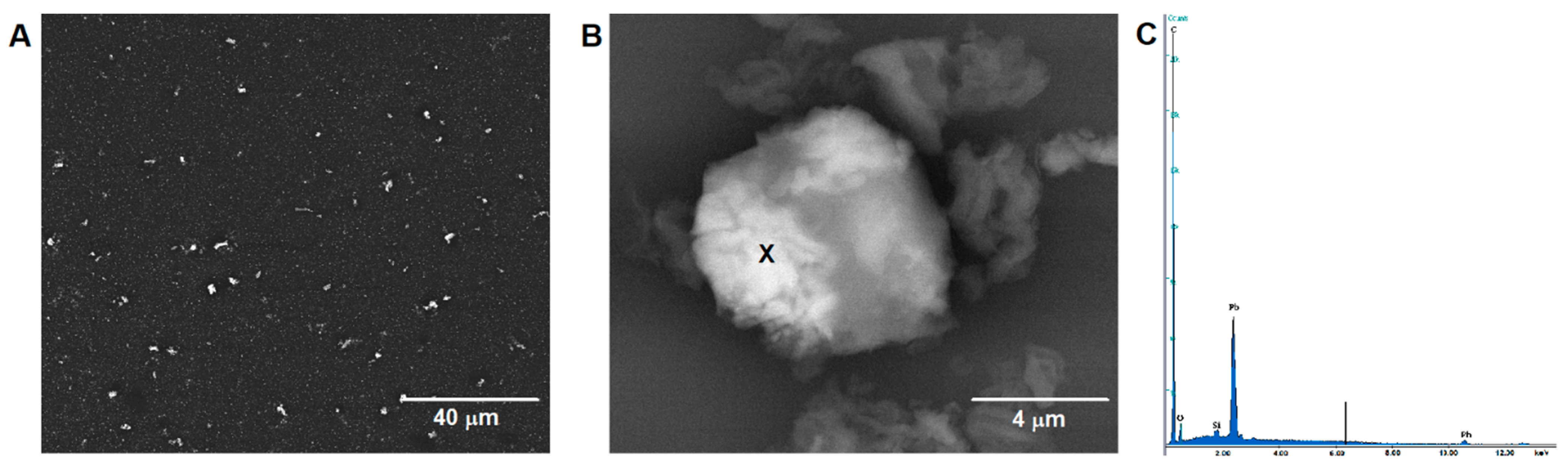
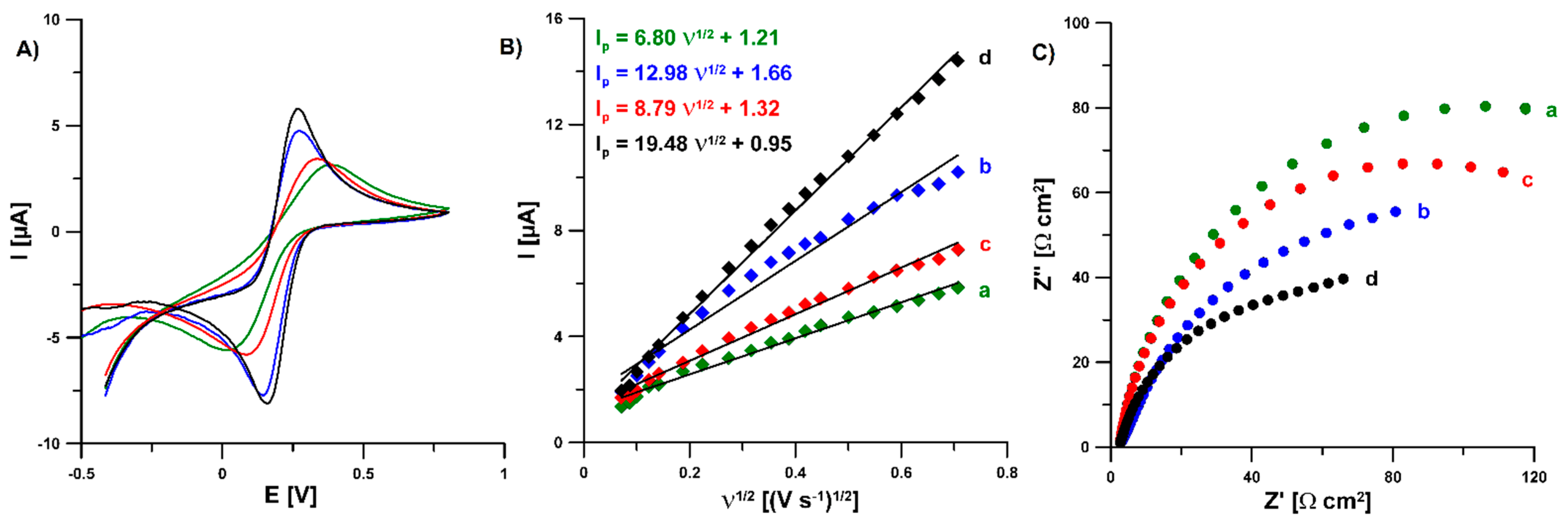
3.2. Characterization of the N-CMK-3/GCE and Pb-N-CMK/GCE
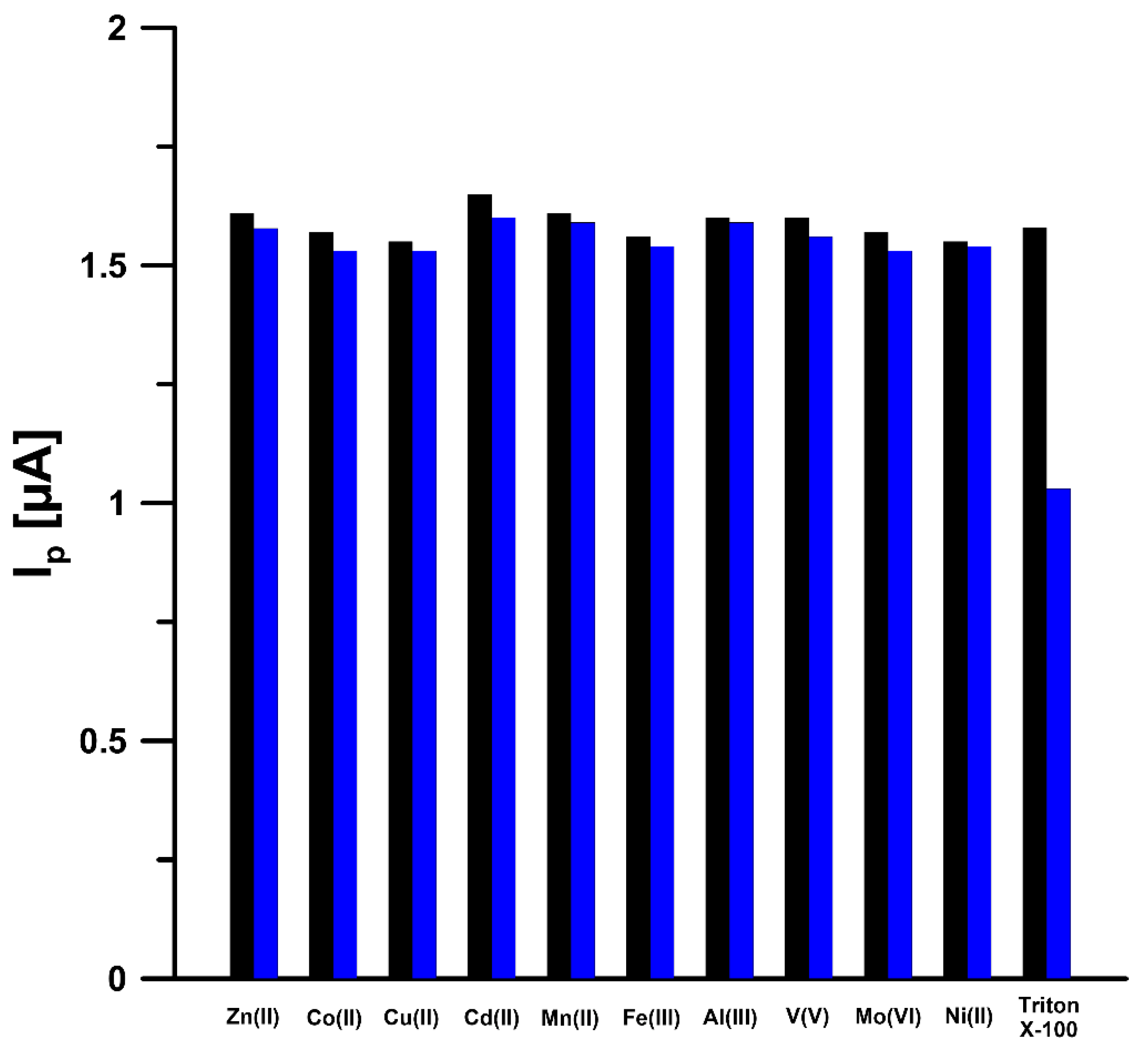
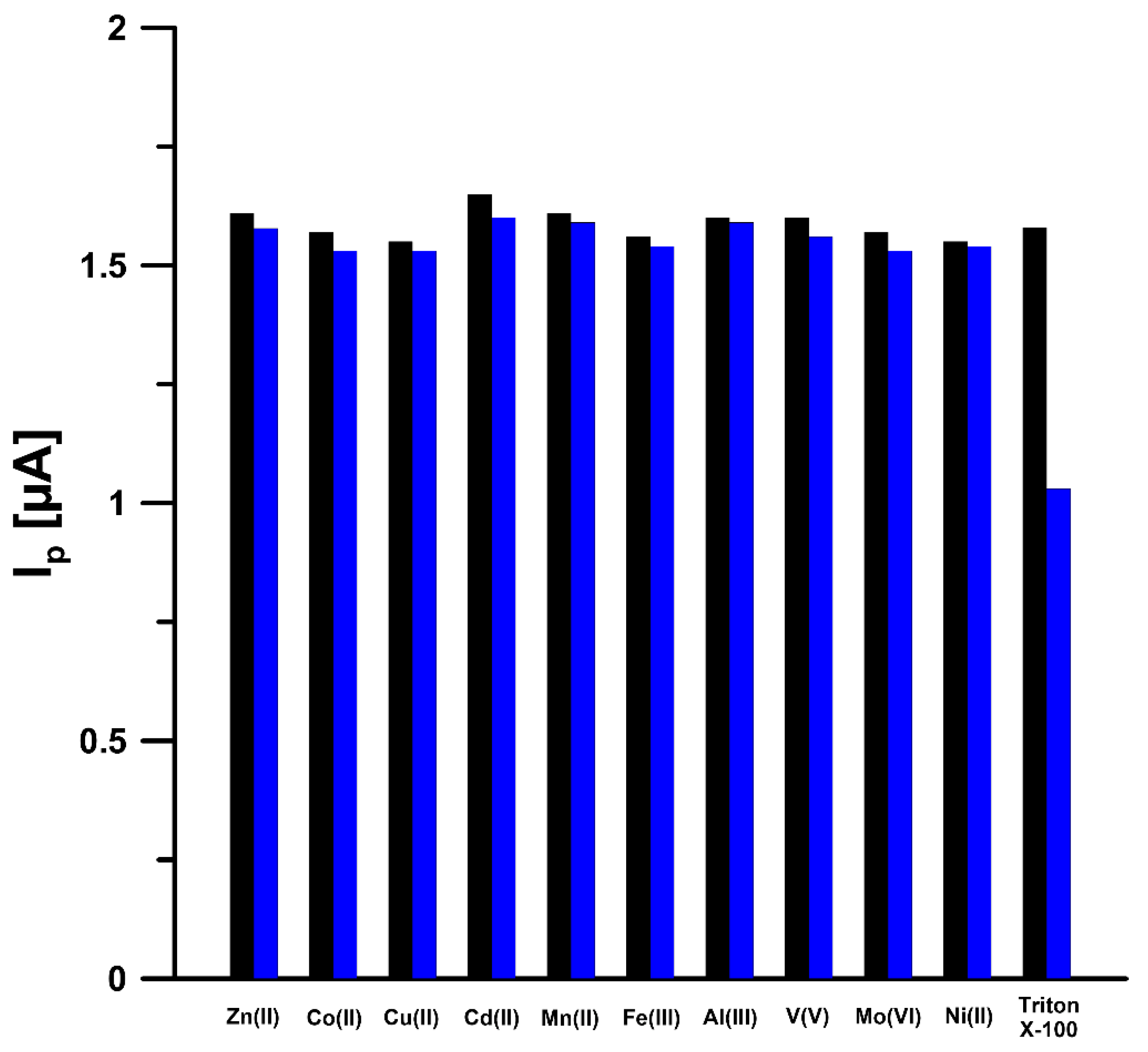
3.3. Optimization of Experimental Conditions
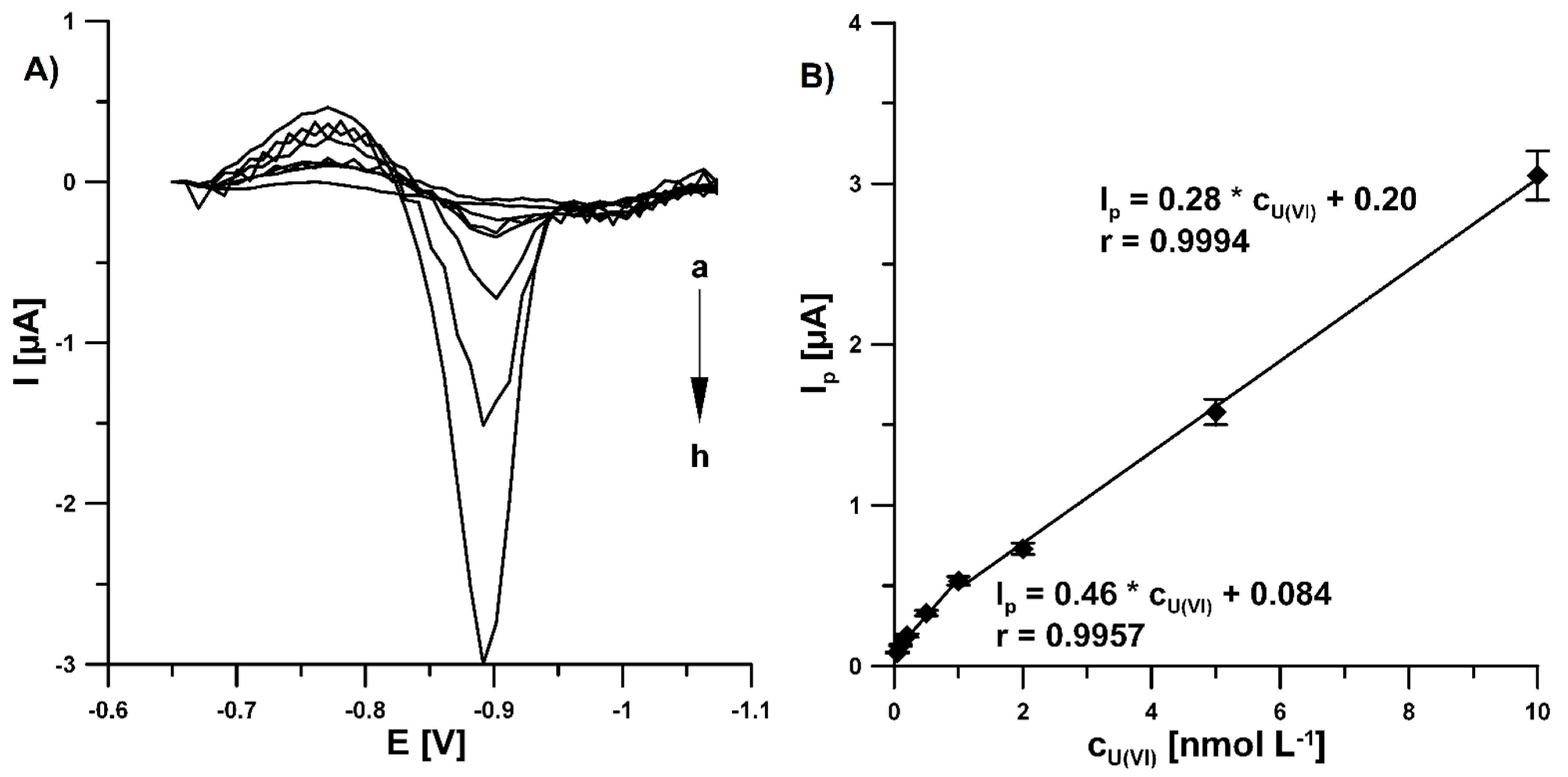
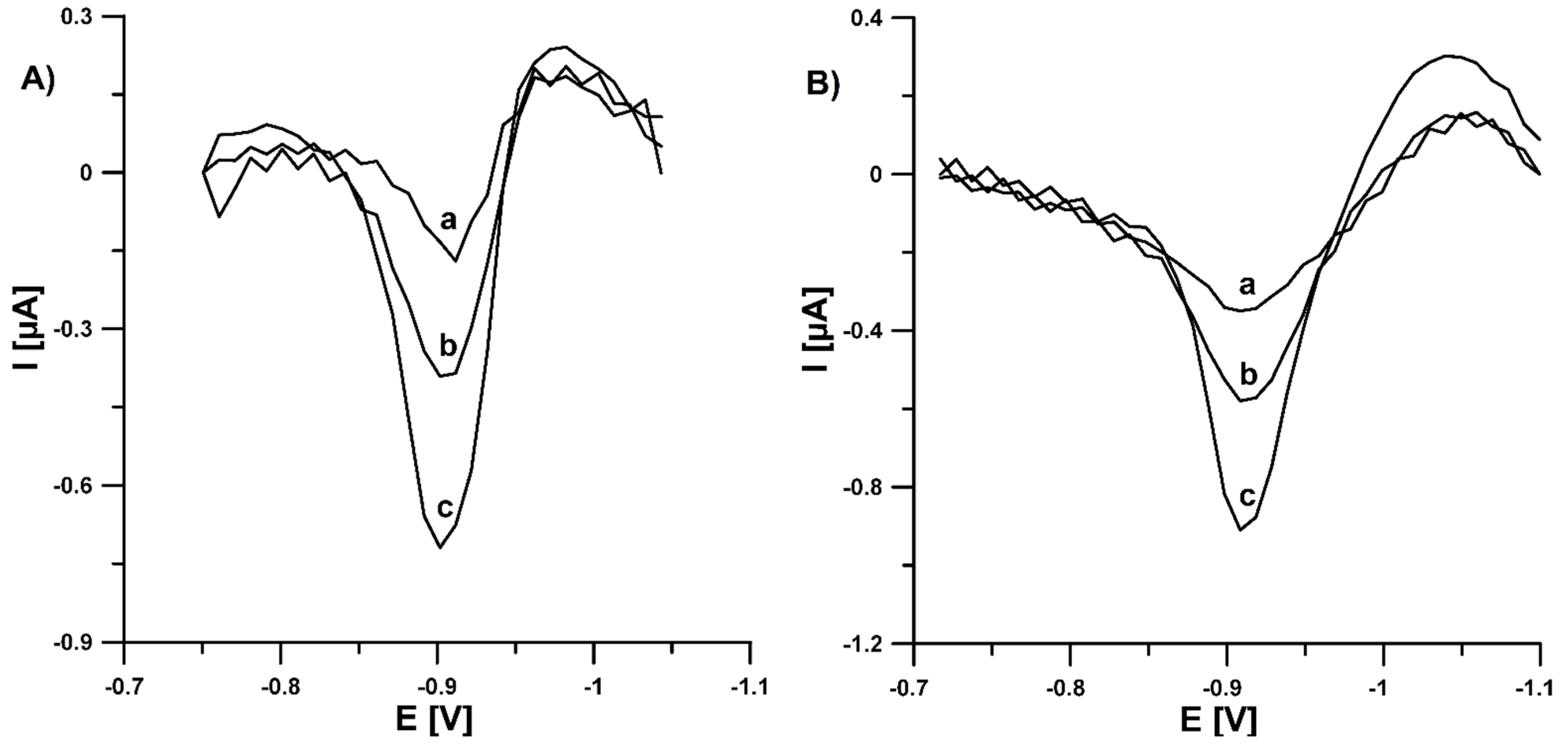
3.4. Analytical Characteristic of Pb-N-CMK-3/GCE towards U(VI)
3.5. Application to Reference Materials Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Xie, Y.; Chen, C.; Ren, X.; Wang, X.; Wang, H.; Wang, X. Emerging natural and tailored materials for uranium-contaminated water treatment and environmental remediation. Prog. Mater. Sci. 2019, 103, 180–234. [Google Scholar] [CrossRef]
- Brugge, D.; de Lemos, J.L.; Oldmixon, B. Exposure pathways and health effects associated with chemical and radiological toxicity of natural uranium: A review. Rev. Environ. Health 2005, 20, 177–194. [Google Scholar] [CrossRef]
- Tosheva, Z.; Stoyanova, K.; Nikolchev, L. Comparison of different methods for uranium determination in water. J. Environ. Radioact. 2004, 72, 47–55. [Google Scholar] [CrossRef]
- Kumar, S.A.; Shenoy, N.S.; Pandey, S.; Sounderajan, S.; Venkateswaran, G. Direct determination of uranium in seawater by laser fluorimetry. Talanta 2008, 77, 422–426. [Google Scholar] [CrossRef]
- Kuczewski, B.; Marquardt, C.M.; Seibert, A.; Geckeis, H.; Kratz, J.V.; Trautmann, N. Separation of plutonium and neptunium species by capillary electrophoresis−inductively coupled plasma-mass spectrometry and application to natural groundwater samples. Anal. Chem. 2003, 75, 6769–6774. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.S.; Teixeira, L.S.G.; dos Santos, W.N.L.; Lemos, V.A.; Godoy, J.M.; Ferreira, S.L.C. Uranium determination using atomic spectrometric techniques: An overview. Anal. Chim. Acta. 2010, 674, 143–156. [Google Scholar] [CrossRef]
- Landsberger, S.; Kapsimalis, R. Comparison of neutron activation analysis techniques for the determination of uranium concentrations in geological and environmental materials. J. Environ. Radioact. 2013, 117, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Solatie, D.; Carbol, P.; Betti, M.; Bocci, F.; Hiernaut, T.; Rondinella, V.V.; Cobos, J. Ion chromatography inductively coupled plasma mass spectrometry (IC-ICP-MS) and radiometric techniques for the determination of actinides in aqueous leachate solutions from uranium oxide. Fresenius J. Anal. Chem. 2000, 368, 88–94. [Google Scholar] [CrossRef]
- Tuovinen, H.; Vesterbacka, D.; Pohjolainen, E.; Read, D.; Solatie, D.; Lehto, J. A comparison of analytical methods for determining uranium and thorium in ores and mill tailings. J. Geochem. Explor. 2015, 148, 174–180. [Google Scholar] [CrossRef]
- Wu, X.; Huang, Q.; Mao, Y.; Wang, X.; Wang, Y.; Hu, Q.; Wang, H.; Wang, X. Sensors for determination of uranium: A review. TrAC Trend Anal. Chem. 2019, 118, 89–111. [Google Scholar] [CrossRef]
- Korolczuk, M.; Tyszczuk, K.; Grabarczyk, M. Determination of uranium by adsorptive stripping voltammetry at a lead film electrode. Talanta 2007, 72, 957–961. [Google Scholar] [CrossRef]
- Tyszczuk-Rotko, K.; Domańska, K.; Czech, B.; Rotko, M. Development simple and sensitive voltammetric procedure for ultra-trace determination of U(VI). Talanta 2017, 165, 474–481. [Google Scholar] [CrossRef]
- Tyszczuk-Rotko, K.; Jędruchniewicz, K. Ultrasensitive sensor for uranium monitoring in water ecosystems. J. Electrochem. Soc. 2019, 166, B837–B844. [Google Scholar] [CrossRef]
- Korolczuk, M.; Grabarczyk, M.; Rutyna, I. An adsorptive stripping voltammetry procedure for ultra-trace determination of U(VI) using double accumulation step on two lead-film working electrodes. Talanta 2014, 130, 342–346. [Google Scholar] [CrossRef]
- Olchowski, R.; Zięba, E.; Giannakoudakis, D.A.; Anastopoulos, I.; Dobrowolski, R.; Barczak, M. Tailoring surface chemistry of sugar-derived ordered mesoporous carbons towards efficient removal of diclofenac from aquatic environments. Materials 2020, 13, 1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Li, Q.; Teng, N.; Long, D.; Ma, C.; Wei, Y.; Wang, J.; Ling, L. Simultaneous micropore development and nitrogen doping of ordered mesoporous carbons for enhanced supercapacitor and Li-S cathode performance. Electrochim. Acta 2016, 214, 231–240. [Google Scholar] [CrossRef]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S.W. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Liu, X.; Liu, Z.; Wang, C.; Liu, G.; Li, Q.; Feng, X. Enhanced photocatalytic activity of g-C3N42D nanosheets through thermal exfoliation using dicyandiamide as precursor. Ceram. Int. 2018, 44, 20613–20619. [Google Scholar] [CrossRef]
- Zhang, J.-B.; Tan, Z.-C.; Meng, S.-H.; Li, S.-H.; Zhang, L.-M. Heat capacity and thermal decomposition of dicyandiamide. Thermochim. Acta. 1997, 307, 11–15. [Google Scholar] [CrossRef]
- Zolfaghari, G.; Esmaili-Sari, A.; Anbia, M.; Younesi, H.; Amirmahmoodi, S.; Ghafari-Nazari, A. Taguchi optimization approach for Pb(II) and Hg(II) removal from aqueous solutions using modified mesoporous carbon. J. Hazard. Mater. 2011, 192, 1046–1055. [Google Scholar] [CrossRef]
- Lakhi, K.S.; Cha, W.S.; Choy, J.-H.; Al-Ejji, M.; Abdullah, A.M.; Al-Enizi, A.M.; Vinu, A. Synthesis of mesoporous carbons with controlled morphology and pore diameters from SBA-15 prepared through the microwave-assisted process and their CO2 adsorption capacity. Microporous Mesoporous Mater. 2016, 233, 44–52. [Google Scholar]
- Sadezky, A.; Muckenhuber, H.; Grothe, H.; Niessner, R.; Pöschl, U. Raman microspectroscopy of soot and related carbonaceous materials: Spectral analysis and structural information. Carbon 2005, 43, 1731–1742. [Google Scholar]
- Paneli, M.; Ouguenoune, H.; David, F.; Bolyos, A. Study of the reduction mechanism and the adsorption properties of uranium (VI)-cupferron complexes using various electrochemical techniques. Anal. Chim. Acta 1995, 304, 177–186. [Google Scholar] [CrossRef]
- Gosser, D.K. Cyclic Voltammetry: Simulation and Analysis of Reaction Mechanism; VCH: New York, NY, USA, 1993. [Google Scholar]
- Mocak, J.; Bond, A.M.; Mitchell, S.; Scollary, G. A statistical overview of standard (IUPAC and ACS) and new procedures for determining the limits of detection and quantification: Application to voltammetric and stripping techniques. Pure Appl. Chem. 1997, 69, 297–328. [Google Scholar]
- Ma, D.; Yuan, Y.; Xiao, X.; Gao, Y.; Li, Y.; Xu, W.; Long, W. A label-free electrochemical biosensor for trace uranium based on DNAzymes and gold nanoparticles. J. Radioanal. Nucl. Chem. 2014, 299, 1911–1919. [Google Scholar]
- Yun, W.; Cai, D.; Jiang, J.; Wang, X.; Liao, J.; Zhang, P.; Sang, G. An ultrasensitive electrochemical biosensor for uranyl detection based on DNAzyme and target-catalyzed hairpin assembly. Microchim. Acta 2016, 183, 1425–1432. [Google Scholar]
- Wen, Y.; Yuan, Y.; Li, L.; Ma, D.; Liao, Q.; Hou, S. Ultrasensitive DNAzyme based amperometric determination of uranyl ion using mesoporous silica nanoparticles loaded with Methylene Blue. Microchim. Acta 2017, 184, 3909–3917. [Google Scholar]
- Konieczki, P.; Namiesnik, J. Quality Assurance and Quality Control in the Analytical Chemical Laboratory: A Practical Approach; CRC Press: Boca Raton, FL, USA, 2009. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters of Porous Structure | CMK-3 | N-CMK-3 |
|---|---|---|
| SBET (m2/g) | 744 | 669 |
| VT (cm3/g) | 0.80 | 0.83 |
| dBJH (nm) | 3.4 | 3.4 |
| Zeta potential/ Raman ID/IG/pHsurf. | CMK-3 | N-CMK-3 |
| ζ (mV) | −20.7 | 7.3 |
| ID/IG (au) | 0.87 | 0.91 |
| pHsurf. (au) | 4.6 | 6.6 |
| Measurement method | CHN | CMK-3 | N-CMK-3 | |
| C (wt. %) | 92.6 | 89.0 | ||
| H (wt. %) | 0.7 | 0.6 | ||
| N (wt. %) | 0.3 | 5.9 | ||
| XPS | C (wt. %) | 92.8 | 95.1 | |
| O (wt. %) | 7.2 | 4.9 | ||
| N (wt. %) | 0.0 | 0.0 | ||
| S (wt. %) | 0.0 | 0.0 | ||
| EDX | C (wt. %) | 93.0 | 94.8 | |
| O (wt. %) | 5.5 | 3.4 | ||
| N (wt. %) | 0.0 | 1.5 | ||
| S (wt. %) | 0.5 | 0.3 |
| Reference Material | Measured Value ± SD (n = 3) (µM) | Information/Certified Value ± SD (µM) | Relative Error (%) c | texp |
|---|---|---|---|---|
| SLEW-3 | 0.00103 ± 0.00038 | 0.00109 a | 5.5% | 0.27 |
| SRM 1640a | 0.109 ± 0.0053 | 0.11 b ± 0.0011 | 0.91% | 0.33 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tyszczuk-Rotko, K.; Olchowski, R.; Kozak, J.; Sekerzh-Zenkovich, O.; Dobrowolski, R. Modified Mesoporous Carbon Material (Pb-N-CMK-3) Obtained by a Hard-Templating Route, Dicyandiamide Impregnation and Electrochemical Lead Particles Deposition as an Electrode Material for the U(VI) Ultratrace Determination. Materials 2021, 14, 6490. https://0-doi-org.brum.beds.ac.uk/10.3390/ma14216490
Tyszczuk-Rotko K, Olchowski R, Kozak J, Sekerzh-Zenkovich O, Dobrowolski R. Modified Mesoporous Carbon Material (Pb-N-CMK-3) Obtained by a Hard-Templating Route, Dicyandiamide Impregnation and Electrochemical Lead Particles Deposition as an Electrode Material for the U(VI) Ultratrace Determination. Materials. 2021; 14(21):6490. https://0-doi-org.brum.beds.ac.uk/10.3390/ma14216490
Chicago/Turabian StyleTyszczuk-Rotko, Katarzyna, Rafał Olchowski, Jędrzej Kozak, Olga Sekerzh-Zenkovich, and Ryszard Dobrowolski. 2021. "Modified Mesoporous Carbon Material (Pb-N-CMK-3) Obtained by a Hard-Templating Route, Dicyandiamide Impregnation and Electrochemical Lead Particles Deposition as an Electrode Material for the U(VI) Ultratrace Determination" Materials 14, no. 21: 6490. https://0-doi-org.brum.beds.ac.uk/10.3390/ma14216490