
Microstructure Evolution and Its Correlation with Performance in Nitrogen-Containing Porous Carbon Prepared by Polypyrrole Carbonization: Insights from Hybrid Calculations

 and
and
Abstract
:1. Introduction
2. Computational Details and Models
2.1. Reax-FF MD Simulations
2.2. DFT-NEB Calculations for Dehydrogenation
2.3. NEGF-DFT Calculations for Electronic Conductance
2.4. Machine Learning for Structural Factors and Tissue-Performance Relationships
3. Results and Discussion
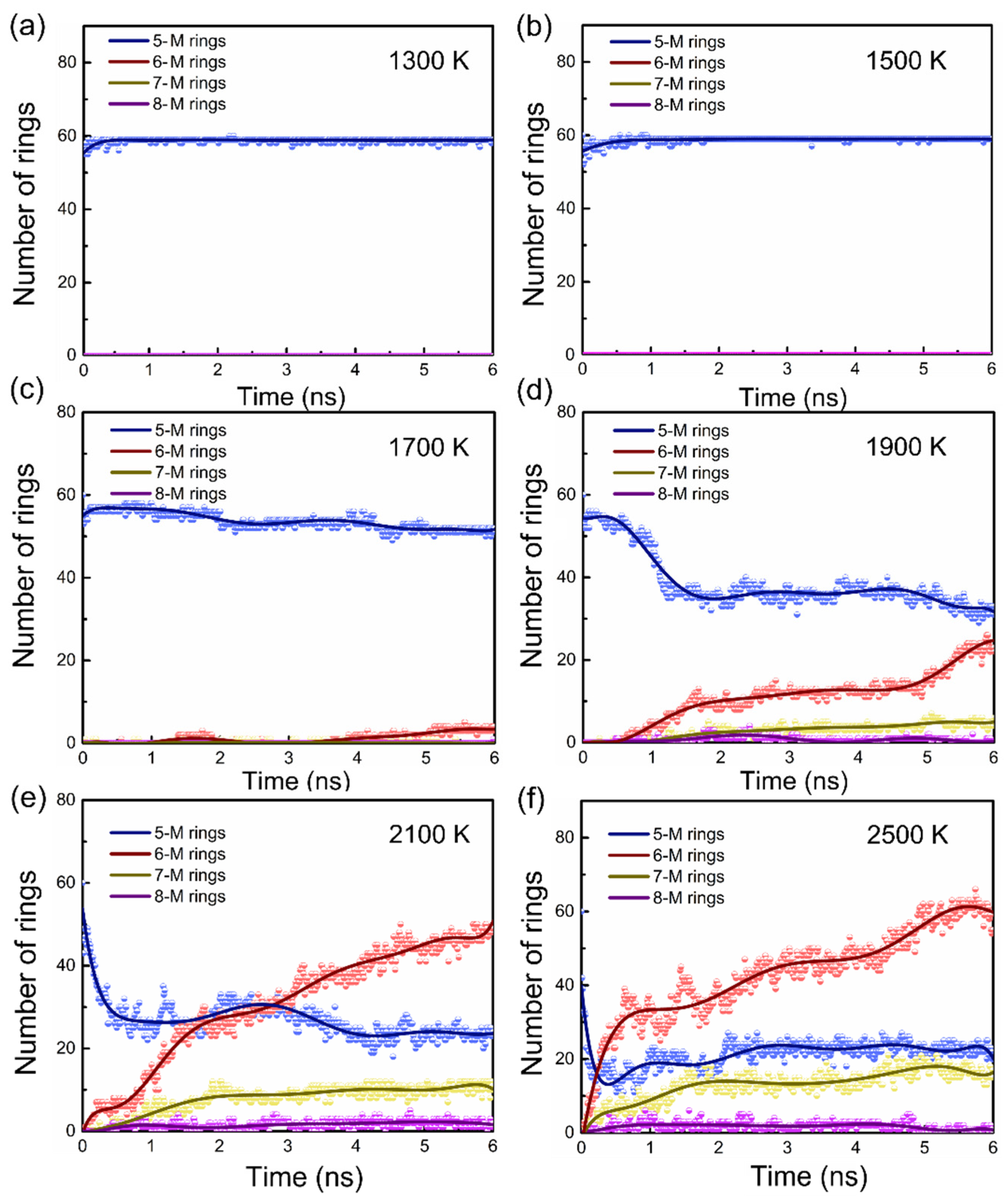
3.1. Tissue Evolution and Critical Temperature during PPy Carbonization
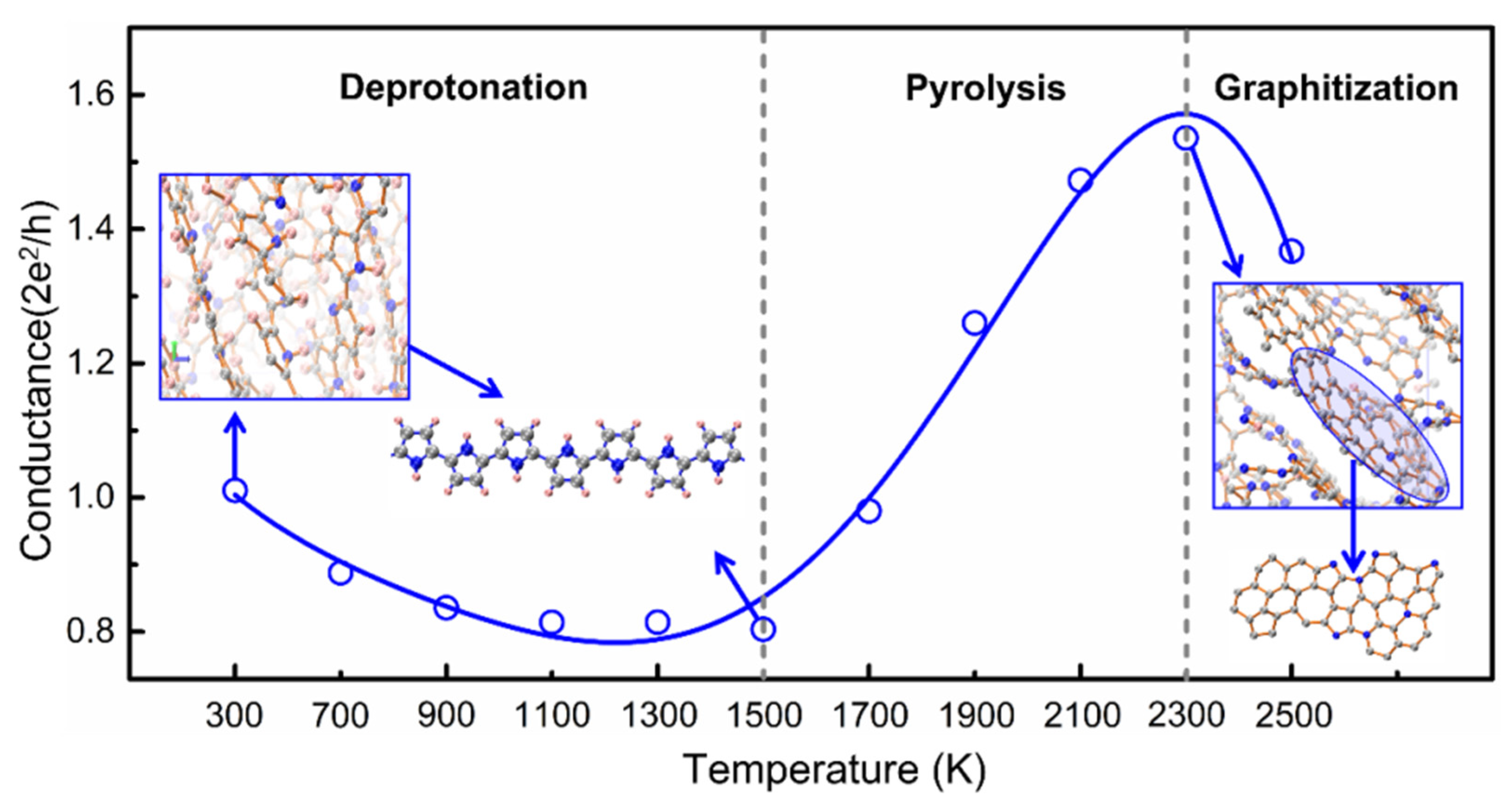
3.2. Tissue Correlation with Electronic Conductance
3.3. Tissue Correlation with Pore Size
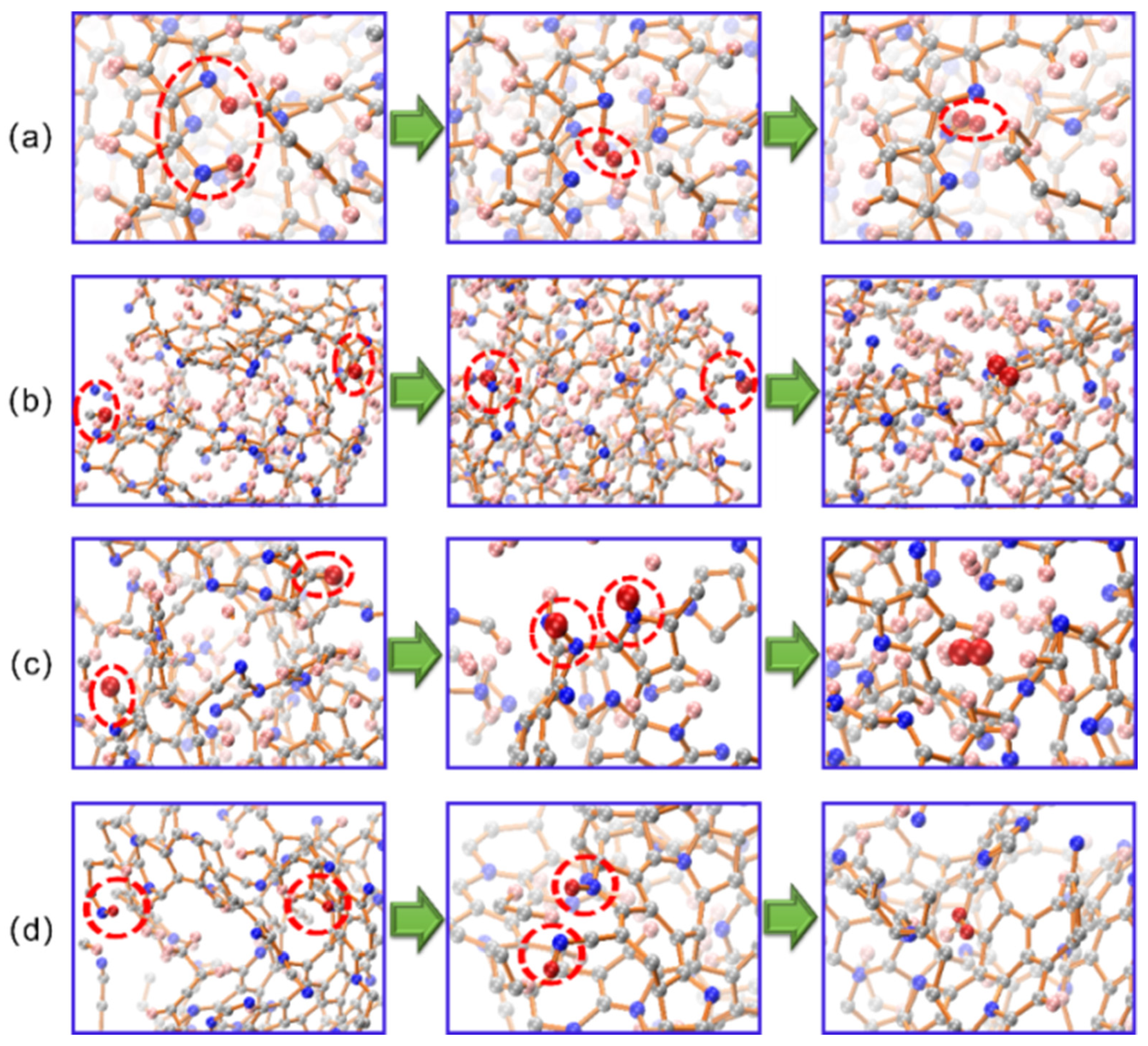
3.4. Deprotonation Mechanism
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vinu, A.; Ariga, K.; Mori, T.; Nakanishi, T.; Hishita, S.; Golberg, D.; Bando, Y. Preparation and Characterization of Well-ordered Hexagonal Mesoporous Carbon Nitride. Adv. Mater. 2005, 17, 1648–1652. [Google Scholar] [CrossRef]
- Tomšík, E.; Morávková, Z.; Stejskal, J.; Trchová, M.; Šálek, P.; Kovárová, J.; Zemek, J.; Cieslar, M.; Prokeš, J. Multi-wall Carbon Nanotubes with Nitrogen-containing Carbon Coating. Chem. Pap. 2013, 67, 1054–1065. [Google Scholar] [CrossRef]
- Oueiny, C.; Berlioz, S.; Perrin, F.X. Carbon Nanotube-polyaniline Composites. Prog. Polym. Sci. 2014, 39, 707–748. [Google Scholar] [CrossRef]
- Adhikari, A.; De, S.; Halder, A.; Pattanayak, S.; Dutta, K.; Mondal, D.; Rana, D.; Ghosh, R.; Bera, N.K.; Chattopadhyay, S.; et al. Biosurfactant Tailored Synthesis of Porous Polypyrrole Nanostructures: A Facile Approach towards CO2 Adsorption and Dopamine Sensing. Synth. Met. 2018, 245, 209–222. [Google Scholar] [CrossRef]
- Wang, T.; Yan, L.; He, Y.; Alhassan, S.I.; Gang, H.; Wu, B.; Jin, L.; Wang, H. Application of Polypyrrole-based Adsorbents in The Removal of Fluoride: A Review. RSC Adv. 2022, 12, 3505–3517. [Google Scholar] [CrossRef]
- Wang, K.; Sun, F.; Su, Y.; Chen, Y.; Gao, J.; Yang, H.; Zhao, G. Natural Template Derived Porous Carbon Nanoplate Architectures with Tunable Pore Configuration for A Full-carbon Sodium-ion Capacitor. J. Mater. Chem. A 2021, 92, 3607–23618. [Google Scholar] [CrossRef]
- Zabrodskii, A.G.; Kompan, M.E.; Malyshkin, V.G.; Sapurina, I.Y. Carbon Supported Polyaniline as Anode Catalyst: Pathway to Platinum-free Fuel Cells. Tech. Phys. Lett. 2006, 32, 758–761. [Google Scholar] [CrossRef] [Green Version]
- Meng, Y.Y.; Voiry, D.; Goswami, A.; Zou, X.X.; Huang, X.X.; Chhowalla, M.; Liu, Z.W.; Asefa, T. N- O-, and S-tridoped Nanoporous Carbons as Selective Catalysts for Oxygen Reduction and Alcohol Oxidation Reactions. J. Am. Chem. Soc. 2014, 136, 13554–13557. [Google Scholar] [CrossRef]
- Meng, Y.Y.; Zou, X.X.; Huang, X.X.; Goswami, A.; Liu, Z.W.; Asefa, T. Polypyrrole-derived Nitrogen and Oxygen Co-doped Mesoporous Carbons as Efficient Metal-free Electrocatalyst for Hydrazine Oxidation. Adv. Mater. 2014, 26, 6510–6516. [Google Scholar] [CrossRef]
- Yuan, X.X.; Sha, H.D.; Ding, X.L.; Kong, H.C.; Lin, H.; Wen, W.; Huang, T.H.; Guo, Z.; Ma, Z.F.; Yang, Y. Comparative Investigation on The Properties of Carbon-supported Cobalt-polypyrrole Pyrolysed at Various Conditions as Electrocatalyst towards Oxygen Reduction Reaction. Int. J. Hydrog. Energy 2014, 39, 15937–15947. [Google Scholar] [CrossRef]
- Yuan, X.X.; Kong, H.C.; He, Y.J.; Ma, Z.F.; Yang, Y.; Li, Q. Effects of Composition on Electrochemical Properties of A Non-precious Metal Catalyst towards Oxygen Reduction Reaction. Int. J. Hydrog. Energy 2014, 39, 16006–16014. [Google Scholar] [CrossRef]
- Rajender, B.; Palaniappan, S. Polyaniline Salts as Polymer-based Solid Acid Catalyst for Low Molecular Weight Poly (Lactic Acid). J. Appl. Polym. Sci. 2014, 131, 41147. [Google Scholar] [CrossRef]
- Silva, R.; Voiry, D.; Chhowalla, M.; Asefa, T. Efficient Metal-free Electrocatalysts for Oxygen Reduction: Polyaniline-derived N- and O-doped Mesoporous Carbons. J. Am. Chem. Soc. 2013, 135, 7823–7826. [Google Scholar] [CrossRef] [PubMed]
- Javier, Q.B.; Carolina, G.G.; Emilia, M.; Diego, C.A. Effect of Carbonization Conditions of Polyaniline on Its Catalytic Activity towards ORR. Some Insights about The Nature of The Active Sites. Carbon 2017, 119, 62–71. [Google Scholar]
- Singh, K.P.; Song, M.Y.; Yu, J.S. Iodine-treated Heteroatom-doped Carbon: Conductivity Driven Electrocatalytic Activity. J. Mater Chem. A 2014, 2, 18115–18124. [Google Scholar] [CrossRef]
- Maldonado, S.; Stevenson, K.J. Influence of Nitrogen Doping on Oxygen Reduction Electrocatalysis at Carbon Nanofiber Electrodes. J. Phys. Chem. B 2005, 109, 4707–4716. [Google Scholar] [CrossRef]
- Wang, G.; Zhang, L.; Zhang, J. A Review of Rlectrode Materials for Electrochemical Supercapacitors. Chem. Soc. Rev. 2012, 41, 797–828. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.M.; Ramachandran, R.; Mani, V.; Saraswathi, R. Recent Aadvancements in Electrode Materials for The High Performance Electrochemical Supercapacitors: A Review. Int. J. Electrochem. Sci. 2014, 9, 4072–4085. [Google Scholar]
- Shen, C.; Sun, Y.; Yao, W.; Lu, Y. Facile Synthesis of Polypyrrole Nanospheres and Their Carbonized Products for Potential Application in High-performance Supercapacitors. Polymer 2014, 55, 2817–2824. [Google Scholar] [CrossRef]
- Kudoh, Y. Properties of Polypyrrole Prepared by Chemical Polymerization Using Aqueous Solution Containing Fe2(SO4)3 and Anionic Surfactant. Synth. Met. 1996, 79, 17–22. [Google Scholar] [CrossRef]
- Ramanaviciene, A.; Kausaite, A.; Tautkus, S.; Ramanavicius, A. Biocompatibility of Polypyrrole Particles: An In-vivo Study in Mice. J. Pharm. Pharmacol. 2007, 59, 311–315. [Google Scholar] [CrossRef] [PubMed]
- Guimard, N.K.; Gomez, N.; Schmidt, C.E. Conducting Polymers in Biomedical Engineering. Prog. Polym. Sci. 2007, 32, 876–921. [Google Scholar] [CrossRef]
- Yuan, X.X.; Ding, X.L.; Wang, C.Y.; Ma, Z.F. Use of Polypyrrole in Catalysis for Low Temperature Fuel Cells. Environ. Sci. 2013, 6, 1105–1124. [Google Scholar]
- Stejskal, J. Conducting Polymers Are Not Just Conducting: A Perspective for Emerging Technology. Polym. Int. 2020, 69, 662–664. [Google Scholar] [CrossRef]
- Stejskal, J. Interaction of Conducting Polymers, Polyaniline and Polypyrrole, with Organic Dyes: Polymer Morphology Control, Dye Adsorption and Photocatalytic Decomposition. Chem. Pap. 2020, 74, 1–54. [Google Scholar] [CrossRef]
- Zhang, W.; Cheng, R.; Bi, H.; Lu, Y.; Ma, L.; He, X. A Review of Porous Carbons Produced by Template Methods for Super Capacitor Applications. New Carbon Mater. 2021, 36, 69–81. [Google Scholar] [CrossRef]
- Ariga, K.; Vinu, A.; Yamauchi, Y.; Ji, Q.; Hill, J.P. Nanoarchitectonics for Mesoporous Materials. Bull. Chem. Soc. Jpn. 2012, 85, 1–32. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Xiong, H.; Xiao, Y. Progress on Synthesis and Applications of Porous Carbon Materials. Int. J. Electrochem. Sci. 2020, 15, 1363–1377. [Google Scholar] [CrossRef]
- Ciric-Marjanovic, G.; Pasti, I.; Gavrilov, N.; Janosevic, A.; Mentus, S. Carbonised Polyaniline and Polypyrrole: Towards Pdvanced Nitrogen-containing Carbon Materials. Chem. Pap. 2013, 67, 781–813. [Google Scholar] [CrossRef]
- Ciric-Marjanovic, G.; Pasti, I.; Mentus, S. One-dimensional Nitrogen-containing Carbon Nanostructures. Prog. Mater. Sci. 2015, 69, 61–182. [Google Scholar] [CrossRef]
- Sapurina, I.; Stejskal, J.; Sedenkova, I.; Trchova, M.; Kovarova, J.; Hromadkova, J.; Kopecka, J.; Cieslar, M.; El-Nasr, A.A.; Ayad, M.M. Catalytic Activity of Polypyrrole Nanotubes Decorated with Noble-metal Nanoparticles and Their Conversion to Carbonized Analogues. Synth. Met. 2016, 214, 14–22. [Google Scholar] [CrossRef]
- Shrestha, L.K.; Shrestha, R.G.; Yamauchi, Y.; Hill, J.P.; Nishimura, T.; Miyazawa, K.; Kawai, T.; Okada, S.; Wakabayashi, K.; Ariga, K. Nanoporous Carbon Tubes from Fullerene Crystals as The π-electron Carbon Source. Angew. Chem. Int. Ed. 2014, 53, 1–6. [Google Scholar]
- Bairi, P.; Shrestha, R.G.; Hill, J.P.; Nishimura, T.; Ariga, K.; Shrestha, L.K. Mesoporous Graphitic Carbon Microtubes Derived from Fullerene C70 Tubes as A High Performance Electrode Material for Advanced Supercapacitors. J. Mater. Chem. A 2016, 4, 13899–13906. [Google Scholar] [CrossRef] [Green Version]
- Bairi, P.; Maji, S.; Hill, J.P.; Kim, J.H.; Ariga, K.; Shrestha, L.K. Mesoporous Carbon Cubes Dderived from Fullerene Crystals as A High Rate Performance Electrode Material for Supercapacitors. J. Mater. Chem. A 2019, 7, 12654–12660. [Google Scholar] [CrossRef]
- Trchova, M.; Konyushenko, E.N.; Stejskal, J.; Kovarova, J.; Ciric-Marjanovic, G. The Conversion of Polyaniline Nanotubes to Nitrogen-containing Carbon Nanotubes and Their Comparison with Multi-walled Carbon Nanotubes. Polym. Degrad. Stab. 2009, 94, 929–938. [Google Scholar] [CrossRef]
- Ciric-Marjanovic, G.; Mentus, S.; Pasti, I.; Gavrilov, N.; Krstic, J.; Travas-Sejdic, J.; Strover, L.T.; Kopecka, J.; Moravkova, Z.; Trchova, M.; et al. Synthesis, Characterization, and Electrochemistry of Nanotubular Polypyrrole and Polypyrrole-derived Carbon Nanotubes. J. Phys. Chem. C 2014, 118, 14770–14784. [Google Scholar] [CrossRef]
- Inagaki, M.; Toyoda, M.; Soneda, Y.; Morishita, T. Nitrogen-doped Carbon Materials. Carbon 2018, 132, 104–140. [Google Scholar] [CrossRef]
- Li, S.S.; Bian, F.; Meng, X.Y.; Zhai, D.; Yang, H.W.; Qin, G.W. Ring Structure Characterization of Nanoporous Carbon Materials Prepared by Thermal Conversion of Fullerenes: Insights from ReaxFF Molecular Dynamics Simulations. Carbon 2022, 189, 484–492. [Google Scholar] [CrossRef]
- Kujundziski, A.P.; Chamovska, D.; Grchev, T. Capacitive Properties of Polypyrrole/Activated Parbon Pomposite. Hem. Ind. 2014, 68, 709–719. [Google Scholar] [CrossRef]
- Kujundziski, A.P.; Chamovska, D.; Cvetkovska, M.; Grchev, T. Electrochemical Study of Electroconducting Composite Material–Polypyrrole/Activated Carbon. Int. J. Electrochem. Sci. 2012, 7, 4099–4113. [Google Scholar]
- Plimpton, S. Fast Parallel Algorithms for Short-range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Stukowski, A. Visualization and Analysis of Atomistic Simulation Data with OVITO-the Open Visualization Tool. Model. Simul. Mater. Sci. Eng. 2010, 18, 2154–2162. [Google Scholar] [CrossRef]
- Willems, T.F.; Rycroft, C.H.; Kazi, M.; Meza, J.C.; Haranczyk, M. Algorithms and Tools for High-throughput Geometry-based Analysis of Crystalline Porous Materials. Micropor. Mesopor. Mater. 2012, 149, 134–141. [Google Scholar] [CrossRef]
- Kowalik, M.; Ashraf, C.; Damirchi, B.; Akbarian, D.; Rajabpour, S.; van Duin, A.C.T. Atomistic Scale Analysis of The Carbonization Process for C/H/O/N-based Polymers with The ReaxFF Reactive Force Field. J. Phys. Chem. B 2019, 123, 5357–5367. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Song, B.; Cao, C.; Zhang, H.; Liu, Q.; Li, K.; Teppen, B.J. Structural Evolution of High-rank Coals During Coalification and Graphitization: X-ray Diffraction, Raman Spectroscopy, High-resolution Transmission Electron Microscopy, and Reactive Force Field Molecular Dynamics Simulation Study. Energy Fuels 2021, 35, 2087–2097. [Google Scholar] [CrossRef]
- Zhu, J.; Gao, Z.; Kowalik, M.; Joshi, K.; Ashraf, C.M.; Arefev, M.I.; Schwab, Y.; Bumgardner, C.; Brown, K.; Burden, D.E.; et al. Unveiling Carbon Ring Structure Formation Mechanisms in Polyacrylonitrile-derived Carbon Fibers. ACS Appl. Mater. Interfaces 2019, 11, 42288–42297. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient Iterative Schemes for Ab Initio Total-energy Calculations Using A Plane-wave Basis Set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of Ab-initio Total Energy Calculations for Metals and Semiconductors Using A Plane-wave Basis Set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab Initio Molecular Dynamics for Open-shell Transition Metals. Phy. Rev. B 1993, 48, 13115–13118. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector Augmented-wave Method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ernzerhof, M.; Scuseria, G.E. Assessment of The Perdew–Burke–Ernzerhof exchange-correlation Functional. J. Chem. Phys. 1999, 110, 5029–5036. [Google Scholar] [CrossRef] [Green Version]
- Monkhorst, H.J.; Pack, J.D. Special Points for Brillouin-zone Integrations. Phys. Rev. B 1976, 13, 12. [Google Scholar] [CrossRef]
- Henkelman, G.; Uberuaga, B.P.; Jónsson, H. A Climbing Image Nudged Elastic Band Method for Finding Saddle Points and Minimum Energy Paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef] [Green Version]
- Henkelman, G.; Jónsson, H. Improved Tangent Estimate in The Nudged Elastic Band Method for Finding Minimum Energy Paths and Saddle Points. J. Chem. Phys. 2000, 113, 9978–9985. [Google Scholar] [CrossRef] [Green Version]
- Bian, F.; Wu, X.; Li, S.; Qin, G.; Meng, X.; Wang, Y.; Yang, H. Role of Transport Polarization in Electrocatalysis: A Case Study of The Ni-cluster/Graphene Interface. J. Mater. Sci. Technol. 2021, 92, 120–128. [Google Scholar] [CrossRef]
- Taylor, J.; Guo, H.; Wang, J. Ab Initio Modeling of Quantum Transport Properties of Molecular Electronic Devices. Phys. Rev. B 2001, 63, 245407. [Google Scholar] [CrossRef] [Green Version]
- Waldron, D.; Haney, P.; Larade, B.; MacDonald, A.; Guo, H. Nonlinear Spin Current and Magnetoresistance of Molecular Tunnel Junctions. Phys. Rev. Lett. 2006, 96, 166804. [Google Scholar] [CrossRef]
- Anisimov, V.I.; Gunnarsson, O. Density-functional Calculation of Effective Coulomb Interactions in Metals. Phys. Rev. B 1991, 43, 7570–7574. [Google Scholar] [CrossRef]
- Soler, J.M.; Artacho, E.; Gale, J.D.; Garcia, A.; Junquera, J.; Ordejon, P.; Sanchez-Portal, D. The SIESTA Method for Ab Initio Order-N Materials Simulation. J. Phys. Condens. Matter. 2002, 14, 2745–2779. [Google Scholar] [CrossRef] [Green Version]
- Ouyang, R.; Curtarolo, S.; Ahmetcik, E.; Scheffler, M.; Ghiringhelli, L.M. SISSO: A Compressed-sensing Method for Identifying The Best low-dimensional Descriptor in An Immensity of Offered Candidates. Phys. Rev. Mater. 2018, 2, 083802. [Google Scholar] [CrossRef]
- Han, Z.K.; Sarker, D.; Ouyang, R.; Mazheika, A.; Gao, Y.; Levchenko, S.V. Single-atom Alloy Catalysts Designed by First-principles Calculations and Artificial Intelligence. Nat. Commun. 2021, 12, 1833. [Google Scholar] [CrossRef] [PubMed]
- Stejskal, J.; Kohl, M.; Trchova, M.; Kolska, Z.; Pekarek, M.; Krivka, I.; Prokes, J. Conversion of Conducting Polypyrrole Nanostructures to Nitrogen-containing Carbons and Its Impact on The Adsorption of Organic Dye. Mater. Adv. 2021, 2, 706–717. [Google Scholar] [CrossRef]
- Saha, B.; Schatz, G.C. Carbonization in Polyacrylonitrile (PAN) Based Carbon Fibers Studied by ReaxFF Molecular Dynamics Simulations. J. Phys. Chem. B 2012, 116, 4684–4692. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Zhang, H.; Li, G.; Zhang, J.; Bouhadja, M.; Liu, Z.; Skelton, A.A.; Barati, M. ReaxFF Molecular Dynamics Simulation for The Graphitization of Amorphous Carbon: A Parametric Study. J. Chem. Theory Comput. 2018, 14, 2322–2331. [Google Scholar] [CrossRef]
- Kopecká, J.; Mrlík, M.; Olejník, R.; Kopecký, D.; Vrnata, M.; Prokeš, J.; Bober, P.; Morávková, Z.; Trchová, M.; Stejskal, J. Polypyrrole Nanotubes and Their Carbonized Analogs: Synthesis, Characterization, Gas Sensing Properties. Sensors 2016, 16, 1917. [Google Scholar] [CrossRef] [Green Version]
- Le, T.H.; Yoon, H. Strategies for Fabricating Versatile Carbon Nanomaterials from Polymer Precursors. Carbon 2019, 152, 796–817. [Google Scholar] [CrossRef]
- Stejskal, J.; Trchova, M.; Bober, P.; Moravkova, Z.; Kopecky, D.; Vrnata, M.; Prokes, J.; Varga, M.; Watzlova, E. Polypyrrole Salts and Bases: Superior Conductivity of Nanotubes and Their Stability Towards the Loss of Conductivity by Deprotonation. RSC Adv. 2016, 6, 88382–88391. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Guestrin, C. XGBoost: A Scalable Tree Boosting System. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016; pp. 785–794. [Google Scholar]
- Zhou, Z.; Lai, C.; Zhang, L.; Qian, Y.; Hou, H.; Reneker, D.H.; Fong, H. Development of Carbon Nanofibers from Aligned Electrospun Polyacrylonitrile Nanofiber Bundles and Characterization of Their Microstructural, Electrical, and Mechanical Properties. Polymer 2009, 50, 2999–3006. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 5-M Ring | 6-M Ring | 7-M Ring | 8-M Ring | |
|---|---|---|---|---|
| N0 | 1.6319 | 2.0311 | 1.8404 | 1.1564 |
| N1 | 0.9319 | 1.2667 | 2.1085 | 1.0176 |
| N2 | 1.0053 | 1.4475 | 1.7509 | 0.9608 |
| N3 | 1.8146 |
| Temperature | Feature | 5-M | 5-MN1 | 5-MN2 | 6-M | 6-MN1 | 6-MN2 | 6-MN3 |
|---|---|---|---|---|---|---|---|---|
| 2100 K | Weights | 0.177 | 0.242 | 0 | 0.174 | 0.092 | 0.046 | 0.026 |
| 2500 K | 0.139 | 0.211 | 0.007 | 0.201 | 0.177 | 0.047 | 0.003 | |
| Temperature | Feature | 7-M | 7-MN1 | 7-MN2 | 8-M | 8-MN1 | 8-MN2 | |
| 2100 K | Weights | 0.046 | 0.056 | 0.056 | 0.043 | 0.036 | 0.006 | |
| 2500 K | 0.068 | 0.044 | 0.031 | 0.017 | 0.031 | 0.024 |
| Temperature | Feature | 5-M | 5-MN1 | 5-MN2 | 6-M | 6-MN1 | 6-MN2 | 6-MN3 |
|---|---|---|---|---|---|---|---|---|
| 2100 K | Weight | 0.187 | 0.224 | 0.018 | 0.140 | 0.132 | 0.052 | 0.032 |
| 2500 K | 0.215 | 0.172 | 0.013 | 0.137 | 0.164 | 0.049 | 0.017 | |
| Temperature | Feature | 7-M | 7-MN1 | 7-MN2 | 8-M | 8-MN1 | 8-MN2 | |
| 2100 K | Weight | 0.052 | 0.029 | 0.052 | 0.026 | 0.033 | 0.023 | |
| 2500 K | 0.051 | 0.066 | 0.035 | 0.028 | 0.032 | 0.021 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, S.; Bian, F.; Wu, X.; Sun, L.; Yang, H.; Meng, X.; Qin, G. Microstructure Evolution and Its Correlation with Performance in Nitrogen-Containing Porous Carbon Prepared by Polypyrrole Carbonization: Insights from Hybrid Calculations. Materials 2022, 15, 3705. https://0-doi-org.brum.beds.ac.uk/10.3390/ma15103705
Li S, Bian F, Wu X, Sun L, Yang H, Meng X, Qin G. Microstructure Evolution and Its Correlation with Performance in Nitrogen-Containing Porous Carbon Prepared by Polypyrrole Carbonization: Insights from Hybrid Calculations. Materials. 2022; 15(10):3705. https://0-doi-org.brum.beds.ac.uk/10.3390/ma15103705
Chicago/Turabian StyleLi, Shanshan, Fang Bian, Xinge Wu, Lele Sun, Hongwei Yang, Xiangying Meng, and Gaowu Qin. 2022. "Microstructure Evolution and Its Correlation with Performance in Nitrogen-Containing Porous Carbon Prepared by Polypyrrole Carbonization: Insights from Hybrid Calculations" Materials 15, no. 10: 3705. https://0-doi-org.brum.beds.ac.uk/10.3390/ma15103705