1. Introduction
Mass spectrometers are widely-used devices (the mass spectrometry market will grow from
$5.3 billion to
$10.5 billion from 2016–2025 [
1]) enabling the identification of proteins from any sample and thus to understand the ongoing biological process in the sample [
2,
3,
4]. The identified proteins can help to optimize biochemical processes, for example those in biogas plants [
5,
6], or diagnose diseases such as inflammatory bowel diseases [
7] or even the spike proteins of the SARS-CoV2 virus [
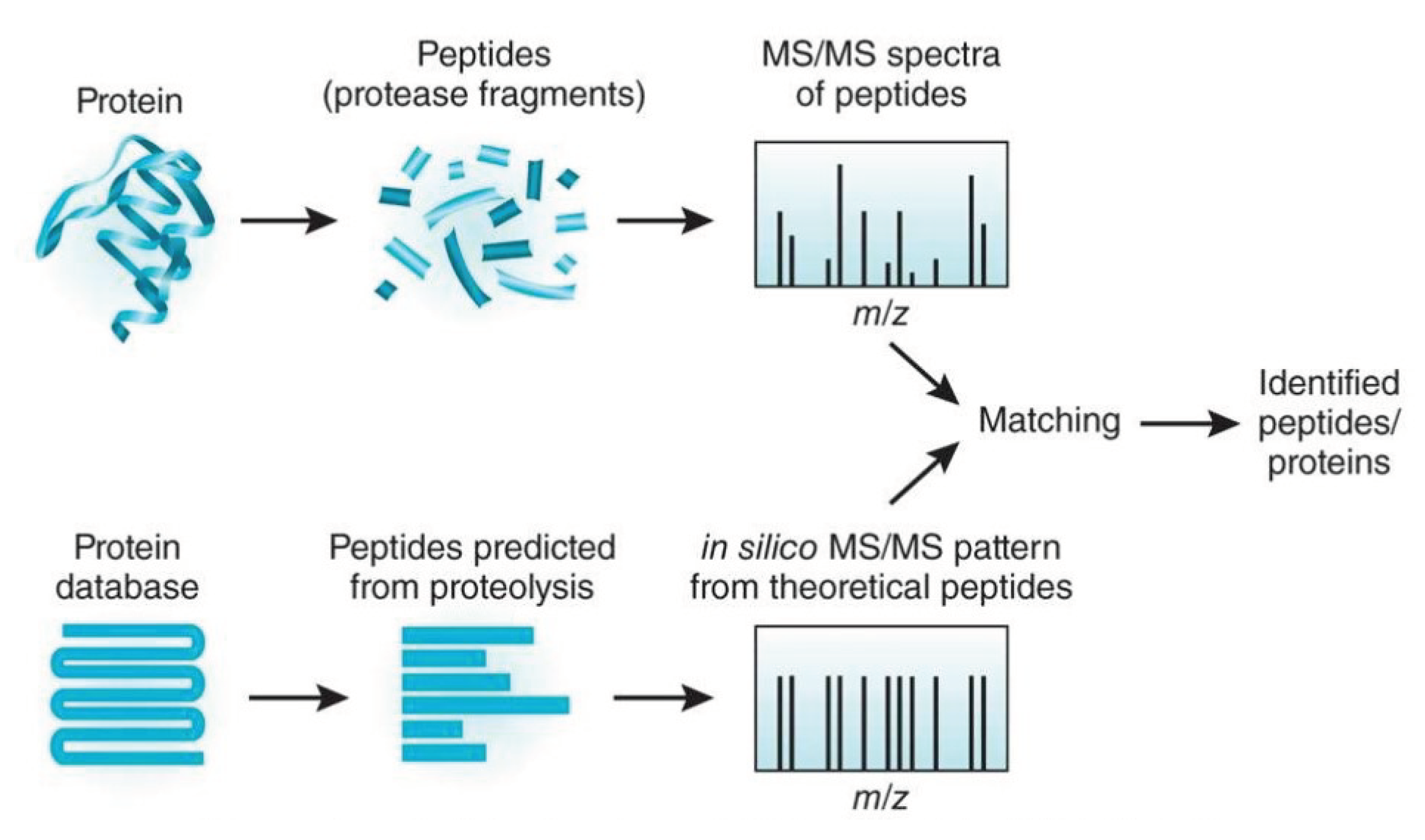
8]. The measurement of a mass spectrometer takes up to two hours and is followed by converting the measured data into a readable format. In order to gain insights into the converted data, an analysis step is required: the protein identification [
9]. The state-of-the-art protein identification approach uses a peptide-centric approach comparing the experimental (spectrum) data with theoretical spectra from a protein sequence database. For this purpose, the proteins are digested into peptides, and theoretical spectra are calculated, resulting in billions of data sets. Subsequently, protein database search algorithms compare the measured spectra with the theoretical spectra to find the highest similarity. As a consequence of all these steps, bioinformatic protein identification takes several hours to complete. In order to speed up the bioinformatic processes, we are currently developing a cloud-based pipeline for protein identification [
10]. One key challenge for this pipeline is the well-performing and efficient storage and retrieval of the proteins. Therefore, we tested and evaluated an index schema to query only suitable candidates of the sequence data and reduce the search area to a minimum [
11,
12], using a column-based index in a distributed database management system (DBMS) that allows streamlining of the analysis step.
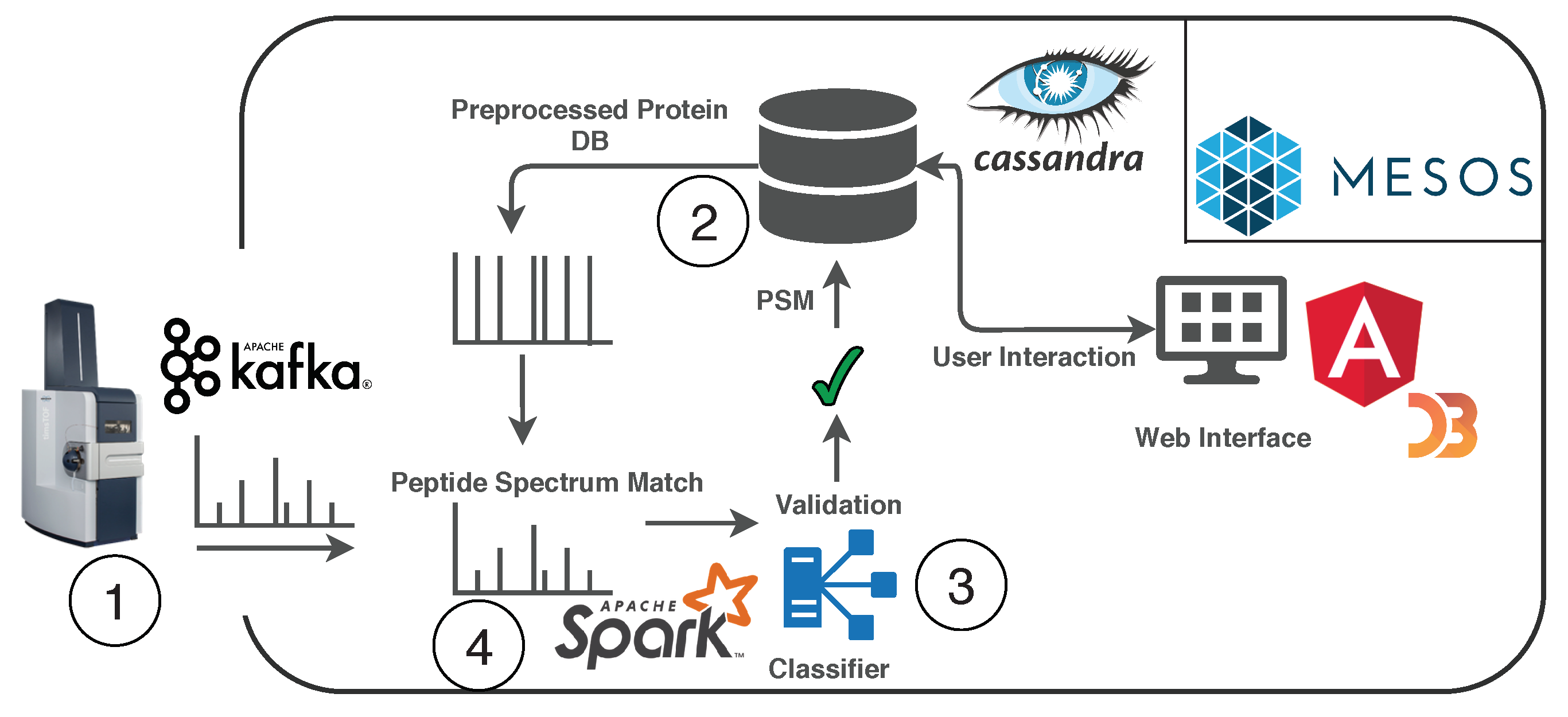
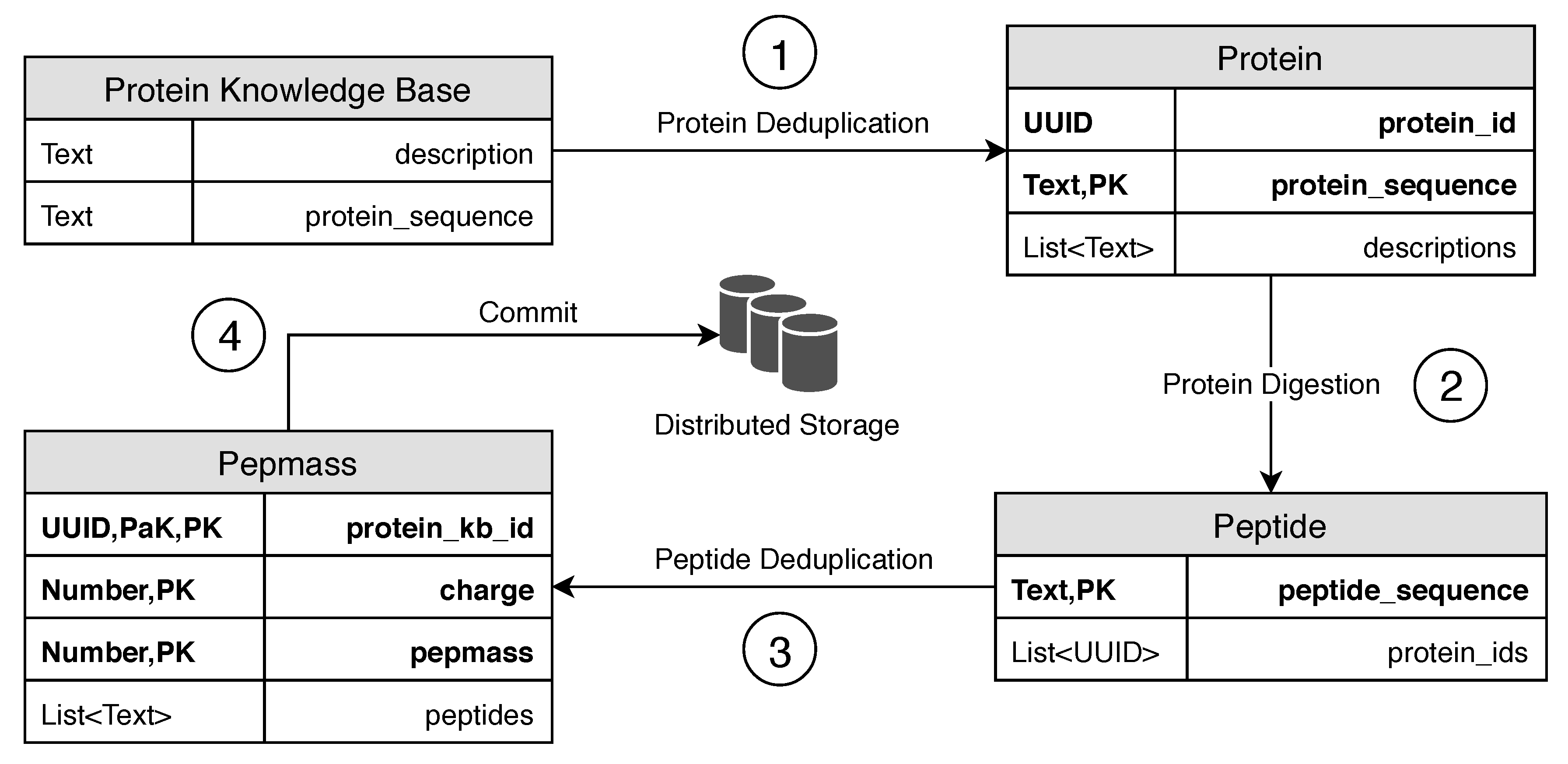
In this work, we present an indexing schema for the sequence data of a protein sequence database, using a column-based index in a distributed database management system (DBMS) that allows streamlining of the analysis step. This step’s key challenges are the memory-efficient and fast transformation of the protein sequence data from the current state-of-the-art FASTA format to the DBMS indexed schema. The transformation involves separating the sequence data, deduplication of the sequence data, and mass calculation steps, which have to be calculated before inserting into the DBMS.
After presenting the transformation process, we describe four methods to aggregate the data into the index structure. The first one is the naive in-memory approach; the second is the structured hard disk approach; the third method uses DBMS queries; and the last one is the radix tree-based method. In the end, we evaluate those methods and show that the radix tree is very efficient for the storage of sequence data and has the best overall performance of all approaches.
In summary, we make the following contributions:
Database schema: We propose a suitable schema for protein sequence data in a real-time cloud system as an extension to our prior paper [
13].
Protein data transformation: We review the process of protein data transformation into the proposed database schema and detail suitable techniques for the transformation steps.
Indexing: We propose a radix tree for efficient deduplication (including an extended description compared to our prior paper [
13]).
Performance comparison: We evaluate the best method to transform the protein sequence data using the four proposed approaches.
This paper is structured as follows: In
Section 2, we explain the basics of the mass spectrometry workflow, the protein identification, the protein data, and the radix tree data structure. In
Section 3, we describe our architecture for the real-time processing of mass spectrometer data and explain the index of the protein data and how to transform this one into an indexed structure. Next, in
Section 4, we present the four methods for the data transformation.
Section 5 presents the evaluation of the methods and is followed by the related work in
Section 6. In the last section,
Section 7, we conclude our work.
4. Implementation
We implemented a protein sequence database transformation as a cloud service, using the Java Jetty application server and the Cassandra DBMS on an Apache Mesos system. Additionally, SQLite was used as the storage for the local structured data. The in-memory approach, the file system approach (SQLite), and the DBMS query approach used standard methods (i.e., Cassandra Query Language (CQL) queries) and data structures (hash maps and sets) for the deduplication and other transformation steps. In this section, we give a brief description of the implementation of the naive approaches and a detailed explanation of our extensions to the radix tree approach [
13].
4.1. Transformation Using a Map Structure
This approach uses a map and a key-value structure for the peptide deduplication and the protein relationship. The maps’ keys are the peptide sequences, and the values are the sets of protein identifiers. For the first implementation, we used an in-memory map and for the second implementation, an SQLite table as the key-value storage.
4.2. Transformation Using DBMS Queries
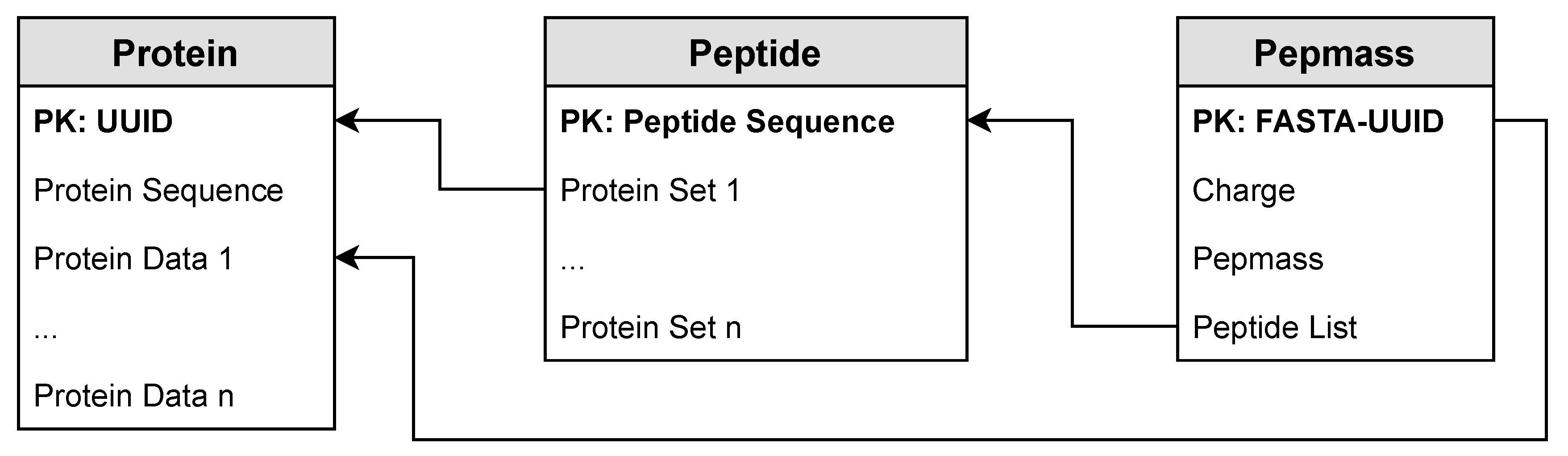
To insert the result data into the database management system (DBMS), we implemented queries to transform the data directly in the DBMS. We used UPDATE queries, which add a new row if the key does not exist, or otherwise append a value to the list in the column. The deduplication of proteins was already implemented using UPDATE queries on the
protein table. In this approach, we implemented additional UPDATE queries for the
peptide table and for the
pepmass table. In
Listing 3, we present these UPDATE queries, which are used in this method to update the
peptide table (Lines 1–2) and the
pepmass table (Lines 3–4).
Listing 3.
Queries to transform protein data from FASTA format to the table schema using the DBMS.
Listing 3.
Queries to transform protein data from FASTA format to the table schema using the DBMS.
4.3. Transformation Using the Extended Radix Tree Structure
A radix tree [
21,
22,
23] is more complex, and hence, we give a detailed explanation of the transformation algorithm (
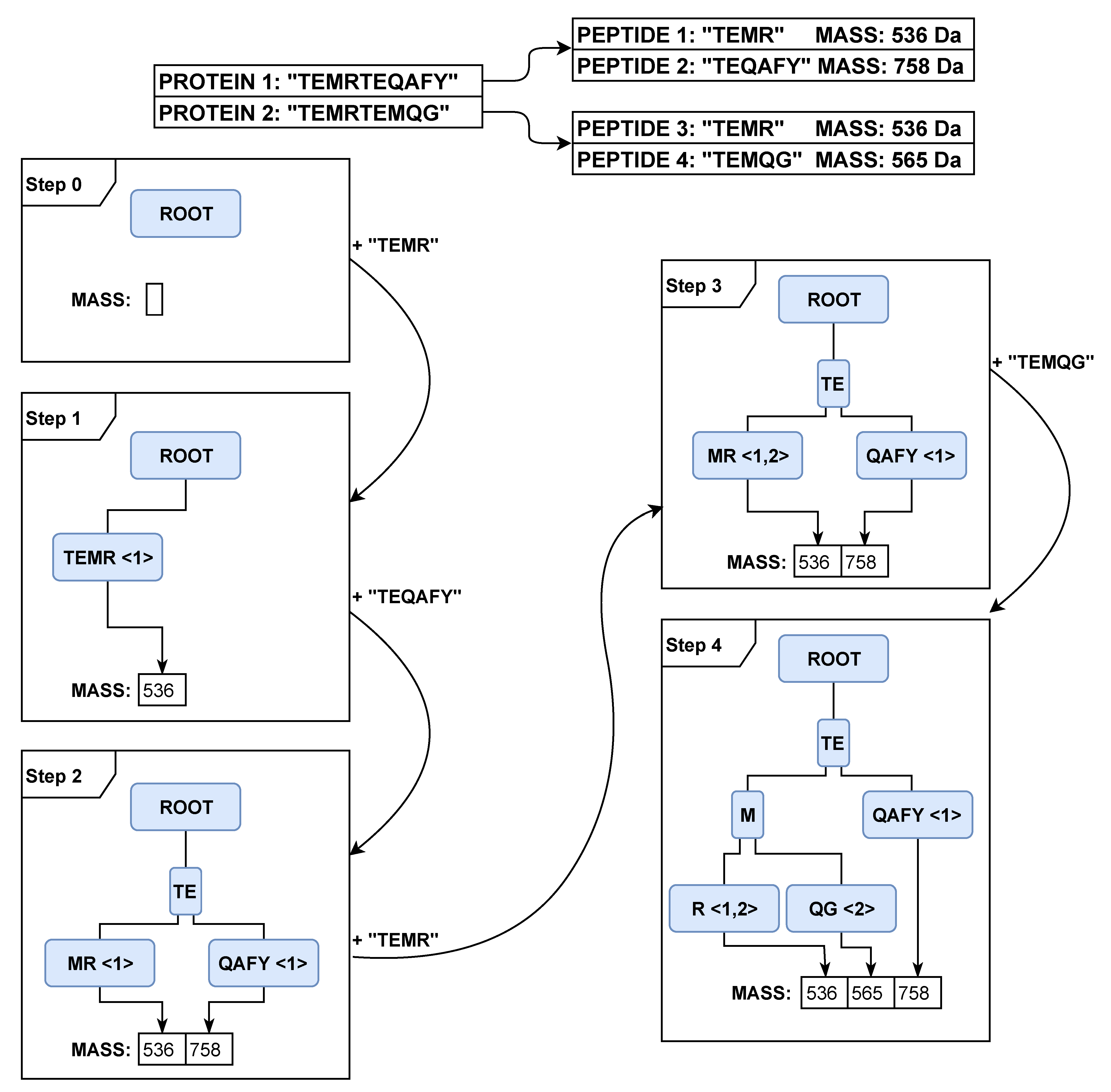
Listing 4) and visualize its application on an example in
Figure 7. At the beginning, a new column is added to the
protein table and to the
peptide table (Line 2). For each protein, an identifier is generated, and the protein descriptions in the protein table are updated (Lines 6 and 7). Next, each protein is digested into peptides, and we generate a list of peptides for each protein (Line 8). In our example, the protein “TEMRTEQAFY” is digested to the peptides “TEMR”, “TEQAFY” and the second protein “TEMRTEMQG” to the peptides “TEMR”, “TEMQG”. Subsequently, each peptide is inserted into the tree with the associated protein identifier (Line 10). At the beginning (
Figure 7, Step 0), the tree and the set of pepmasses are empty. In Step 1, we add the peptide “TEMR” to the tree. The end node gets a relation to the protein to which it belongs and a pointer to the set of pepmasses. In Step 2, we add the peptide “TEQAFY” to the tree. Because both peptides share the prefix “TE”, the first node is split, and two end nodes are created. Because the mass is different from other values in the set of pepmasses, a new entry in the set of pepmasses is created. In Step 3, we add the peptide “TEMR” to the tree. The complete sequence is already available, and only a new relation to another protein is created in the last node. In the last step, we add the peptide “TEMQG” to the tree. All the shared prefixes create an additional node, and only the suffix nodes are left with the relation to the protein that the peptide comes from and a pointer to the unique pepmasses.
Listing 4.
Transformation algorithm to transform protein data from FASTA format into the table schema using the radix tree data structure.
Listing 4.
Transformation algorithm to transform protein data from FASTA format into the table schema using the radix tree data structure.
4.4. Our Radix Tree Adaptation
The difference from the original radix tree by De La Briandais [
21] is our usage of the pessimistic path compression and lazy expansion of tree nodes that we adapted from Leis et al. [
22]. However, we do not use special adaptive node types and rely on the node16 design by Leis et al. [
22], because it matches our fan-out. Furthermore, our main use case is the efficient deduplication of proteins, which has a high priority on insertion, while the tree is traversed only once at the end of the process.
Another difference to other radix trees is that we are not indexing the peptides (i.e., adding a reference to the storage location of the peptides on disk). Instead, we link the protein relationship to the peptides. Hence, in order to adapt the radix tree to our use case of peptide deduplication, we extend the usual radix tree using a set of protein identifiers in each node. Hence, the tree contains all information for the peptide table (Line 13). The link to the proteins is needed to identify the proteins, based on the peptide identification. As a positive side effect, the peptide sequences are automatically deduplicated.
The next step is to calculate the masses for each peptide. Therefore, an additional map with the pepmass as the key and a list of the pointers to the end nodes in the radix tree as the value are created. For each of the end nodes, the pepmasses are calculated (Line 14), focusing first on the different modifications and second on the three charges. For each mass, the peptide sequences are calculated from the end node recursively. Beginning with the end node, which represents the end of a peptide sequence, one needs to traverse over all nodes to the top of the tree to get the sequence completely. The last step is to insert the data from the map with masses and the peptide sequences into the
pepmass table (Line 16). As we show in the example in
Figure 7, the radix tree needs only a few nodes to store multiple peptide sequences, while the map approach adds a new entry for each different sequence. If only one letter is different, only one additional node in the radix tree structure is added, which is more compressed than a whole additional entry in the map.
5. Evaluation
The evaluation compares resource consumption and the runtime of all four approaches for the peptide deduplication step. As test datasets, we used the entire UniProt/SwissProt protein database and a subset containing only proteins from the species Homo sapiens.
The Homo sapiens data set (
Table 5) comprises 4794 proteins and requires a storage size of 2.88 MB. It results in 484,479 overall peptides and 248,996 non-redundant peptides. Finally, these peptides result in a radix tree structure with 295,602 nodes. UniProt/SwissProt has a 255 MB storage size and contains 556,196 proteins and 37,403,696 peptides. After deduplication, only 23,254,068 peptides are left using 28,481,207 nodes in the radix tree.
5.1. Time Evaluation
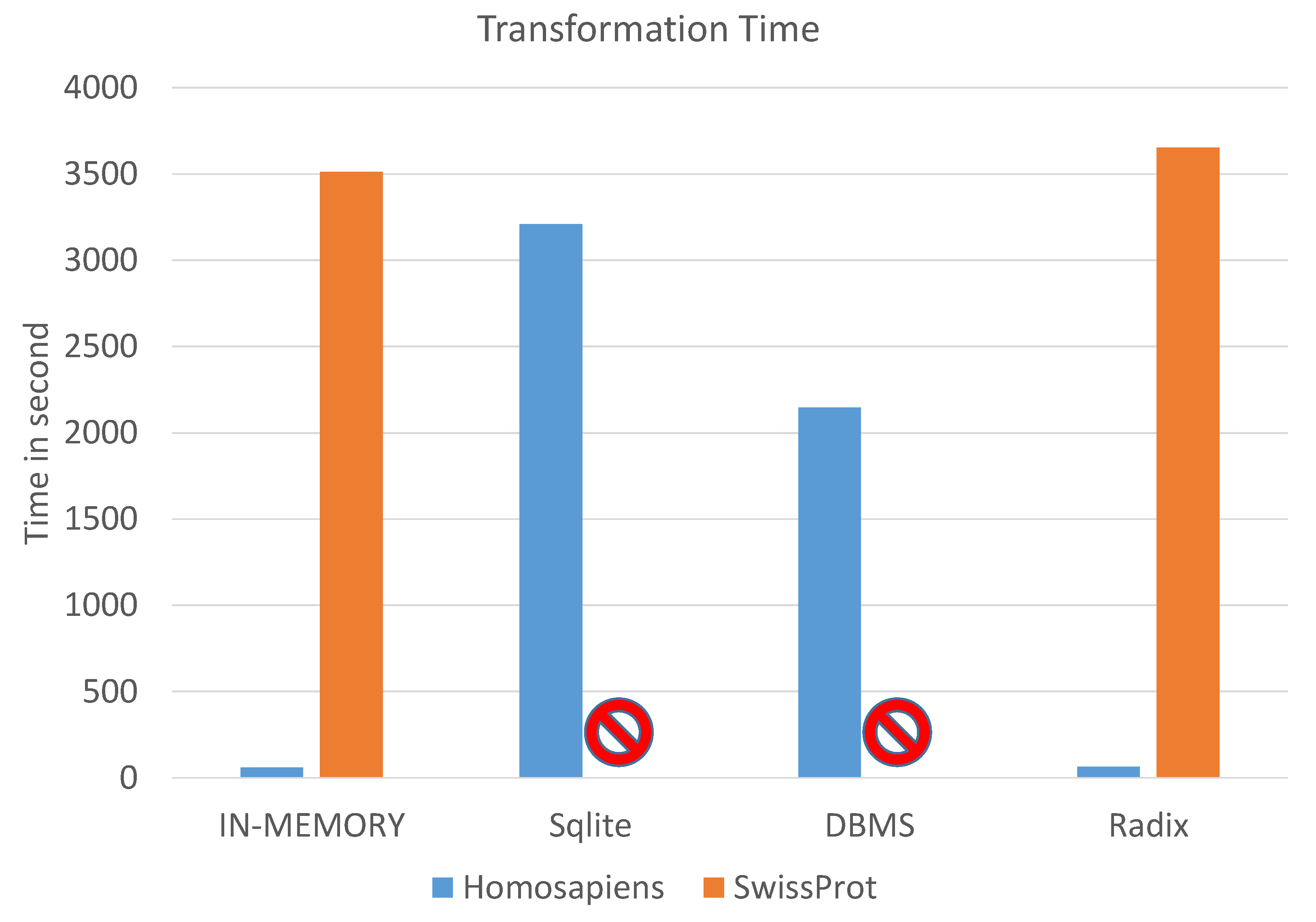
In order to identify the fastest indexing strategy, we repeated each measurement 50 times and averaged the runtimes. We visualize the runtimes for each strategy on each of the two protein databases in
Figure 8.
5.1.1. Homo sapiens Data Set
For the Homo sapiens protein database (represented by the blue bars in
Figure 8), the naive in-memory approach was five seconds faster than the radix tree method. Furthermore, we observed that the SQLite and the DBMS approach are very slow, requiring one half to one hour of runtime. This was caused by the high number of writes on the slow hard disk.
5.1.2. UniProtKB/SwissProt Data Set
The naive approach required for the indexing of the UniProtKB/SwissProt protein database (represented by the orange bars in
Figure 8) 58 min and the radix tree 60 min. The differences come from the calculation of the sequence recursively from the end node. In the radix tree structure, the mass is calculated traversing over the nodes, while in the naive approach, the whole sequence is accessible. This is not needed in the naive approach. The SQLite and the DBMS approaches took days on the UniProtKB/SwissProt data and are not applicable for such big protein sequence databases. Hence, only the naive in-memory approach and the radix tree approach seem to be promising.
5.2. Memory Consumption
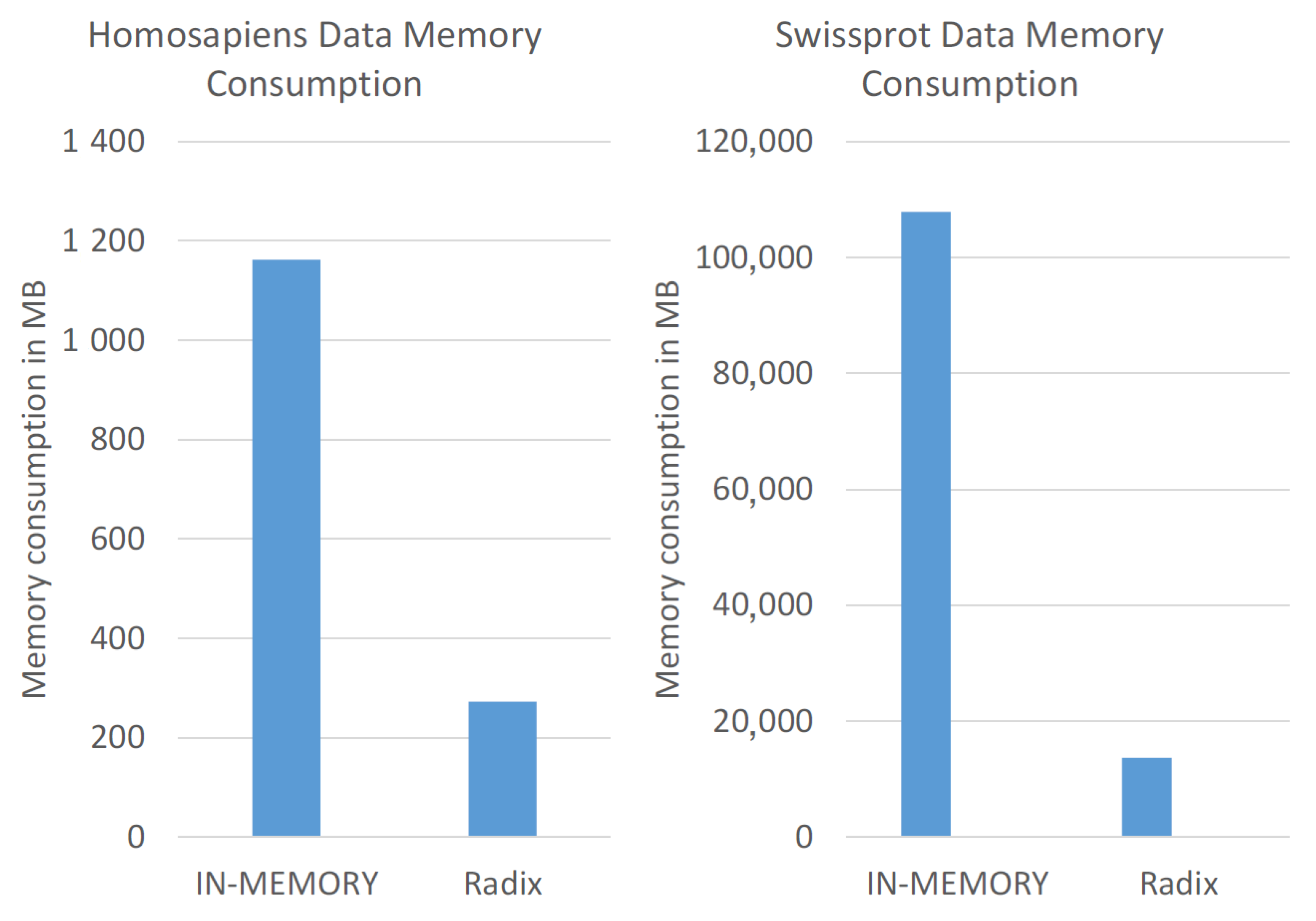
We only consider the in-memory approach and the radix tree method for the memory consumption evaluation in
Figure 9, because the other two approaches required too long computation times. The in-memory approach required around 1 GB for the Homo sapiens data set and over 100 GB for the UniProtKB/SwissProt data set. In contrast, the radix tree approach consumed less than 300 MB for the Homo sapiens data set and around 14 GB for the UniProtKB/SwissProt data set. Moreover, the memory consumption of the radix tree increased between the data sets by a factor of 50, while the naive in-memory approach by a factor of 92. Hence, the radix tree method combines in-memory speed and efficient data compression for sequence data and is, therefore, beneficial for storing the peptide sequence data.
5.3. Result Summary
The evaluation shows that radix trees are beneficial for peptide sequence data and could be transformed into our system. In our in-depth investigation, we could identify three benefits of the radix tree structure for peptide sequences. Firstly, most of the differences between peptides are in the first amino acid position. Hence, in higher levels of the tree, the sequences end up in an end node. Due to this fact, the tree consumes less memory. Secondly, many of the differences are only due to one letter. A single-letter difference results in only one additional node in the tree compared to two separate peptide sequences being stored in the naive approach. Thirdly, the insertion of new data into the tree automatically resolves redundancy, which is needed for further processing. Due to its minimized memory consumption, it is even possible to process the data in the RAM, reaching a considerable performance boost compared to its competitors.
6. Related Work
This section presents related work: the protein data indexing approach of Andromeda and the use case of the radix tree structure as a storage structure for sequence data. The protein search engine Andromeda, part of the MaxQuant Software suite, uses an indexing method on pepmasses to reduce the search space during the identification. This approach points to proteins in the protein sequence database files [
24]. Nevertheless, the data are still in a FASTA file. In contrast, we structure and transform them completely. Furthermore, we remove redundancies over all the protein databases uploaded into our system.
The software MetaProteomeAnalyzer proposes a relational structure to store the results in a relational database management system [
25]. In order to process the data, the MetaProteomeAnalyzer loads the whole data in a map structure into the memory, which required many resources of the system and corresponded to our in-memory map approach.
Enrico Siragusa proposed the radix tree as a structure to store DNA sequences as a preprocessing step in his thesis [
26], which is similar to our approach. In our work, however, we use the radix tree firstly to store the peptide sequences, and secondly, we extend the end nodes with the relationship to the proteins.
7. Conclusions
In our work, we investigate how to transform protein sequence databases into the final index schema. Therefore, we implemented the four approaches, in-memory map, HDD map, database engine, and radix tree, evaluated their performance, and summed up the results in
Table 6. The extended radix tree is the best structure for the peptide sequence data to transform the protein data into the index schema. The radix tree for peptide sequences combines the almost best performance with a minimal memory consumption across all competitors.
In the future, we will apply the radix tree as a storage system for protein databases and build a query engine around it. Consequently, we assume that we can speed up the current processes based on the FASTA file by using this tree structure. For the application as a cloud service, the protein data need to be uploaded before the later identification process can start.


 ,
, 














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
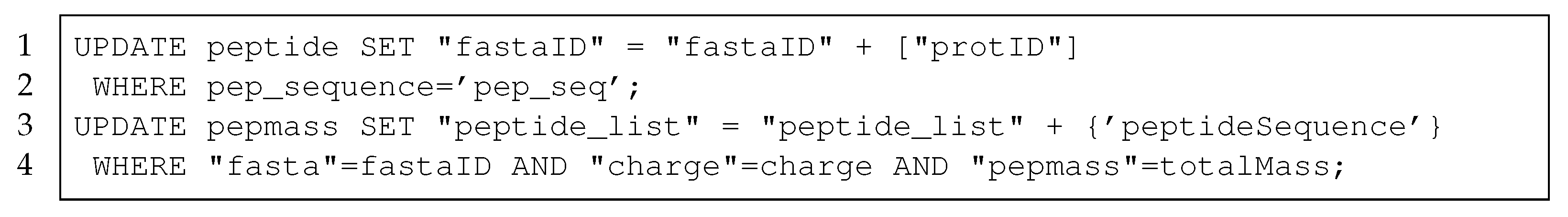{kind=link}
{kind=link}