Transcriptome Analysis of Elm (Ulmus pumila) Fruit to Identify Phytonutrients Associated Genes and Pathways


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and RNA Extraction
2.2. Library Construction and De Novo Assembly
2.3. Calculation of Genes’ Expression and Enrichment Analyses in U. pumila
2.4. qRT-PCR Analysis
3. Results
3.1. Transcriptome Profiling of U. pumila
3.2. Functional Annotations of Unigenes in U. pumila
3.3. Differentially Expressed Genes (DEGs) Calculation in U. pumila
3.4. GO, MapMan, and KEGG Enrichment Result of DEGs in U. pumila
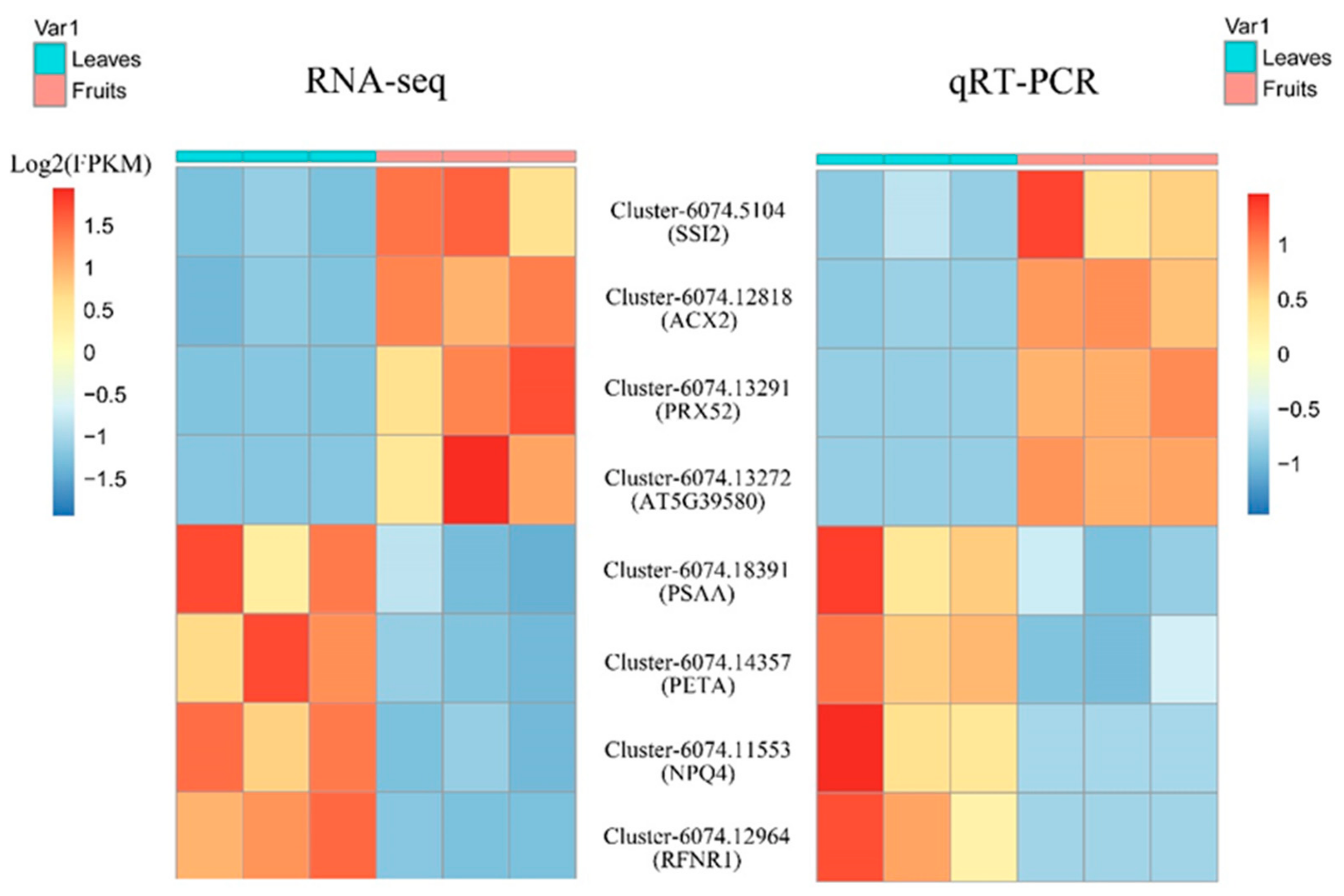
3.5. Real-Time Quantitative PCR Validation
3.6. qRT-PCR Analysis for Phytonutrient-Associated Genes in Different Stages of Fruit Development
4. Discussion
5. Conclusions
Data Archiving Statement
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kris-Etherton, P.M.; Hecker, K.D.; Bonanome, A.; Coval, S.M.; Binkoski, A.E.; Hilpert, K.F.; Griel, A.E.; Etherton, T.D. Bioactive compounds in foods: Their role in the prevention of cardiovascular disease and cancer. Am. J. Med. 2002, 113, 71–88. [Google Scholar] [CrossRef]
- Lampe, J.W. Health effects of vegetables and fruit: Assessing mechanisms of action in human experimental studies. Am. J. Clin. Nutr. 1999, 70, 475s–490s. [Google Scholar] [CrossRef]
- Neto, C.C. Cranberry and blueberry: Evidence for protective effects against cancer and vascular diseases. Mol. Nutr. Food Res. 2010, 51, 652–664. [Google Scholar] [CrossRef]
- Fernie, A.R.; Schauer, N. Metabolomics-assisted breeding: A viable option for crop improvement? Trends Genet. 2009, 25, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Chen, Y.; Wang, X.; Gong, G.; Li, G.; Li, C. Consumption of fruit, but not vegetables, may reduce risk of gastric cancer: Results from a meta-analysis of cohort studies. Eur. J. Cancer 2014, 50, 1498–1509. [Google Scholar] [CrossRef]
- Everitt, A.V.; Hilmer, S.N.; Brandmiller, J.C.; Jamieson, H.A.; Truswell, A.S.; Sharma, A.P.; Mason, R.S.; Morris, B.J.; Le, C.D. Dietary approaches that delay age-related diseases. Clin. Interv. Aging 2006, 1, 11–31. [Google Scholar] [CrossRef] [PubMed]
- Bendinelli, B.; Masala, G.; Saieva, C.; Salvini, S.; Calonico, C.; Sacerdote, C.; Agnoli, C.; Grioni, S.; Frasca, G.; Mattiello, A. Fruit, vegetables, and olive oil and risk of coronary heart disease in Italian women: The EPICOR Study. Am. J. Clin. Nutr. 2011, 94, 287–288. [Google Scholar] [CrossRef]
- Wiegrefe, S.J.; Sytsma, K.J.; Guries, R.P. Phylogeny of elms (Ulmus, Ulmaceae): Molecular evidence for a sectional classification. Syst. Bot. 1994, 19, 590–612. [Google Scholar] [CrossRef]
- Ghosh, C.; Yang, S.H.; Hwang, S.G. Methanol extract of Ulmus pumila. L exerts potent anti-inflammatory effects in murine macrophages and mouse skin. FASEB J. 2013, 27, 1093. [Google Scholar]
- Wang, D.; Xia, M.Y.; Cui, Z. New triterpenoids isolated from the root bark of Ulmus pumila L. Chem. Pharm. Bull. 2006, 54, 775–778. [Google Scholar] [CrossRef]
- Feng, Z.T.; Deng, Y.Q.; Fan, H.; Sun, Q.J.; Sui, N.; Wang, B.S. Effects of NaCl stress on the growth and photosynthetic characteristics of Ulmus pumila L. seedlings in sand culture. Photosynthetica 2014, 52, 313–320. [Google Scholar] [CrossRef]
- Mu, D.Y.; Zwiazek, J.J.; Li, Z.Q.; Zhang, W.Q. Genotypic variation in salt tolerance of Ulmus pumila plants obtained by shoot micropropagation. Acta Physiol. Plant 2016, 38, 188. [Google Scholar] [CrossRef]
- Zhu, J.F.; Yang, X.Y.; Liu, Z.X.; Zhang, H.X. Identification and Target Prediction of MicroRNAs in Ulmus pumila L. Seedling Roots under Salt Stress by High-Throughput Sequencing. Forests 2016, 7, 318. [Google Scholar] [CrossRef]
- Zhou, Z.H.; Shao, H.J.; Han, X.; Wang, K.J.; Gong, C.P.; Yang, X.B. The extraction efficiency enhancement of polyphenols from Ulmus pumila L. barks by trienzyme-assisted extraction. Ind. Crop Prod. 2017, 97, 401–408. [Google Scholar] [CrossRef]
- Yu, S.L. Nutrition and health effects of elm fruits. Food Nutr. China 2009, 9, 60–62. [Google Scholar]
- Li, Y.; Wang, Y.; Xue, H.; Pritchard, H.W.; Wang, X.F. Changes in the mitochondrial protein profile due to ROS eruption during ageing of elm (Ulmus pumila L.) seeds. Plant Physiol. Biochem. 2017, 114, 72–87. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, Y.; Xue, H.; Pritchard, H.W.; Wang, X.F. Reactive oxygen species-provoked mitochondria-dependent cell death during ageing of elm (Ulmus pumila L.) seeds. Plant J. 2015, 81, 438–452. [Google Scholar] [CrossRef] [PubMed]
- Yuan, F.; Lyu, M.J.A.; Leng, B.Y.; Zhu, X.G.; Wang, B.S. The transcriptome of NaCl-treated Limonium bicolor leaves reveals the genes controlling salt secretion of salt gland. Plant Mol. Biol. 2016, 91, 241–256. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, Q.; Zhai, H.; Li, Y.; Wang, X.; Liu, Q.; He, S. Transcript profile analysis reveals important roles of jasmonic acid signalling pathway in the response of sweet potato to salt stress. Sci. Rep. UK 2017, 7, 40819. [Google Scholar] [CrossRef] [Green Version]
- Yuan, F.; Lyu, M.J.A.; Leng, B.Y.; Zheng, G.Y.; Feng, Z.T.; Li, P.H.; Zhu, X.G.; Wang, B.S. Comparative transcriptome analysis of developmental stages of the Limonium bicolor leaf generates insights into salt gland differentiation. Plant Cell Environ. 2015, 38, 1637–1657. [Google Scholar] [CrossRef]
- Xu, J.J.; Li, Y.Y.; Ma, X.L.; Ding, J.F.; Wang, K.; Wang, S.S.; Tian, Y.; Zhang, H.; Zhu, X.G. Whole transcriptome analysis using next-generation sequencing of model species Setaria viridis to support C-4 photosynthesis research. Plant Mol. Biol. 2013, 83, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Li, J.P.; Yuan, F.; Yang, Z.; Wang, B.S.; Chen, M. Transcriptome profiling of genes involved in photosynthesis in Elaeagnus angustifolia L. under salt stress. Photosynthetica 2018, 56, 998–1009. [Google Scholar] [CrossRef]
- Yang, S.; Li, L.; Zhang, J.L.; Geng, Y.; Guo, F.; Wang, J.G.; Meng, J.J.; Sui, N.; Wan, S.B.; Li, X.G. Transcriptome and Differential Expression Profiling Analysis of the Mechanism of Ca2+ Regulation in Peanut (Arachis hypogaea) Pod Development. Front. Plant Sci. 2017, 8, 1609. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wang, Y.; Wei, X.C.; Zhao, X.; Wang, B.S.; Sui, N. Transcription Profiles of Genes Related to Hormonal Regulations Under Salt Stress in Sweet Sorghum. Plant Mol. Biol. Rep. 2017, 35, 586–599. [Google Scholar] [CrossRef]
- Du, M.F.; Ding, G.J.; Cai, Q.O. The Transcriptomic Responses of Pinus massoniana to Drought Stress. Forests 2018, 9, 326. [Google Scholar] [CrossRef]
- Cai, Q.F.; Li, B.; Lin, F.R.; Huang, P.; Guo, W.Y.; Zheng, Y.Q. De Novo Sequencing and Assembly Analysis of Transcriptome in Pinus bungeana Zucc. ex Endl. Forests 2018, 9, 156. [Google Scholar] [CrossRef]
- Zhao, D.Q.; Zhang, X.Y.; Fang, Z.W.; Wu, Y.Q.; Tao, J. Physiological and Transcriptomic Analysis of Tree Peony (Paeonia section Moutan DC.) in Response to Drought Stress. Forests 2019, 10, 135. [Google Scholar] [CrossRef]
- Wu, H.X.; Jia, H.M.; Ma, X.W.; Wang, S.B.; Yao, Q.S.; Xu, W.T.; Zhou, Y.G.; Gao, Z.S.; Zhan, R.L. Transcriptome and proteomic analysis of mango (Mangifera indica Linn) fruits. J. Proteom. 2014, 105, 19–30. [Google Scholar] [CrossRef]
- Munoz-Espinoza, C.; Di Genova, A.; Correa, J.; Silva, R.; Maass, A.; Gonzalez-Aguero, M.; Orellana, A.; Hinrichsen, P. Transcriptome profiling of grapevine seedless segregants during berry development reveals candidate genes associated with berry weight. BMC Plant Biol. 2016, 16, 104. [Google Scholar] [CrossRef]
- Sweetman, C.; Wong, D.C.J.; Ford, C.M.; Drew, D.P. Transcriptome analysis at four developmental stages of grape berry (Vitis vinifera cv. Shiraz) provides insights into regulated and coordinated gene expression. BMC Genom. 2012, 13, 691. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Moran, Y.; Levin, J.Z.; Thompson, D.A.; Ido, A.; Xian, A.; Lin, F.; Raktima, R.; Qiandong, Z. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644. [Google Scholar] [CrossRef] [PubMed]
- NCBI Sequence Read Archive (SRA) Database. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/sra (accessed on 27 August 2019).
- Bo, L.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar]
- Haas, B.J.; Alexie, P.; Moran, Y.; Manfred, G.; Blood, P.D.; Joshua, B.; Matthew Brian, C.; David, E.; Bo, L.; Matthias, L. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Thimm, O.; Blasing, O.; Gibon, Y.; Nagel, A.; Meyer, S.; Kruger, P.; Selbig, J.; Muller, L.A.; Rhee, S.Y.; Stitt, M. MAPMAN: A user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 2010, 37, 914–939. [Google Scholar] [CrossRef]
- Chen, X.; Xizeng, M.; Jiaju, H.; Yang, D.; Jianmin, W.; Shan, D.; Lei, K.; Ge, G.; Chuan-Yun, L.; Liping, W. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, 316–322. [Google Scholar]
- Latocha, P. The Nutritional and Health Benefits of Kiwiberry (Actinidia arguta)—A Review. Plant Food Hum. Nutr. 2017, 72, 325–334. [Google Scholar] [CrossRef]
- Aune, D.; Giovannucci, E.; Boffetta, P.; Fadnes, L.T.; Keum, N.; Norat, T.; Greenwood, D.C.; Riboli, E.; Vatten, L.J.; Tonstad, S. Fruit and vegetable intake and the risk of cardiovascular disease, total cancer and all-cause mortality-a systematic review and dose-response meta-analysis of prospective studies. Int. J. Epidemiol. 2017, 46, 1029–1056. [Google Scholar] [CrossRef]
- Block, G.; Patterson, B.; Subar, A. Fruit, vegetables, and cancer prevention: A review of the epidemiological evidence. Nutr. Cancer 1992, 18, 1–29. [Google Scholar] [CrossRef]
- Steinmetz, K.A.; Potter, J.D. Vegetables, fruit, and cancer prevention: A review. J. Am. Diet Assoc. 1996, 96, 1027–1039. [Google Scholar] [CrossRef]
- Dukic, M.; Dunisijevic-Bojovic, D.; Samuilov, S. The Influence of Cadmium and Lead on Ulmus Pumila L. Seed Germination and Early Seedling Growth. Arch. Biol. Sci. 2014, 66, 253–259. [Google Scholar] [CrossRef]
- Ghosh, C.; Chung, H.Y.; Nandre, R.M.; Lee, J.H.; Jeond, T.I.; Kim, I.S.; Yang, S.H.; Hwang, S.G. An active extract of Ulmus pumila inhibits adipogenesis through regulation of cell cycle progression in 3T3-L1 cells. Food Chem. Toxicol. 2012, 50, 2009–2015. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Xi, W.M.; Rahmlow, A.; Kong, H.Y.; Zhang, Z.; Shangguan, Z.P. Effects of forest plantation types on leaf traits of Ulmus pumila and Robinia pseudoacacia on the Loess Plateau, China. Ecol. Eng. 2016, 97, 416–425. [Google Scholar] [CrossRef]
- Yu, K.; Xu, Q.; Da, X.; Guo, F.; Ding, Y.; Deng, X. Transcriptome changes during fruit development and ripening of sweet orange (Citrus sinensis). BMC Genom. 2012, 13, 10. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wang, H.; Yi, H.; Zhai, W.; Wang, G.; Fu, Q. Transcriptome profiling of Cucumis melo fruit development and ripening. Hortic. Res. 2016, 3, 16014. [Google Scholar] [CrossRef] [PubMed]
- Li, R.J.; Gao, X.; Li, L.M.; Liu, X.L.; Wang, Z.Y.; Lü, S.Y. De novo assembly and characterization of the fruit transcriptome of Idesia polycarpa reveals candidate genes for lipid biosynthesis. Front. Plant Sci. 2016, 7, 801. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, J.; Teixeira, P. Development of probiotic fruit juice powders by spray-drying: A review. Food Rev. Int. 2017, 33, 335–358. [Google Scholar] [CrossRef]
- Seymour, G.B.; Ostergaard, L.; Chapman, N.H.; Knapp, S.; Martin, C. Fruit development and ripening. Annu. Rev. Plant Biol. 2013, 64, 219–241. [Google Scholar] [CrossRef]
- Kumar, R.; Khurana, A.; Sharma, A.K. Role of plant hormones and their interplay in development and ripening of fleshy fruits. J. Exp. Bot. 2014, 65, 4561–4575. [Google Scholar] [CrossRef]
- Zhu, Y.M.; Zheng, P.; Varanasi, V.; Shin, S.B.; Main, D.; Curry, E.; Mattheis, J.P. Multiple plant hormones and cell wall metabolism regulate apple fruit maturation patterns and texture attributes. Tree Genet. Genomes 2012, 8, 1389–1406. [Google Scholar] [CrossRef]
- Xue, S.; Dong, M.; Liu, X.; Xu, S.; Pang, J.; Zhang, W.; Weng, Y.; Ren, H. Classification of fruit trichomes in cucumber and effects of plant hormones on type II fruit trichome development. Planta 2019, 249, 407–416. [Google Scholar] [CrossRef]
- Jiang, Y.; Joyce, D.C.; Macnish, A.J. Effect of Abscisic Acid on Banana Fruit Ripening in Relation to the Role of Ethylene. J. Plant Growth Regul. 2000, 19, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Balk, E.M.; Raman, G.; Tatsioni, A.; Chung, M.; Lau, J.; Rosenberg, I.H. Vitamin B6, B12, and folic acid supplementation and cognitive function: A systematic review of randomized trials. Arch. Intern. Med. 2007, 167, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Mathers, J.C. Plant foods for human health: Research challenges. Proc. Nutr. Soc. 2006, 65, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Luthje, S.; Deswal, R.; Agrawal, G.K. Plant-based Foods: Seed, Nutrition and Human Health. Proteomics 2015, 15, 1638. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Maximova, S.N.; Guiltinan, M.J. Characterization of a stearoyl-acyl carrier protein desaturase gene family from chocolate tree, Theobroma cacao L. Front. Plant Sci. 2015, 6, 239. [Google Scholar] [CrossRef] [PubMed]
- Bryant, F.M.; Munoz-Azcarate, O.; Kelly, A.A.; Beaudoin, F.; Kurup, S.; Eastmond, P.J. ACYL-ACYL CARRIER PROTEIN DESATURASE2 and 3 Are Responsible for Making Omega-7 Fatty Acids in the Arabidopsis Aleurone. Plant Physiol. 2016, 172, 154–162. [Google Scholar] [CrossRef]
- Chi, X.; Yang, Q.; Pan, L.; Chen, M.; He, Y.; Yang, Z.; Yu, S. Isolation and characterization of fatty acid desaturase genes from peanut (Arachis hypogaea L.). Plant Cell Rep. 2011, 30, 1393–1404. [Google Scholar] [CrossRef]
- Gerber, M. Omega-3 fatty acids and cancers: A systematic update review of epidemiological studies. Br. J. Nutr. 2012, 107, S228–S239. [Google Scholar] [CrossRef]
- Weber, H. Fatty acid-derived signals in plants. Trends Plant Sci. 2002, 7, 217–224. [Google Scholar] [CrossRef]
- Hemaiswarya, S.; Doble, M. Combination of phenylpropanoids with 5-fluorouracil as anti-cancer agents against human cervical cancer (HeLa) cell line. Phytomedicine 2013, 20, 151–158. [Google Scholar] [CrossRef]
- Rochfort, S.; Parker, A.J.; Dunshea, F.R. Plant bioactives for ruminant health and productivity. Phytochemistry 2008, 69, 299–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Marchand, L. Cancer preventive effects of flavonoids—A review. Biomed. Pharmacother. 2002, 56, 296–301. [Google Scholar] [CrossRef]
- Romagnolo, D.F.; Selmin, O.I. Flavonoids and cancer prevention: A review of the evidence. J. Nutr. Gerontol. Geriatr. 2012, 31, 206–238. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, S.S.C.; Subramanian, I.P.; Subramanian, S.P. Isolation, characterization of syringin, phenylpropanoid glycoside from, Musa paradisiaca, tepal extract and evaluation of its antidiabetic effect in streptozotocin-induced diabetic rats. Biomed. Prev. Nutr. 2014, 4, 105–111. [Google Scholar] [CrossRef]
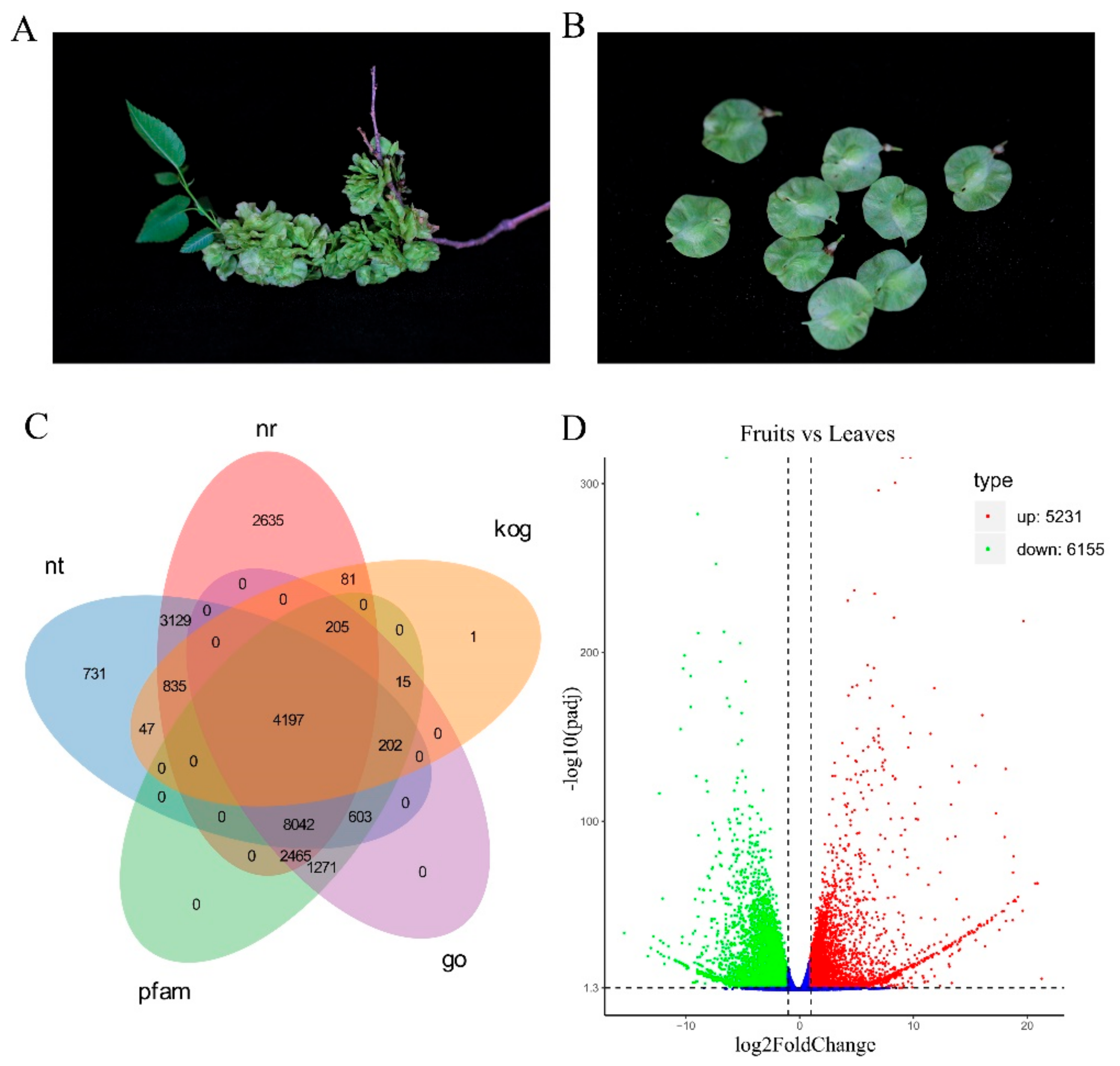



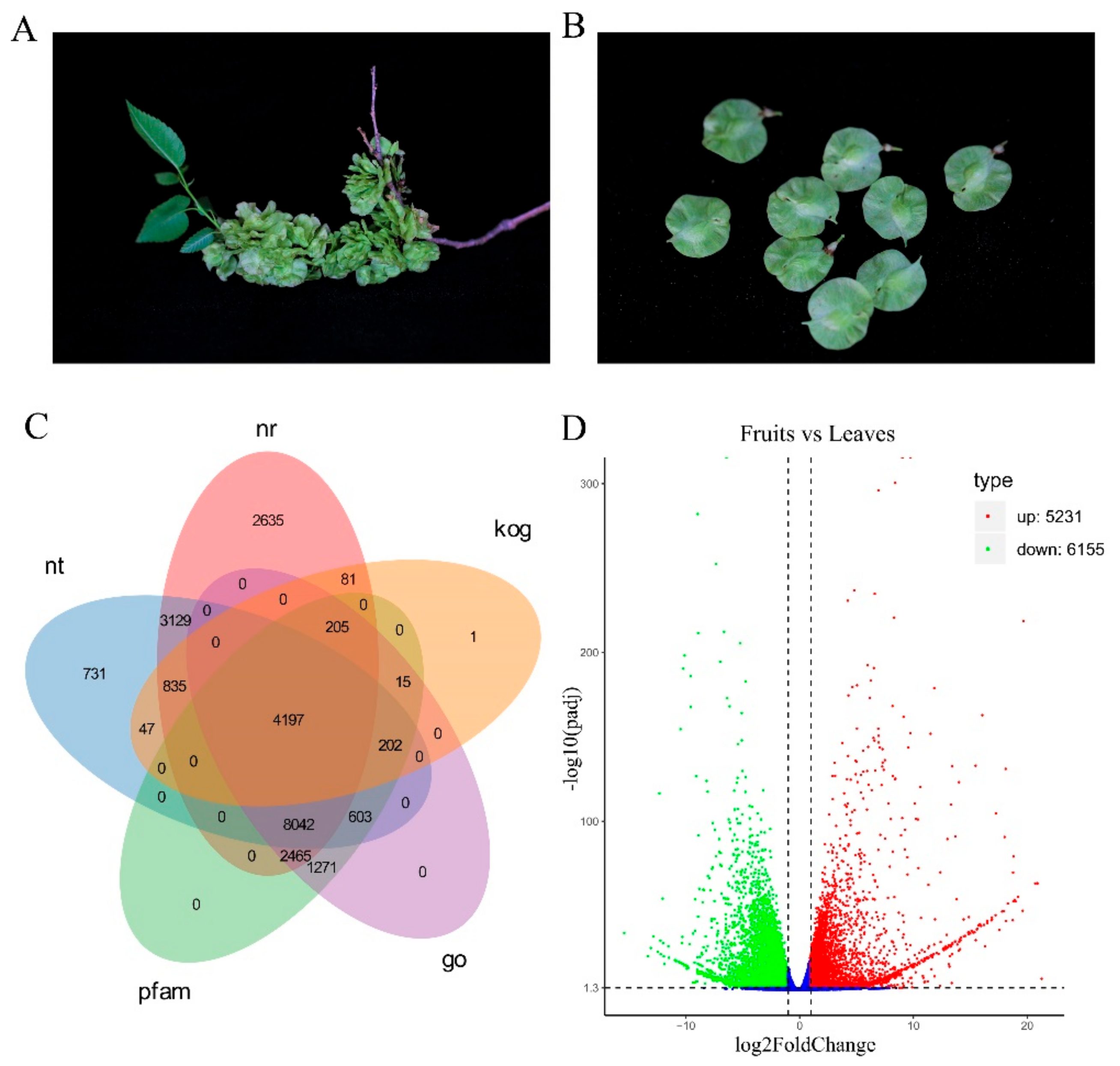{kind=link}
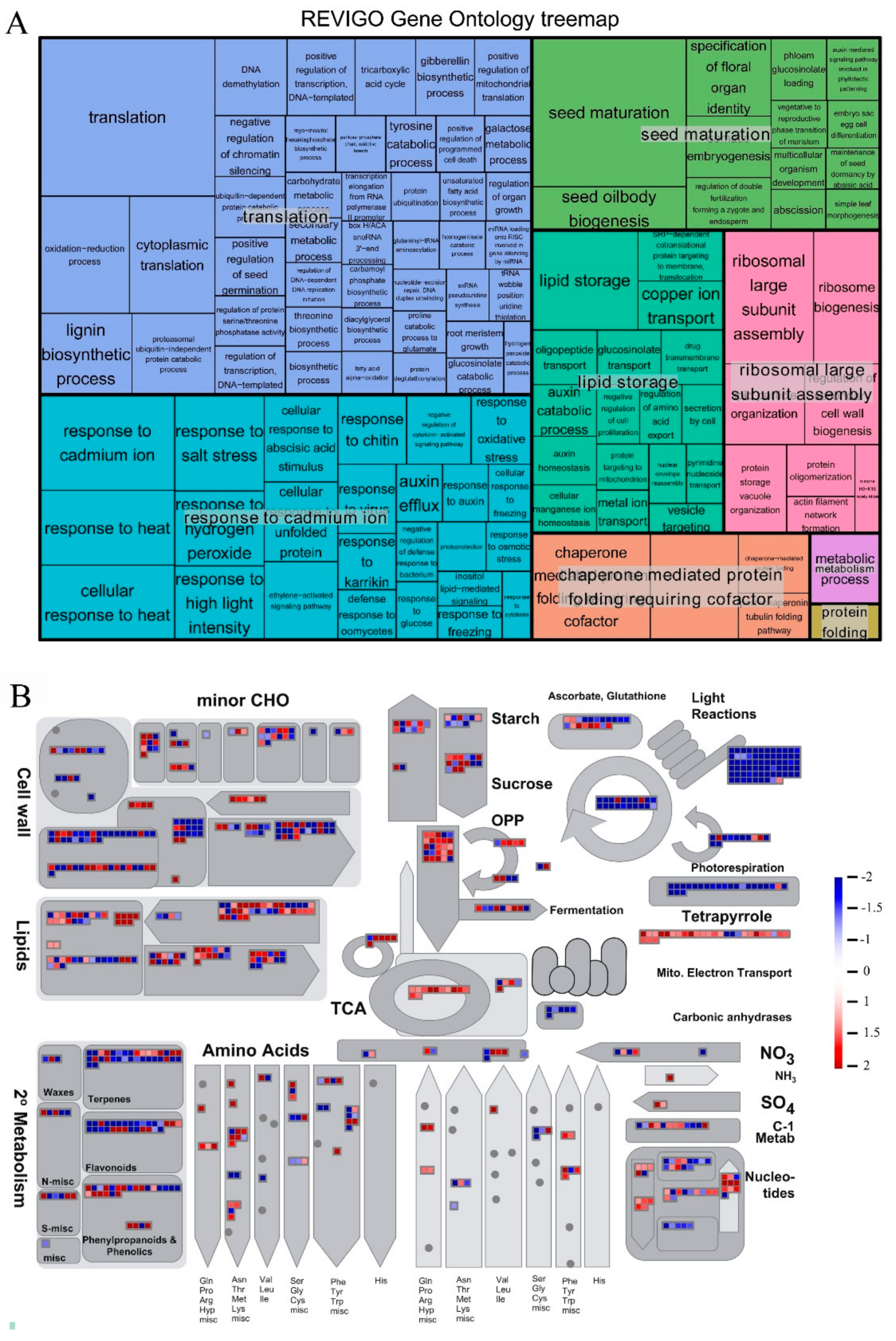
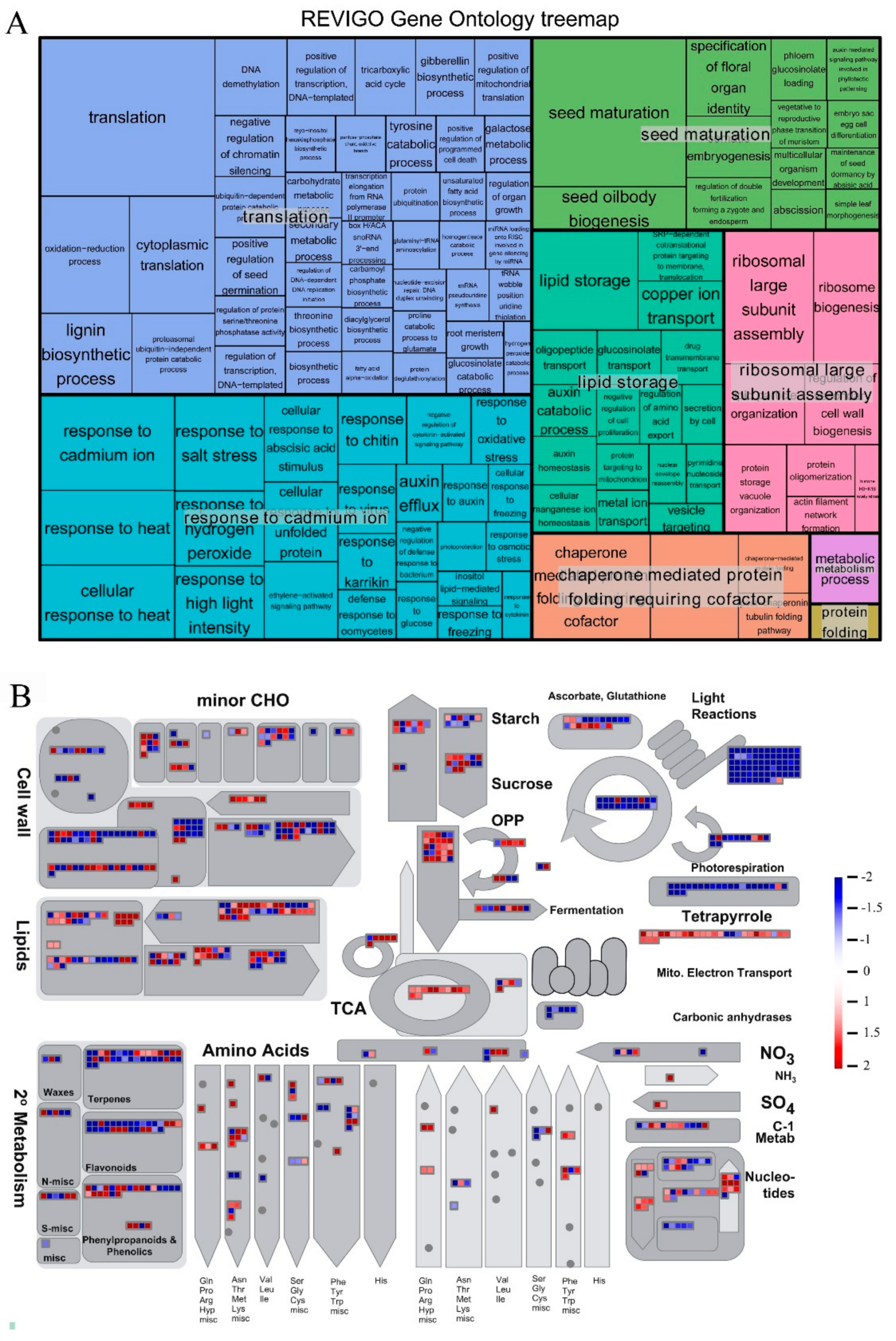{kind=link}
{kind=link}
{kind=link}
| Sample Name | Sample Description | Total Reads | Total Mapped | Ratio of Mapped Reads |
|---|---|---|---|---|
| Up_F_1 | Fruits replication 1 | 51,869,958 | 41,402,822 | 79.82% |
| Up_F_2 | Fruits replication 2 | 54,374,336 | 44,276,232 | 81.43% |
| Up_F_3 | Fruits replication 3 | 63,237,860 | 51,316,902 | 81.15% |
| Up_L_1 | Leaves replication 1 | 66,499,546 | 53,135,314 | 79.90% |
| Up_L_2 | Leaves replication 2 | 64,015,920 | 50,704,484 | 79.21% |
| Up_L_3 | Leaves replication 3 | 53,666,908 | 42,942,126 | 80.02% |
| Gene ID | L2fc | Padj | Arabidopsis ID | Gene Description |
|---|---|---|---|---|
| Upregulated | ||||
| Cluster-6074.11735 | 9.880 | 0.00 | AT5G40420 | oleosin 2 |
| Cluster-6074.11841 | 9.147 | 0.00 | ||
| Cluster-6074.12126 | 13.102 | 0.00 | AT2G25890 | |
| Cluster-6074.12191 | 18.879 | 0.00 | AT5G44120 | CRUCIFERINA |
| Cluster-6074.12375 | 13.383 | 0.00 | ||
| Cluster-6074.13108 | 18.27 | 0.00 | AT1G03890 | |
| Cluster-6074.12362 | 8.535 | 3.45 × 10−301 | AT1G62710 | beta vacuolar processing enzyme |
| Cluster-6074.13152 | 7.047 | 9.73 × 10−297 | AT5G12380 | annexin 8 |
| Cluster-6074.12791 | 14.590 | 6.74 × 10−295 | AT4G25140 | oleosin 1 |
| Cluster-6074.12340 | 4.909 | 1.58 × 10−237 | AT5G49360 | beta-xylosidase 1 |
| Cluster-6074.11747 | 6.724 | 1.65 × 10−235 | AT5G12380 | annexin 8 |
| Cluster-6074.10421 | 4.358 | 2.17 × 10−231 | AT1G21410 | |
| Cluster-6074.12488 | 8.434 | 2.01 × 10−221 | AT4G37370 | cytochrome P450, family 81, subfamily D, polypeptide 8 |
| Cluster-6074.12147 | 19.796 | 1.96 × 10−219 | AT1G03890 | |
| Cluster-6074.13683 | 6.109 | 4.23 × 10−193 | AT1G04560 | |
| Downregulated | ||||
| Cluster-6074.9536 | −6.306 | 0.00 | ||
| Cluster-6074.1319 | −8.832 | 8.48 × 10−283 | AT2G45180 | |
| Cluster-6074.23644 | −7.223 | 2.72 × 10−253 | AT1G20030 | |
| Cluster-6074.18869 | −6.508 | 6.11 × 10−213 | AT5G20740 | |
| Cluster-6074.1201 | −8.801 | 2.92 × 10−212 | AT4G11650 | osmotin 34 |
| Cluster-6074.20011 | −5.099 | 3.94 × 10−206 | AT2G22540 | short vegetative phase |
| Cluster-6074.25984 | −9.979 | 6.56 × 10−199 | ||
| Cluster-6074.18265 | −6.868 | 2.59 × 10−195 | AT5G35630 | glutamine synthetase 2 |
| Cluster-6074.1293 | −10.101 | 4.36 × 10−191 | ||
| Cluster-6074.22654 | −9.467 | 9.42 × 10−187 | AT5G59190 | |
| Cluster-6074.1730 | −4.625 | 1.19 × 10−183 | AT5G22430 | |
| Cluster-6074.24974 | −6.239 | 1.35 × 10−173 | AT5G67150 | |
| Cluster-6074.16553 | −6.034 | 9.25 × 10−169 | AT3G54420 | homolog of carrot EP3-3 chitinase |
| Cluster-6074.1245 | −9.437 | 2.26 × 10−168 | ||
| Cluster-6074.21282 | −4.960 | 8.82 × 10−165 | AT4G15440 | hydroperoxide lyase 1 |
| Item | Pathway | Annotated Gene Number | Enriched Gene Number |
|---|---|---|---|
| Amino acid | Glycine, serine and threonine metabolism | 66 | 29 |
| Alanine, aspartate and glutamate metabolism | 46 | 17 | |
| Arginine biosynthesis | 36 | 13 | |
| Tyrosine metabolism | 52 | 17 | |
| Cysteine and methionine metabolism | 99 | 29 | |
| Cyanoamino acid metabolism | 56 | 16 | |
| Beta-Alanine metabolism | 44 | 12 | |
| Valine, leucine and isoleucine degradation | 48 | 13 | |
| Fatty acid | Glycosphingolipid biosynthesis-globo series | 10 | 4 |
| Fatty acid biosynthesis | 65 | 25 | |
| Selenocompound metabolism | 22 | 7 | |
| Biosynthesis of unsaturated fatty acids | 31 | 9 | |
| Glycerophospholipid metabolism | 86 | 24 | |
| Glycerolipid metabolism | 72 | 19 | |
| Natural compounds | Diterpenoid biosynthesis | 23 | 11 |
| Zeatin biosynthesis | 18 | 7 | |
| Phenylpropanoid biosynthesis | 147 | 49 | |
| Isoquinoline alkaloid biosynthesis | 31 | 8 | |
| Vitamin | Biotin metabolism | 19 | 7 |
| Vitamin B6 metabolism | 14 | 5 | |
| Ascorbate and aldarate metabolism | 60 | 17 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Zhang, X.; Li, M.; Wang, N.; Qu, X.; Fan, S. Transcriptome Analysis of Elm (Ulmus pumila) Fruit to Identify Phytonutrients Associated Genes and Pathways. Forests 2019, 10, 738. https://0-doi-org.brum.beds.ac.uk/10.3390/f10090738
Zhang L, Zhang X, Li M, Wang N, Qu X, Fan S. Transcriptome Analysis of Elm (Ulmus pumila) Fruit to Identify Phytonutrients Associated Genes and Pathways. Forests. 2019; 10(9):738. https://0-doi-org.brum.beds.ac.uk/10.3390/f10090738
Chicago/Turabian StyleZhang, Luoyan, Xuejie Zhang, Mengfei Li, Ning Wang, Xiaojian Qu, and Shoujin Fan. 2019. "Transcriptome Analysis of Elm (Ulmus pumila) Fruit to Identify Phytonutrients Associated Genes and Pathways" Forests 10, no. 9: 738. https://0-doi-org.brum.beds.ac.uk/10.3390/f10090738