Distance from the Forest Edge Influences Soil Fungal Communities Colonizing a Reclaimed Soil Borrow Site in Boreal Mixedwood Forest

Abstract
:1. Introduction
2. Materials and Methods
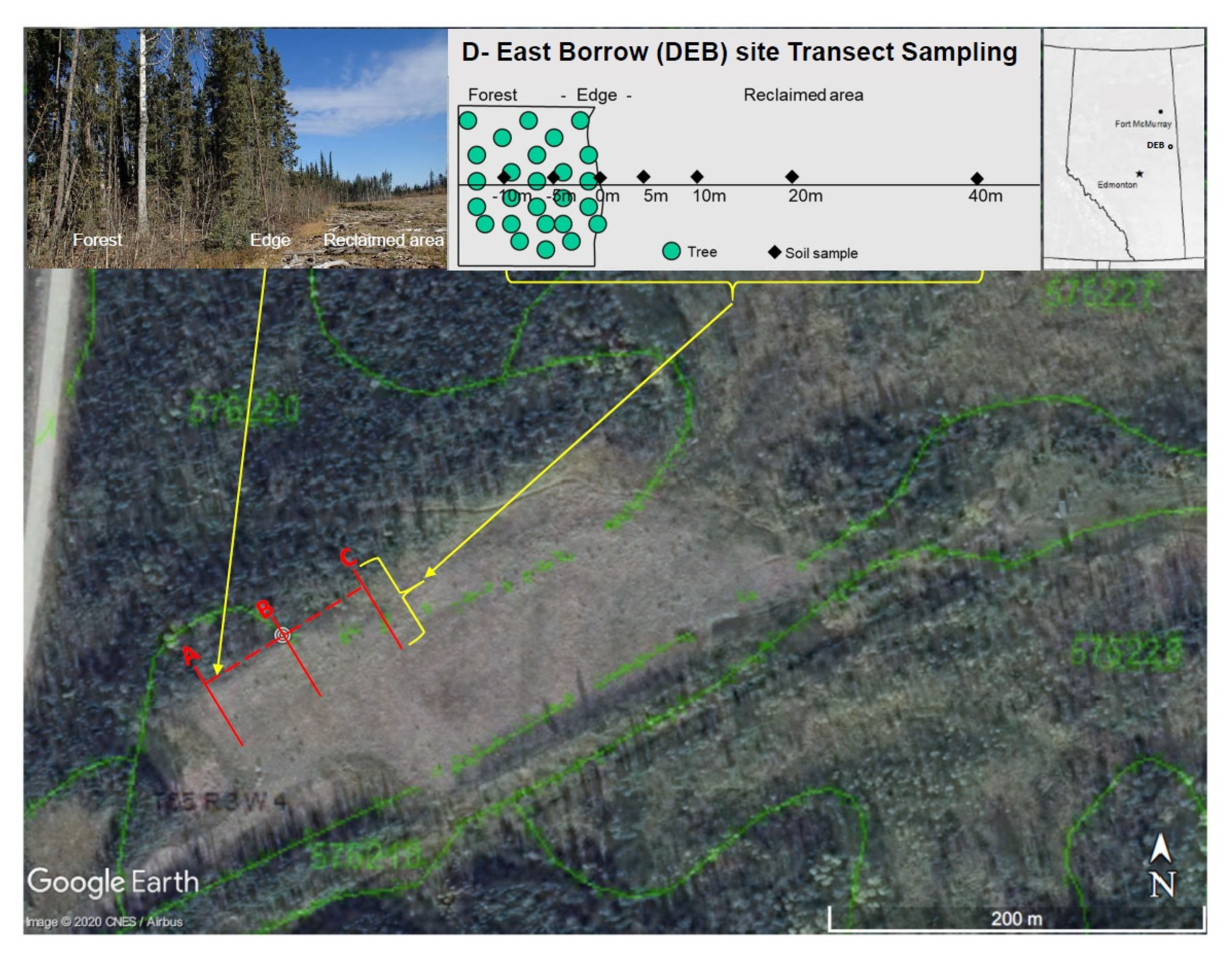
2.1. Site Description, Transect Layout, and Sample Collection
2.2. Culturing and DNA-Based Identification of Fungi from Aspen Roots
2.3. DNA Extraction and Bioinformatic Analysis from Aspen Root Tissue Using the Roche 454 Platform
2.4. DNA Extraction and Bioinformatic Analysis from Forest Floor Organic and Mineral Soil Samples Using the Illumina Platform
2.5. Nutrient Analysis
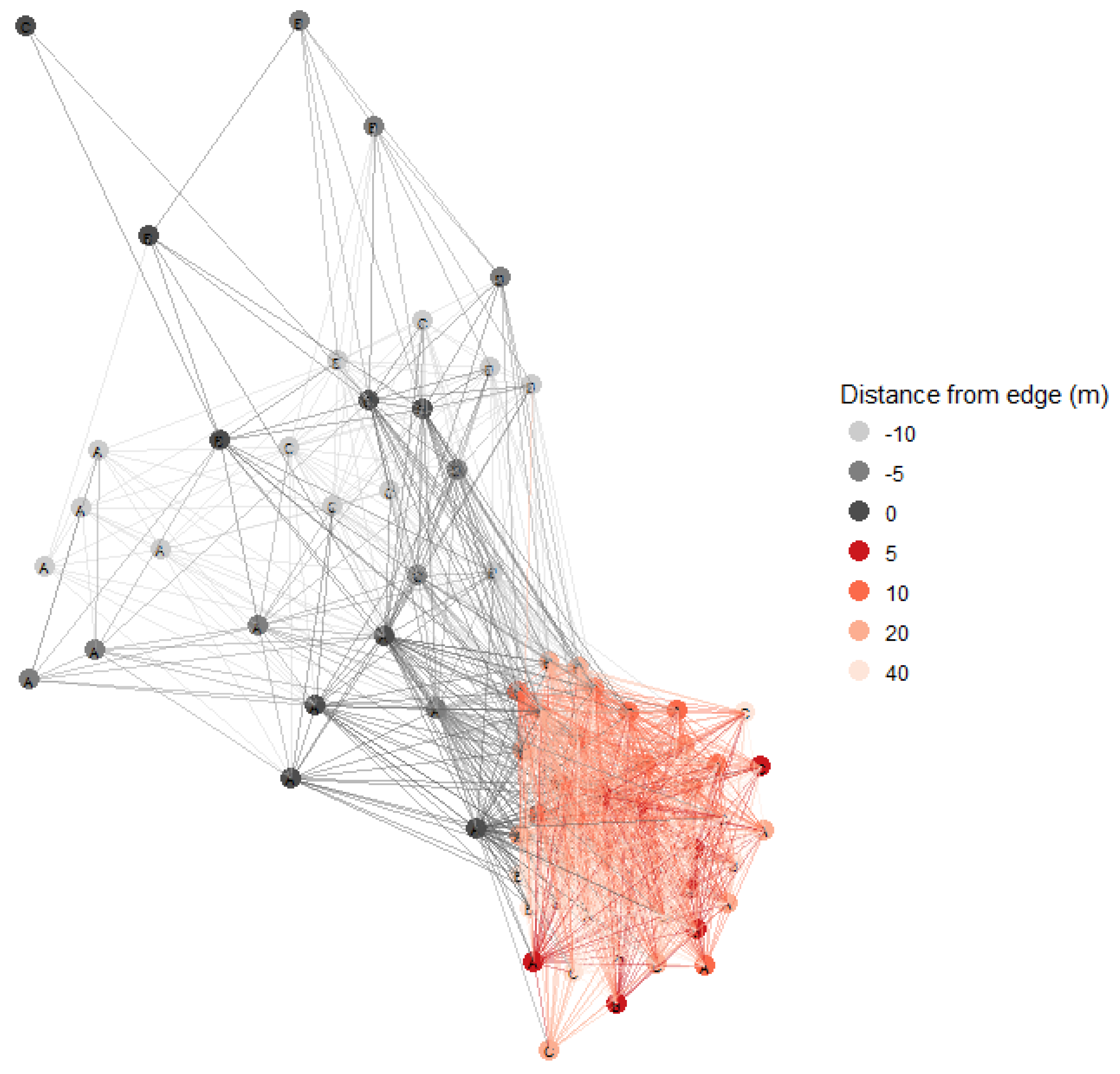
2.6. Statistical Analyses
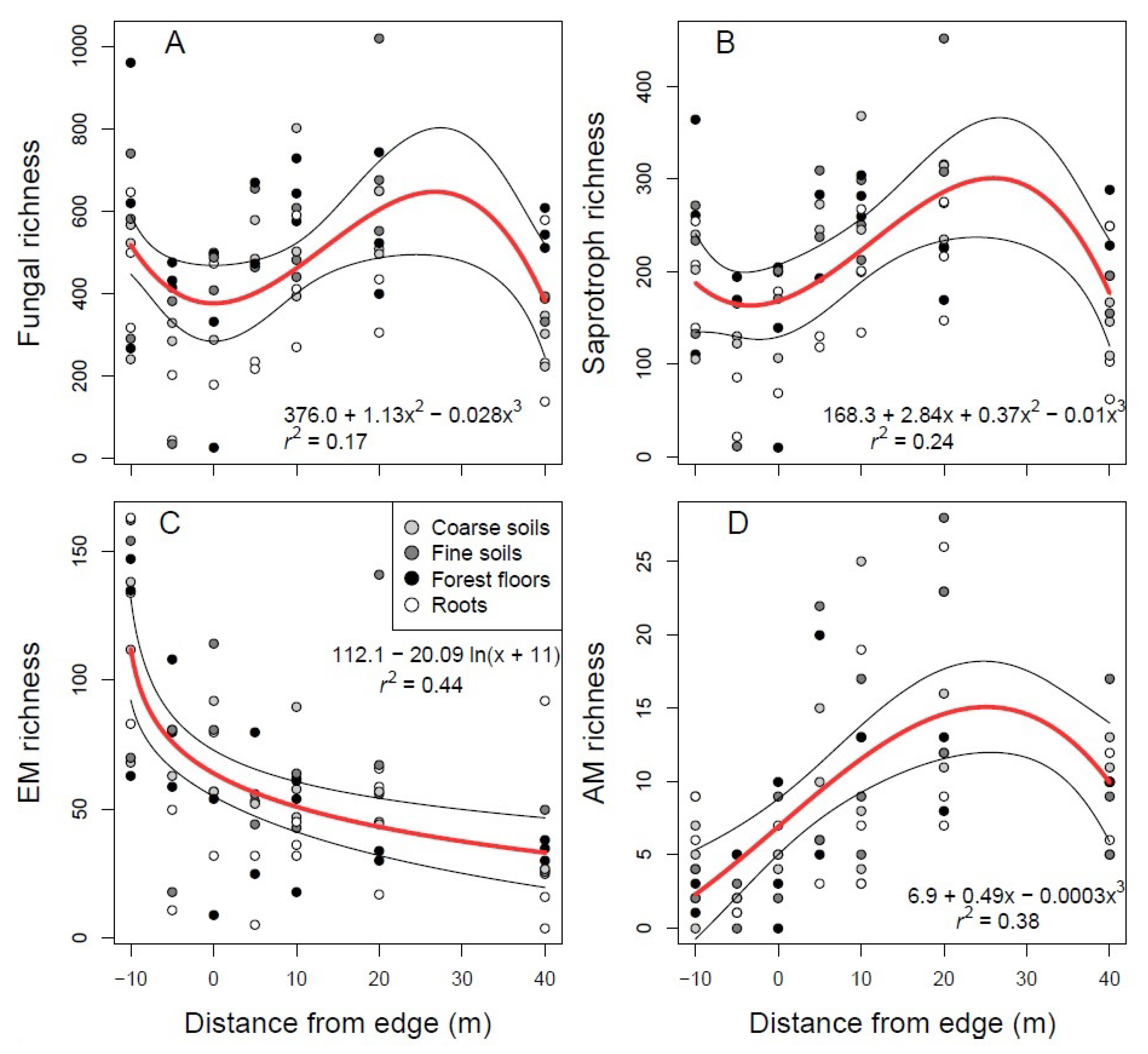
3. Results
3.1. Fungi Cultured from Aspen Roots
3.2. Fungi from Aspen Roots Identified by Roche 454 Analysis
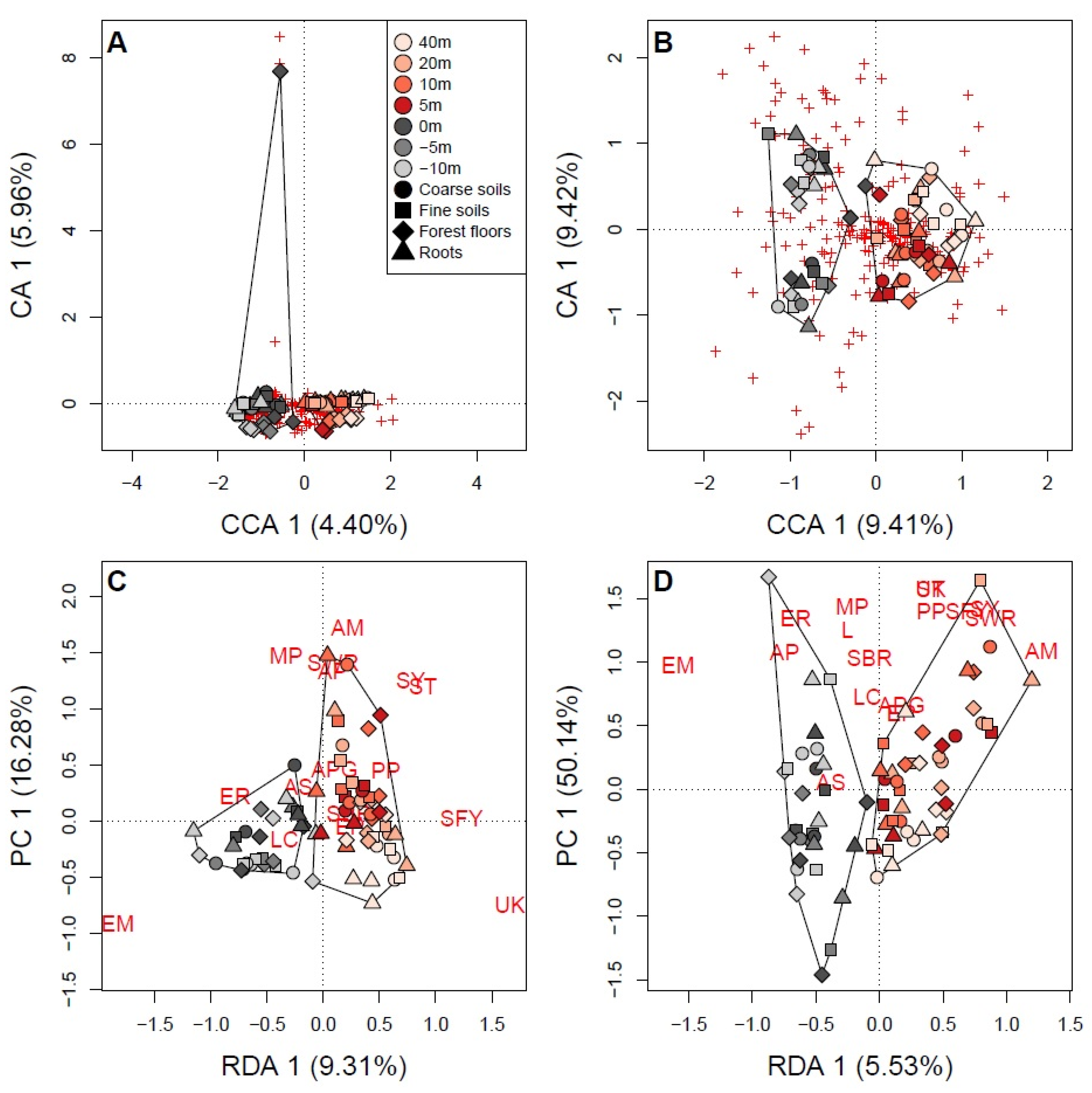
3.3. Soil Fungal Communities Identified by Illumina Analysis
4. Discussion
4.1. Mycorrhizal Taxa
4.2. Saprotrophs and Soil Nutrients
4.3. Plant Pathogens and Seedlings
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jeffries, P.; Gianinazzi, S.; Perotto, S.; Turnau, K.; Barea, J.-M. The contribution of arbuscular mycorrhizal fungi in sustainable maintenance of plant health and soil fertility. Biol. Fertil. Soils 2003, 37, 1–16. [Google Scholar] [CrossRef]
- Berch, S.M.; Monreal, M.A.; Kernaghan, G. Mycorrhizas in Canadian forest and agricultural ecosystems. In Advances in Mycorrhizal Science and Technology; Khasa, D., Piché, Y., Coughlan, A.P., Eds.; NRC Research Press: Ottawa, ON, Canada, 2009; pp. 1–13. [Google Scholar]
- Visser, S. Ectomycorrhizal fungal succession in jack pine stands following wildfire. New Phytol. 1995, 129, 389–401. [Google Scholar] [CrossRef]
- Walbert, K.; Ramsfield, T.D.; Ridgway, H.J.; Jones, E.E. Ectomycorrhiza of Pinus radiata (D. Don 1836) in New Zealand―An above- and below-ground assessment. Australas. Mycol. 2010, 29, 7–16. [Google Scholar]
- Peay, K.G.; Schubert, M.G.; Nguyen, N.H.; Bruns, T.D. Measuring ectomycorrhizal fungal dispersal: Macroecological patterns driven by microscopic propagules. Mol. Ecol. 2012, 21, 4122–4136. [Google Scholar] [CrossRef] [PubMed]
- Teste, F.P.; Simard, S.W.; Durall, D.M. Role mycorrhizal networks and tree proximity in ectomycorrhizal colonization of planted seedlings. Fungal Ecol. 2009, 2, 21–30. [Google Scholar] [CrossRef]
- Lilleskov, E.A.; Bruns, T.D. Spore dispersal of a resupinate ectomycorrhizal fungus, Tomentella sublilacina, via soil food webs. Mycologia 2005, 97, 762–769. [Google Scholar] [CrossRef] [PubMed]
- Schickmann, S.; Urban, A.; Kräutler, K.; Nopp-Mayr, U.; Hackländer, K. The interrlelationship of mycophagous small mammals and ectomycorrhizal fungi in primeval, disturbed and managed Central European mountainous forests. Oecologia 2012, 170, 395–409. [Google Scholar] [CrossRef] [Green Version]
- Fleming, L.V. Effects of soil trenching and coring on the formation of ectomycorrhizas on birch seedlings grown around mature trees. New Phyt. 1984, 98, 143–153. [Google Scholar] [CrossRef]
- Province of Alberta, Oil Sands Facts and Statistics. Available online: https://www.alberta.ca/oil-sands-facts-and-statistics.aspx (accessed on 25 March 2020).
- Johnson, E.A.; Miyanishi, K. Creating new landscapes and ecosystems the Alberta oil sands. Ann. N. Y. Acad. Sci. 2008, 1134, 120–145. [Google Scholar] [CrossRef]
- Jiang, Q.; Thornton, B.; Russel-Houston, J.; Spence, S. Review of thermal recovery technologies for the Clearwater and Lower Grand Rapids formations in Cold Lake, Alberta. J. Can. Petrol. Technol. 2010, 49, 57–68. [Google Scholar] [CrossRef] [Green Version]
- Powter, C.B.; Chymko, N.R.; Dinwoodie, G.; Howat, D.; Janz, A.; Puhlmann, R.; Richens, T.; Watson, D.; Sinton, H.; Ball, J.K.; et al. Regulatory history of Alberta’s industrial land conservation and reclamation program. Can. J. Soil Sci. 2012, 92, 39–51. [Google Scholar] [CrossRef]
- Pinno, B.D.; Errington, R.C. Maximizing natural trembling aspen seedling establishment on a reclaimed boreal oil sands site. Ecol. Restor. 2015, 33, 43–50. [Google Scholar] [CrossRef] [Green Version]
- Audet, P.; Pinno, B.D.; Thiffault, E. Reclamation of boreal forest after oil sands mining: Anticipating novel challenges in novel environments. Can. J. For. Res. 2014, 45, 364–371. [Google Scholar] [CrossRef] [Green Version]
- Howell, D.M.; MacKenzie, M.D. Using bioavailable nutrients and microbial dynamics to assess soil type and placement depth in reclamation. Appl. Soil Ecol. 2017, 116, 87–97. [Google Scholar] [CrossRef]
- Das Gupta, S.; Kirby, W.; Pinno, B.D. Effects of stockpiling and organic matter addition on nutrient bioavailability in reclamation soils. Soil Sci. Soc. Am. J. 2019, 83, S27–S41. [Google Scholar]
- Dhar, A.; Comeau, P.G.; Vassov, R. Effects of cover soil stockpiling on plant community development following reclamation of oil sands sites in Alberta. Restor. Ecol. 2019, 27, 352–360. [Google Scholar] [CrossRef]
- deBortoli, L.A.; Pinno, B.D.; MacKenzie, M.D.; Li, E.H.Y. Plant community composition and tree seedling establishment in response to seeding and weeding treatments on different reclamation cover soils. Can. J. For. Res. 2019, 49, 836–843. [Google Scholar] [CrossRef] [Green Version]
- Tremblay, P.-Y.; Thiffault, E.; Pinno, B.D. Effects of land reclamation practices on the productivity of young trembling aspen and white spruce on a reclaimed oil sands mining site in northern Alberta. New For. 2019, 50, 911–942. [Google Scholar] [CrossRef]
- Bois, G.; Piché, Y.; Fung, M.Y.P.; Khasa, D.P. Mycorrhizal inoculum potentials of pure reclamation materials and revegetated tailing sands from the Canadian oil sand industry. Mycorrhiza 2005, 15, 149–158. [Google Scholar] [CrossRef]
- Bois, G.; Coughlan, A.P. Ectomycorrhizal inoculation for boreal forest ecosystem restoration following oil sand extraction: The need for an initial three-step screening process. In Advances in Mycorrhizal Science and Technology; Khasa, D., Piché, Y., Coughlan, A.P., Eds.; NRC Research Press: Ottawa, ON, Canada, 2009; pp. 129–137. [Google Scholar]
- Neuenkamp, L.; Prober, S.M.; Price, J.N.; Zobel, M.; Standish, R.J. Benefits of mycorrhizal inoculation to ecological restoration depend on plant functional type, restoration context and time. Fungal Ecol. 2019, 40, 140–149. [Google Scholar] [CrossRef]
- Danielson, R.M.; Visser, S.; Parkinson, D. Microbial activity and mycorrhizal potential of four overburden types used in the reclamation of extracted oil sands. Can. J. Soil Sci. 1983, 63, 363–375. [Google Scholar] [CrossRef] [Green Version]
- Outerbridge, R.A.; Trofymow, J.A. Diversity of ectomycorrhizae on experimentally planted Douglas-fir seedlings in variable retention forestry sites on southern Vancouver Island. Can. J. Bot. 2004, 82, 1671–1681. [Google Scholar] [CrossRef]
- Trofymow, J.A.; Shay, P.-E.; Myrholm, C.L.; Tomm, B.; Bérubé, J.A.; Ramsfield, T.D. Fungi associated with tree species at an Alberta oil sands reclamation area, as determined by sporocarp assessments and high-throughput DNA sequencing. Appl. Soil Ecol. 2020, 147, 103359. [Google Scholar] [CrossRef]
- Gardes, M.; Bruns, T.D. ITS primers with enhanced specificity for basidiomycetes―Application to the identification of mycorrhizae and rusts. Mol. Ecol. 1993, 2, 113–118. [Google Scholar] [CrossRef]
- White, T.J.; Bruns, T.D.; Lee, S.B.; Taylor, J.W. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In PCR Protocols: A Guide to Methods and Applications; Innis, M., Gelfand, D., Shinsky, J., White, T., Eds.; Academic Press: San Diego, CA, USA, 1990; pp. 315–322. [Google Scholar]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Reeder, J.; Knight, R. The ‘rare biosphere’: A reality check. Nat. Methods 2009, 6, 636. [Google Scholar] [CrossRef]
- Huse, S.M.; Welch, D.M.; Morrison, H.G.; Sogin, M.L. Ironing out the wrinkles in the rare biosphere through improved OTU clustering. Environ. Microbiol. 2010, 12, 1889–1898. [Google Scholar] [CrossRef] [Green Version]
- Kunin, V.; Engelbrektson, A.; Ochman, H.; Hugenholtz, P. Wrinkles in the rare biosphere: Pyrosequencing errors can lead to artificial inflation of diversity estimates. Environ. Microbiol. 2010, 12, 118–123. [Google Scholar] [CrossRef] [Green Version]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, R.H.; Kristiansson, E.; Ryberg, M.; Hallenberg, N.; Larsson, K.-H. Intraspecific ITS variability in the kingdom Fungi as expressed in the international sequence databases and its implications for molecular species identification. Evol. Bioinform. Online 2008, 4, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Schoch, C.L.; Seifert, K.A.; Huhndorf, S.; Robert, V.; Spouge, J.L.; Levesque, C.A.; Chen, W. Fungal Barcoding Consortium, Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. USA 2012, 109, 6241–6246. [Google Scholar] [CrossRef] [Green Version]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tedersoo, L.; Bahram, M.; Põlme, S.; Kõljalg, U.; Yorou, N.S.; Wijesundera, R.; Villarreal Ruiz, L.; Vasco-Palacios, A.M.; Thu, P.Q.; Suija, A.; et al. Global diversity and geography of soil fungi. Science 2014, 346, 1256688. [Google Scholar] [CrossRef] [Green Version]
- Bérubé, J.A.; Gagné, P.N.; Ponchart, J.P.; Tremblay, É.D.; Bilodeau, G.J. Detection of Diplodia corticola spores in Ontario and Québec based on High Throughput Sequencing (HTS) methods. Can. J. Plant. Pathol. 2018, 40, 378–386. [Google Scholar] [CrossRef]
- Masella, A.P.; Bartram, A.K.; Truszkowski, J.M.; Brown, D.G.; Neufeld, J.D. PANDAseq: Paired-end assembler for Illumina sequences. BMC Bioinform. 2012, 13, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gagné, P.N.; Bérubé, J.A. Illumicut, a C++ Program Specifically Designed to Efficiently Detect and Remove Forward and Revers Sequencing Primers in Paired-End Reconstructed Sequences. 2017. Available online: https://github.com/Patg13/Illumicut (accessed on 14 February 2020).
- Gagné, P.N.; Bérubé, J.A. HomopRemover, a Program Designed to Efficiently Remove Sequences Containing Very Long Homopolymers. 2017. Available online: https://github.com/Patg13/HomopRemover (accessed on 14 February 2020).
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Kalra, Y.P.; Maynard, D.G. Methods Manual for Forest Soil and Plant Analysis; Information Report NOR-X-319E; Forestry Canada, Northwest Region, Northern Forestry Centre: Edmonton, AB, Canada, 1991.
- R Core Team. R: A Language and Environment for Statistical Computing; Version 3.5.1; R Foundation for Statistical Computing: Vienna, Austria, 2017. [Google Scholar]
- Chiu, C.H.; Wang, Y.T.; Walther, B.A.; Chao, A.N. An improved nonparametric lower bound of species richness via a modified Good-Turing frequency formula. Biometrics 2014, 70, 671–682. [Google Scholar] [CrossRef]
- O’Hara, R.B. Species richness estimators: How many species can dance on the head of a pin? J. Anim. Ecol. 2005, 74, 375–386. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. Version 1.24.2. PLoS ONE 2013, 8, 11. [Google Scholar] [CrossRef] [Green Version]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; Stevens, M.H.H.; et al. Vegan: Community Ecology Package, R Package, Version 2.4-5. 2017. Available online: https://CRAN.R-project.org/package=vegan (accessed on 14 February 2020).
- Legendre, P.; Birks, H.J.B. From classical to canonical ordination. In Tracking Environmental Change Using Lake Sediments, Volume 5: Data Handling and Numerical Techniques; Birks, H.J.B., Lotter, A.F., Juggins, S., Smol, J.P., Eds.; Springer: Dordrecht, The Netherlands, 2012; pp. 201–248. [Google Scholar]
- Borcard, D.; Legendre, P.; Avois-Jacquet, C.; Tuomisto, H. Dissecting the spatial structure of ecological data at multiple scales. Ecology 2004, 85, 1826–1832. [Google Scholar] [CrossRef] [Green Version]
- Godínez-Domínguez, E.; Freire, J. Information-theoretic approach for selection of spatial and temporal models of community organization. Mar. Ecol. Prog. Ser. 2003, 253, 17–24. [Google Scholar] [CrossRef]
- Fagan, W.F.; Cantrell, R.S.; Cosner, C. How habitat edges change species interactions. Am. Nat. 1999, 153, 165–182. [Google Scholar] [CrossRef] [PubMed]
- Ries, L.; Fletcher, R.J.; Battin, J.; Sisk, T.D. Ecological responses to habitat edges: Mechanisms, models, and variability explained. Annu. Rev. Ecol. Evol. Syst. 2004, 35, 491–522. [Google Scholar] [CrossRef] [Green Version]
- Harper, K.A.; MacDonald, S.E.; Burton, P.J.; Chen, J.; Brosofske, K.D.; Saunders, S.C.; Euskirchen, E.S.; Roberts, D.; Jaiteh, M.S.; Esseen, P.-E. Edge influence on forest structure and composition in fragmented landscapes. Conserv. Biol. 2005, 19, 768–782. [Google Scholar] [CrossRef]
- Green, J.L.; Holmes, A.J.; Westoby, M.; Briscoe, O.I.; Dangerfield, M.; Gillings, M.; Geattie, A.J. Spatial scaling of microbial eukaryote diversity. Nature 2004, 432, 747–750. [Google Scholar] [CrossRef]
- Martiny, J.B.H.; Bohannan, B.J.M.; Brown, J.H.; Colwell, R.K.; Fuhrman, J.A.; Green, J.L.; Horner-Devine, M.C.; Kane, M.; Adams Krumins, J.; Kuske, C.R.; et al. Microbial biogeography: Putting microorganisms on the map. Nat. Rev. Microbiol. 2006, 4, 102–112. [Google Scholar] [CrossRef]
- Ramette, A.; Tiedje, J.M. Multiscale responses of microbial life to spatial distance and environmental heterogeneity in a patchy ecosystem. Proc. Natl. Acad. Sci. USA 2007, 104, 2761–2766. [Google Scholar] [CrossRef] [Green Version]
- Shay, P.E.; Winder, R.S.; Trofymow, J.A. Nutrient-cycling microbes in coastal Douglas-fir forests: Regional-scale correlation between communities, in situ climate, and other factors. Front. Microbiol. 2015, 6, 13. [Google Scholar] [CrossRef] [Green Version]
- Nacke, H.; Goldmann, K.; Schöning, I.; Pfeiffer, B.; Kaiser, K.; Castillo-Villamizar, G.A.; Schrumph, M.; Buscot, F.; Daniel, R.; Wubet, T. Fine spatial scale variation of soil microbial communities under European Beech and Norway Spruce. Front. Microbiol. 2016, 7, 2067. [Google Scholar] [CrossRef] [Green Version]
- Anderson, I.C.; Cairney, J.W.G. Ectomycorrhizal fungi: Exploring the mycelial frontier. FEMS Microbiol. Rev. 2007, 31, 388–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agerer, R. Exploration types of ectomycorrhizae. A proposal to classify ectomycorrhizal mycelial systems according to their patterns of differentiation and putative ecological importance. Mycorrhiza 2001, 11, 107–114. [Google Scholar] [CrossRef]
- Outerbridge, R.A.; Trofymow, J.A. Forest management and maintenance of ectomycorrhizae: A case study of green tree retention in south-coastal British Columbia. BC J. Ecosyst. Manag. 2009, 10, 59–80. [Google Scholar]
- Burgess, T.; Dell, B.; Malajczuk, N. Variation in mycorrhizal development and growth stimulation by 20 Pisolithus isolates inoculated on to Eucalyptus grandis W. Hill ex Maiden. New Phytol. 1994, 127, 731–739. [Google Scholar] [CrossRef] [Green Version]
- Schweitzer, J.A.; Bailey, J.K.; Fischer, D.G.; LeRoy, C.J.; Lonsdorf, E.V.; Whitham, T.G.; Hart, S.C. Plant-soil-microorganism interactions: Heritable relationship between plant genotype and associated soil microorganisms. Ecology 2008, 89, 773–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoeksema, J.D.; Piculell, B.J.; Thompson, J.N. Within-population genetic variability in mycorrhizal interactions. Commun. Integr. Biol. 2009, 2, 110–112. [Google Scholar] [CrossRef]
- Opelt, K.; Chobot, V.; Hadacek, F.; Schonmann, S.; Eberl, L.; Berg, G. Investigations of the structure and function of bacterial communities associated with Sphagnum mosses. Environ. Microbiol. 2007, 9, 2795–2809. [Google Scholar] [CrossRef]
- Moltzan, B.D.; Blenis, P.V.; Hiratsuka, Y. Temporal occurrence and impact of Scytalidium uredinicola, a mycoparasite of western gall rust. Can. J. Plant Pathol. 2001, 23, 384–390. [Google Scholar] [CrossRef]
- Brimner, T.A.; Boland, G.J. A review of the non-target effects of fungi used to biologically control plant diseases. Agric. Ecosyst. Environ. 2003, 100, 3–16. [Google Scholar] [CrossRef]
- Kaewchai, S.; Soytong, K.; Hyde, K. Mycofungicides and fungal biofertilizers. Fungal Divers 2009, 38, 25–50. [Google Scholar]
- Jumpponen, A.; Trappe, J.M. Dark septate endophytes: A review of facultative biotrophic root-colonizing fungi. New Phytol. 1998, 140, 295–310. [Google Scholar] [CrossRef]
- Booth, M.S.; Stark, J.M.; Rastetter, E. Controls on nitrogen cycling in terrestrial ecosystems: A synthetic analysis of literature data. Ecol. Monogr. 2005, 75, 139–157. [Google Scholar] [CrossRef] [Green Version]
- Belser, L.W. Population ecology of nitrifying bacteria. Annu. Rev. Microbiol. 1979, 33, 309–333. [Google Scholar] [CrossRef]
- Schimel, J.; Bennett, J. Nitrogen mineralization: Challenges of a changing paradigm. Ecology 2004, 85, 591–602. [Google Scholar] [CrossRef]
- Kranabetter, J.M.; Durall, D.M.; MacKenzie, W.H. Diversity and species distribution of ectomycorrhizal fungi along productivity gradients of a southern boreal forest. Mycorrhiza 2009, 19, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Mills, K.E.; Bever, J.D. Maintenance of diversity within plant communities: Soil pathogens as agents of negative feedback. Ecology 1998, 79, 1595–1601. [Google Scholar] [CrossRef]
- Packer, A.; Clay, K. Soil pathogens and spatial patterns of seedling mortality in a temperate tree. Nature 2000, 404, 278. [Google Scholar] [CrossRef]
- Hudson, P.J.; Dobson, A.P.; Lafferty, K.D. Is a healthy ecosystem one that is rich in parasites? Trends Ecol. Evol. 2006, 21, 381–385. [Google Scholar] [CrossRef]
- Schmidt, P.-A.; Bálint, M.; Greshake, B.; Bandow, C.; Römbke, J.; Schmitt, I. Illumina metabarcoding of a soil fungal community. Soil Biol. Biochem. 2013, 65, 128–132. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fungal Ta xa Cultured | Representative Culture | Representative GenBank Accession | # of Aspen | TF * | Pyro. Aspen Roots | Illumina Soils | ||
|---|---|---|---|---|---|---|---|---|
| Species | Genus | Species | Genus | |||||
| Cylindrocarpon olidum | NoF 3158 | MT294406 | 3 | PP | N | 0 | N | 2 |
| Fusarium acuminatum | NoF 3159 | MT294407 | 1 | SB | N | 2 | N | 13 |
| Ilyonectria crassa † | NoF 3124 | MT294410 | 1 | SB | N | 2 | N | 2 |
| Tolypocladium inflatum‡ | NoF 3144 | MT294423 | 1 | SB | Y | 1 | N | 1 |
| Lachnum pygmaeum | NoF 3127 | MT294411 | 2 | ST | N | 2 | Y | 7 |
| Mycena epipterygia | NoF 3165 | MT294413 | 1 | ST | N | 5 | N | 34 |
| Mycena leptocephala | NoF 3163 | MT294414 | 1 | ST | N | 5 | N | 34 |
| Nodulisporium sp. | NoF 3148 | MT294416 | 1 | ST | NA | 0 | NA | 0 |
| Oidiodendron echinulatum | NoF 3152 | MT294417 | 1 | ST | N | 1 | N | 13 |
| Porodaedalea pini | NoF 3172 | MT294420 | 1 | ST | N | 0 | N | 0 |
| Scedosporium minutisporum | NoF 3135 | MT294422 | 1 | ST | N | 0 | N | 1 |
| Trichocladium opacum | NoF 3133 | MT294424 | 1 | ST | Y | 1 | Y | 2 |
| Cadophora finlandica | NoF 3116 | MT294403 | 5 | ST ‖ | N | 1 | Y | 18 |
| Cladophialophora chaetospira | NoF 3119 | MT294404 | 1 | ST ‖ | Y | 1 | Y | 7 |
| Cryptosporiopsis ericae | NoF 3122 | MT294405 | 1 | ST ‖ | N | 1 | N | 0 |
| Leptodontidium orchidicola | NoF 3121 | MT294412 | 3 | ST ‖ | N | 0 | N | 4 |
| Oidiodendron pilicola | NoF 3120 | MT294418 | 2 | ST ‖ | N | 1 | Y | 13 |
| Phialocephala fortinii | NoF 3123 | MT294419 | 11 | ST ‖ | Y | 2 | Y | 14 |
| Rhizoscyphus ericae§ | NoF 3128 | MT294421 | 2 | ST ‖ | Y | 1 | Y | 2 |
| Acremonium sp. | NoF 3147 | MT294401 | 2 | UK | NA | 1 | NA | 16 |
| Auriculariales order | NoF 3160 | MT294402 | 1 | UK | NA | NA | NA | NA |
| Helotiales order | NoF 3169 | MT294408 | 1 | UK | NA | NA | NA | NA |
| Hypocreales order | None | MT294409 | 1 | UK | NA | NA | NA | NA |
| Nectriaceae family | NoF 3145 | MT294415 | 2 | UK | NA | NA | NA | NA |
| Response Variable | All Taxa | Highly Variable Taxa | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Conditions | Conditions | ||||||||
| None | None | Transect | Seedling Species | No. of Seedlings | Dom. Tree Species | Nutrients | PCNMs * | ||
| Rel. abun. | All soil fractions † | 0.001 4.04% | 0.001 4.22% | 0.001 4.24% | 0.001 3.07% | NA | 0.001 4.23% | 0.001 3.10% | 0.001 2.11% |
| Forest floor | 0.017 6.72% | 0.024 6.61% | NA | NA | NA | NA | 0.467 5.33% | NA | |
| Coarse soils | 0.004 9.91% | 0.004 10.27% | NA | NA | NA | NA | 0.245 6.44% | NA | |
| Fine soils | 0.001 10.82% | 0.003 11.41% | NA | NA | NA | NA | 0.119 7.26% | 0.005 10.70% | |
| Roots | 0.002 8.89% | 0.004 8.98% | NA | NA | NA | NA | 0.189 6.49% | NA | |
| Pres./abs. | All soil fractions † | 0.001 4.60% | 0.001 9.09% | 0.001 9.19% | 0.001 7.90% | 0.001 8.74% | 0.001 9.08% | 0.005 2.34% | 0.001 2.64% |
| Forest floor | 0.001 8.97% | 0.001 12.14% | NA | NA | NA | 0.001 12.12% | 0.390 4.54% | 0.140 5.90% | |
| Coarse soils | 0.001 9.52% | 0.001 17.43% | NA | NA | NA | 0.002 17.29% | 0.027 8.10% | 0.230 5.86% | |
| Fine soils | 0.002 9.26% | 0.002 16.01% | NA | NA | NA | 0.001 15.77% | 0.067 6.80% | 0.028 8.86% | |
| Roots | 0.008 8.13% | 0.023 8.99% | NA | NA | NA | 0.027 8.91% | 0.678 4.52% | 0.943 3.44% | |
| RN | Forest Interior | TF * | RN | Reclaimed Area | TF * |
|---|---|---|---|---|---|
| 107,334 | Amphinema sp. ML-1 | EM | 10,056 | Podospora intestinacea | ST |
| 71,115 | Russula fragilis var. fragilis | EM | 9094 | Leptospora rubella | UK |
| 55,212 | Tricholoma platyphyllum | EM | 9035 | Psathyrella abieticola | ST |
| 52,951 | Tomentella fusco-cinerea | EM | 8030 | Phlebia sp. DLL2011-1 | SWR |
| 38,287 | Laccaria sp. AWW564 | EM | 7092 | Hypholoma capnoides | ST |
| 30,365 | Cortinarius colymbadinus | EM | 6491 | Paraphoma sp. L13 † | UK |
| 29,191 | Piloderma sphaerosporum† | EM | 5167 | Plectosphaerella sp. FPGLXJ06 | PP |
| 14,544 | Hygrophorus sp. EL-2014 | EM | 4995 | Epulorhiza sp. SO 035 | UK |
| 13,770 | Sebacina sp. Seb13I | EM | 4053 | Cadophora sp. 9232S2 † | ST |
| 12,855 | Cortinarius paragaudis | EM | 3511 | Trogia venenata | ST |
| 12,308 | Trechispora stellulata | ST | 2747 | Seimatosporium vitis | PP |
| 9853 | Sistotrema sp. PC14 | ST | 1113 | Trichoderma aureoviride | ST |
| 5241 | Mycena amicta | ST | 1071 | Rhizoctonia sp. 70B | SBR |
| 4377 | Inocybe fulvipes | EM | 1009 | Dendrosporium sp. 1 RB-2011 | ST |
| 3870 | Clavariadelphus sachalinensis | ST | |||
| 3707 | Tomentella sp. 4 RT-2012 | EM | |||
| 3618 | Sistotrema oblongisporum | ST | |||
| 3481 | Tricholoma saponaceum var. saponaceum | EM | |||
| 2593 | Tomentella sp. YM1903 | EM | |||
| 2137 | Tomentella subtestacea | EM | |||
| 1981 | Thyronectria coryli | UK | |||
| 1880 | Inocybe calida | EM | |||
| 1400 | Clavariadelphus ligula | ST | |||
| 1389 | Sclerotinia nivalis | PP | |||
| 659 | Hypochnicium albostramineum | SWR | |||
| 118 | Tricholoma magnivelare | EM |
| Response Variable | Conditions | |||||||
|---|---|---|---|---|---|---|---|---|
| None | Transect | Seedling Species | No. of Seedlings | Dom. Tree Species | Nutrients | PCNMs * | ||
| Rel. abun. | All soil fractions † | 0.001 8.89% | 0.001 9.06% | 0.001 5.87% | 0.001 7.12% | NA | 0.007 2.52% | 0.210 1.50% |
| Forest floor | 0.001 13.39% | NA | NA | NA | NA | 0.203 5.76% | 0.247 5.98% | |
| Coarse soils | 0.011 14.84% | NA | 0.042 9.19% | NA | NA | 0.041 8.82% | NA | |
| Fine soils | 0.003 16.11% | NA | 0.012 11.26% | NA | NA | 0.065 8.11% | 0.146 8.42% | |
| Roots | 0.034 10.84% | NA | 0.198 5.72% | NA | NA | 0.039 10.13% | 0.007 11.97% | |
| No. of taxa | All soil fractions † | 0.007 5.01% | 0.005 4.85% | 0.012 3.98% | NA | 0.006 4.96% | 0.103 2.45% | 0.056 2.48% |
| Forest floor | 0.114 10.14% | NA | NA | NA | NA | 0.288 5.17% | NA | |
| Coarse soils | 0.088 11.17% | NA | NA | NA | NA | 0.002 18.00% | 0.083 10.45% | |
| Fine soils | 0.546 4.30% | NA | NA | NA | NA | NA | 0.228 5.60% | |
| Roots | 0.426 5.16% | 0.286 4.34% | NA | NA | NA | 0.662 2.87% | 0.229 4.66% | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramsfield, T.; Shay, P.-E.; Trofymow, T.; Myrholm, C.; Tomm, B.; Gagné, P.; Bérubé, J. Distance from the Forest Edge Influences Soil Fungal Communities Colonizing a Reclaimed Soil Borrow Site in Boreal Mixedwood Forest. Forests 2020, 11, 427. https://0-doi-org.brum.beds.ac.uk/10.3390/f11040427
Ramsfield T, Shay P-E, Trofymow T, Myrholm C, Tomm B, Gagné P, Bérubé J. Distance from the Forest Edge Influences Soil Fungal Communities Colonizing a Reclaimed Soil Borrow Site in Boreal Mixedwood Forest. Forests. 2020; 11(4):427. https://0-doi-org.brum.beds.ac.uk/10.3390/f11040427
Chicago/Turabian StyleRamsfield, Tod, Philip-Edouard Shay, Tony Trofymow, Colin Myrholm, Bradley Tomm, Patrick Gagné, and Jean Bérubé. 2020. "Distance from the Forest Edge Influences Soil Fungal Communities Colonizing a Reclaimed Soil Borrow Site in Boreal Mixedwood Forest" Forests 11, no. 4: 427. https://0-doi-org.brum.beds.ac.uk/10.3390/f11040427