
Genome Wide Association Study Identifies Candidate Genes Related to the Earlywood Tracheid Properties in Picea crassifolia Kom.

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Wood Tracheid Traits Phenotyping
2.3. DNA Isolation, Construction of SLAF Library, and Genome Sequencing
2.4. Population Genetic Analyses and Linkage Disequilibrium
2.5. Association Genetics Analysis
3. Results
3.1. Sequencing Results
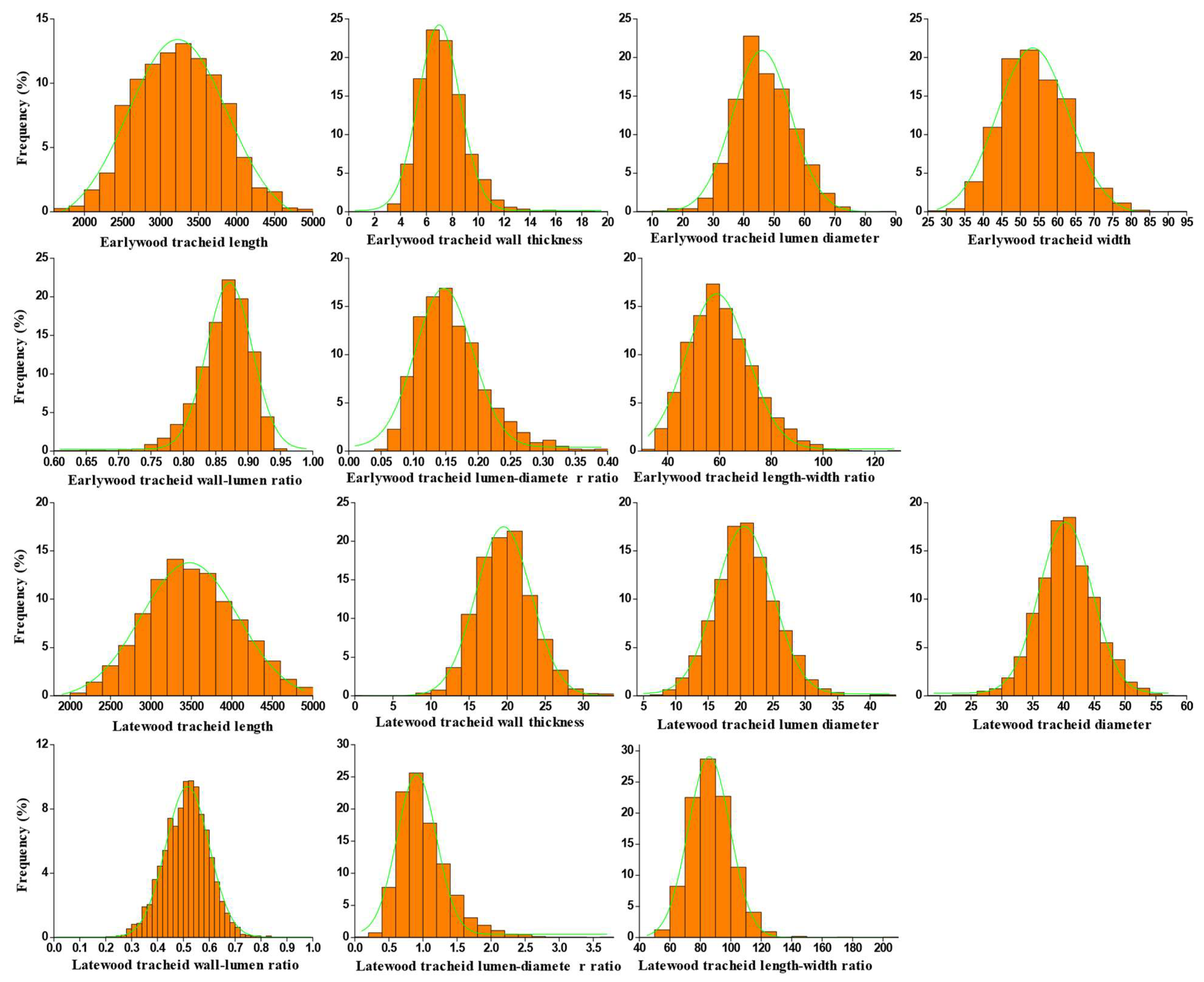
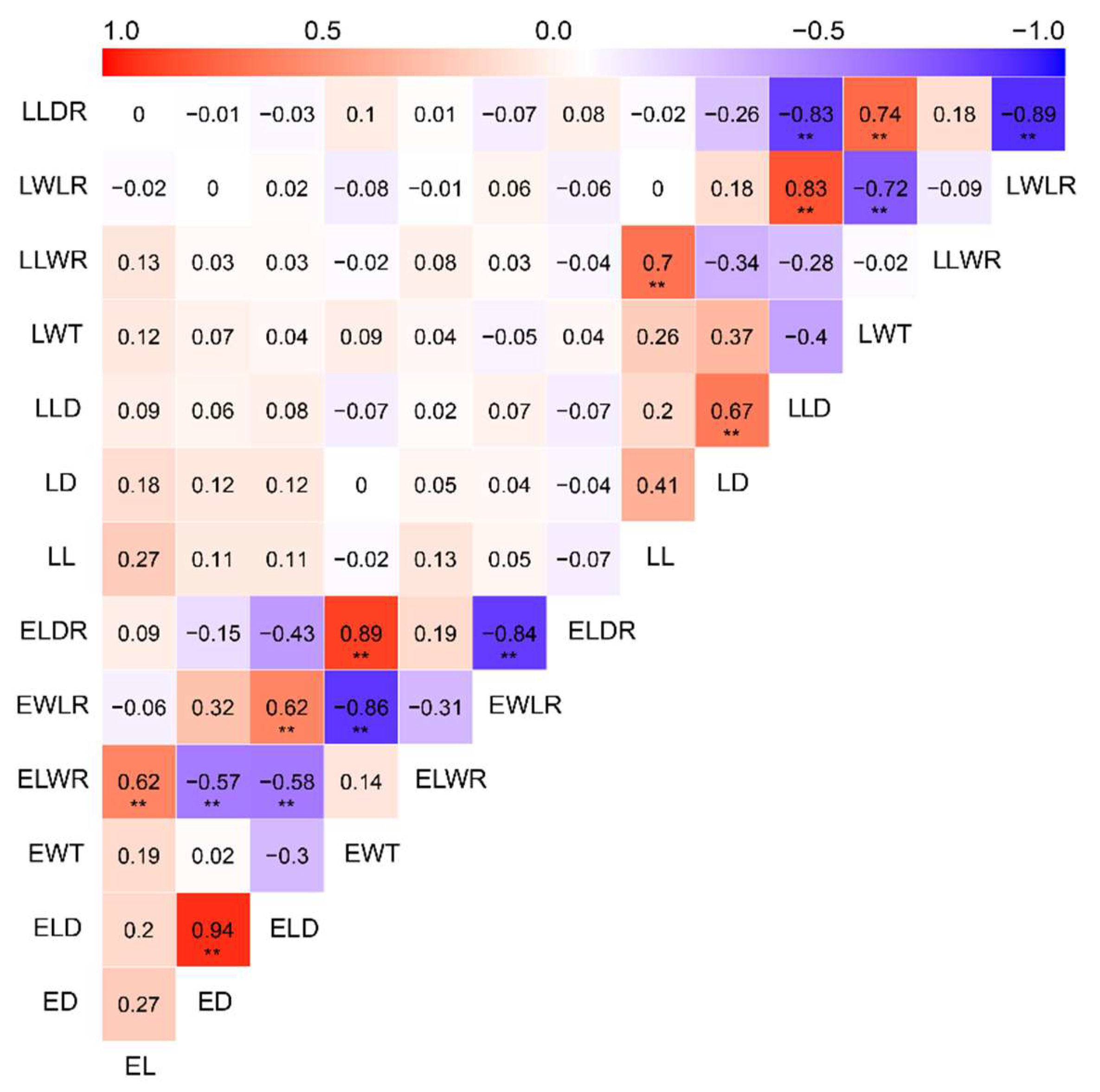
3.2. Phenotypic Characterization of 14 Wood Tracheid Traits
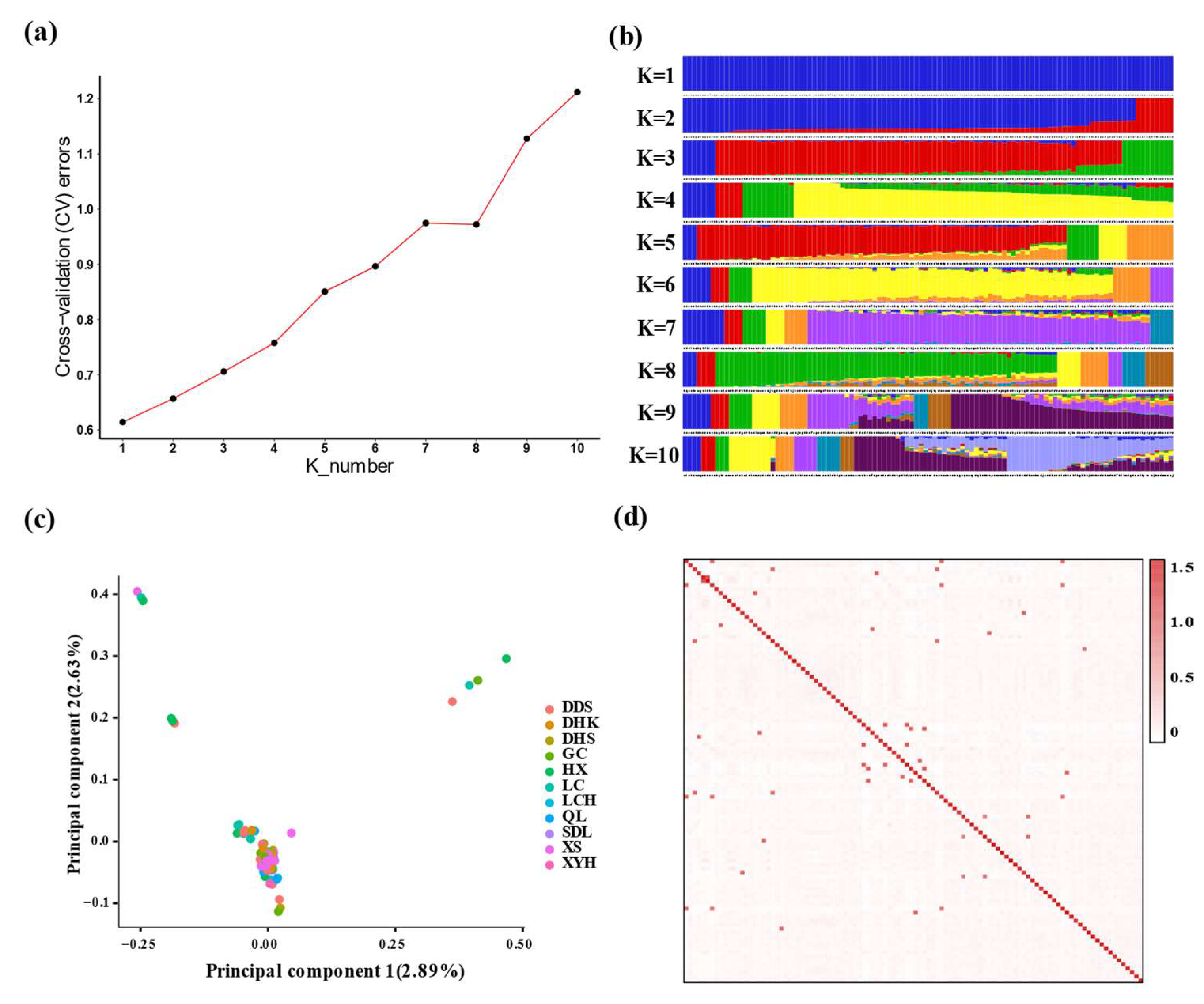
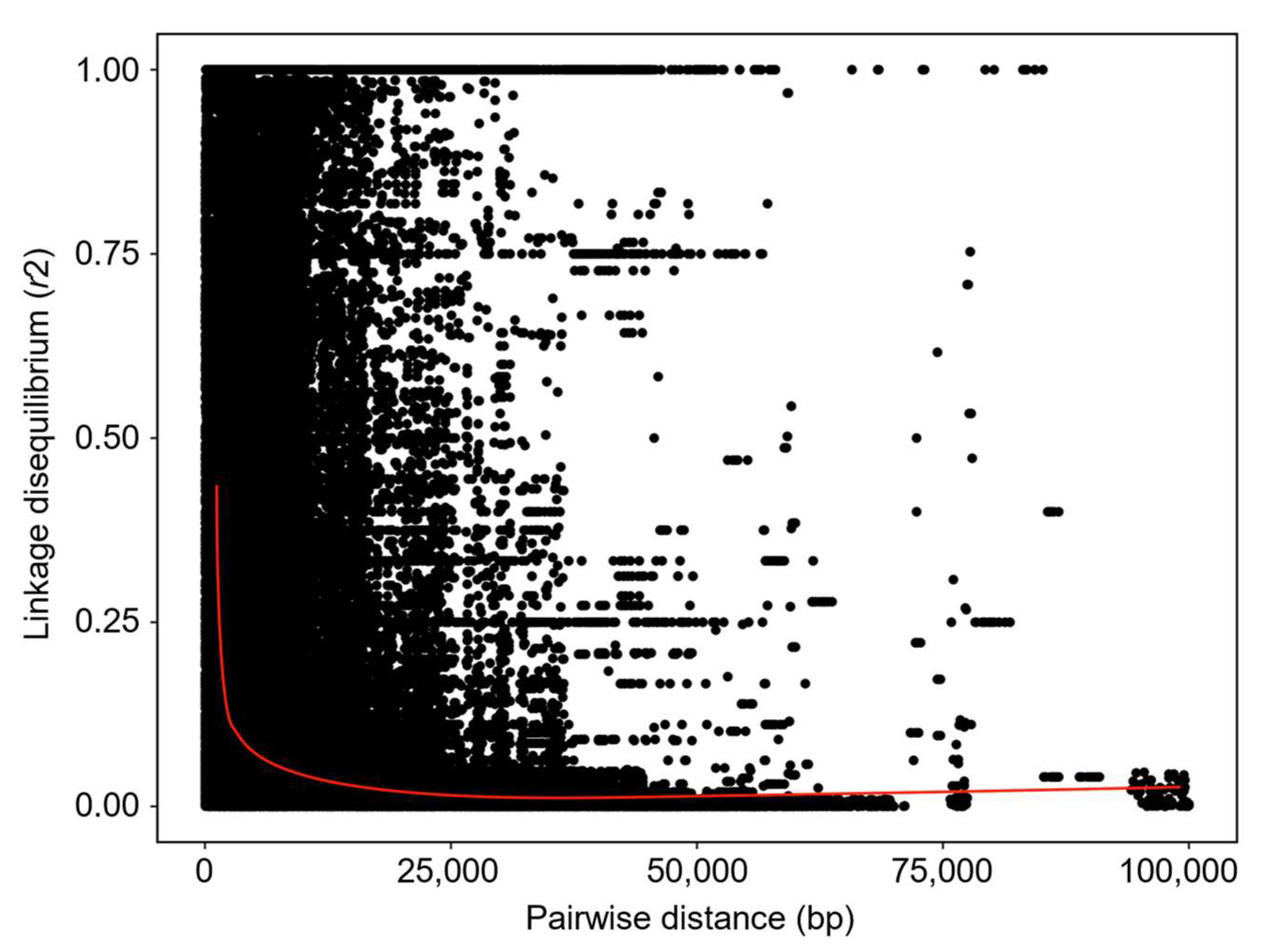
3.3. Analysis of Population Structure and Linkage Disequilibrium
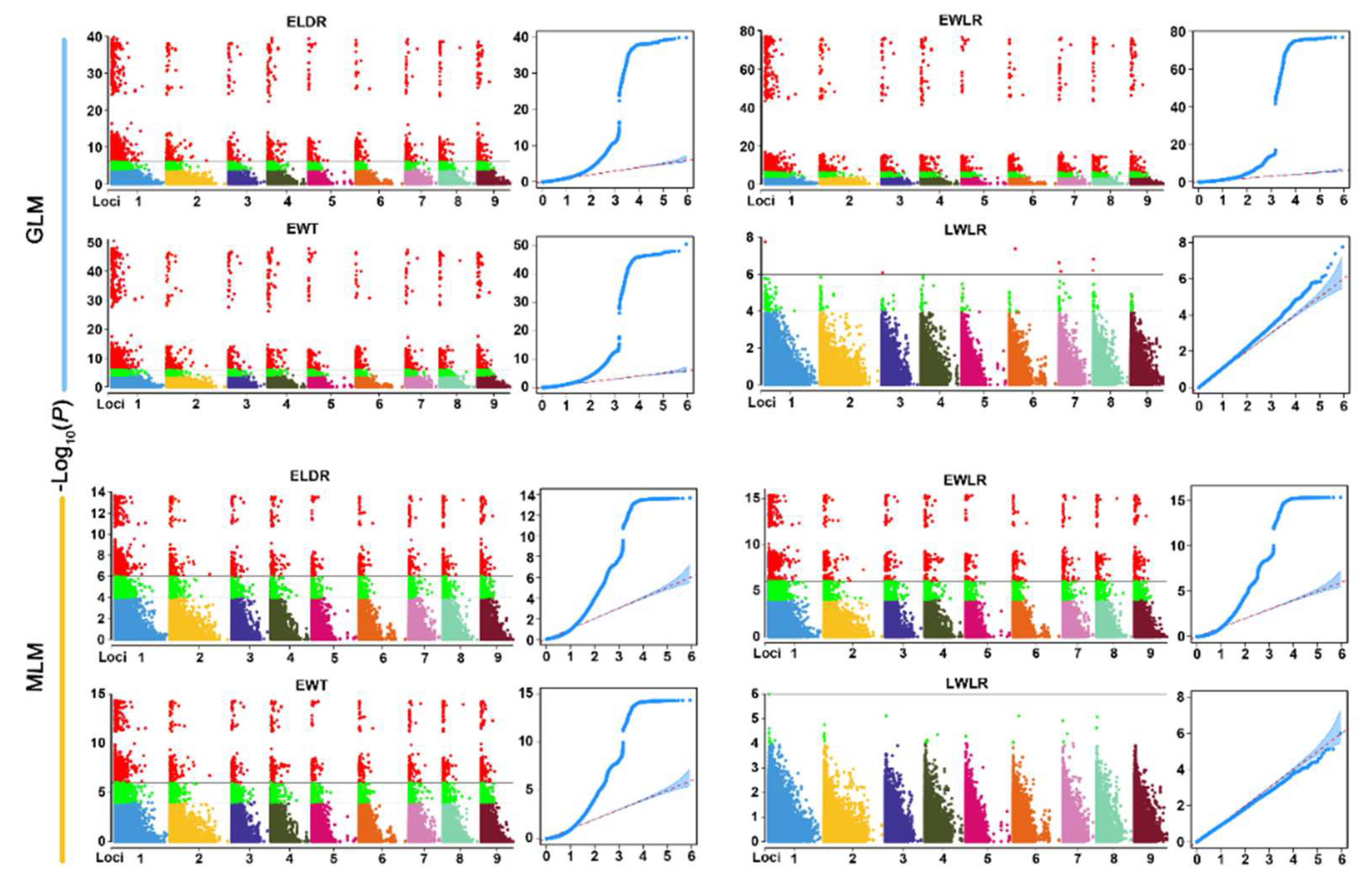
3.4. Association Analysis of 14 Wood Tracheid Traits
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xu, J.M.; Lu, J.X.; Evans, R.; Downes, G.M. Climatic signal in cellulose microfibril angle and tracheid radial diameter of Picea crassifolia at different altitudes of the Tibetan plateau, northwest China. Wood Sci. Technol. 2015, 49, 1307–1318. [Google Scholar] [CrossRef]
- Tian, Q.Y.; He, Z.B.; Xiao, S.C.; Peng, X.M.; Ding, A.J.; Lin, P.F. Response of stem radial growth of Qinghai spruce (Picea crassifolia) to environmental factors in the Qilian Mountains of China. Dendrochronologia 2017, 44, 76–83. [Google Scholar] [CrossRef]
- Zhu, T.Q.; Hu, J.W.; Qi, S.X.; Ouyang, F.Q.; Kong, L.S.; Wang, J.H. Transcriptome and morpho-physiological analyses reveal factors regulating cone bud differentiation in Qinghai spruce (Picea crassifolia Kom.). Trees 2021, 35, 1151–1166. [Google Scholar] [CrossRef]
- Gao, J.N.; Yang, B.; Peng, X.M.; Rossi, S. Tracheid development under a drought event producing intra-annual density fluctuations in the semi-arid China. Agric. For. Meteorol. 2021, 308–309, 108572. [Google Scholar] [CrossRef]
- Song, W.Q.; Mu, C.C.; Zhang, Y.D.; Zhang, X.; Li, Z.S.; Zhao, H.Y. Moisture-driven changes in the sensitivity of the radial growth of Picea crassifolia to temperature, northeastern Tibetan plateau. Dendrochronologia 2020, 64, 125761. [Google Scholar] [CrossRef]
- Yamashita, S.; Yoshida, M.; Takayama, S.; Okuyama, T. Stem-righting mechanism in gymnosperm trees deduced from limitations in compression wood development. Ann. Bot. 2007, 99, 487–493. [Google Scholar] [CrossRef] [Green Version]
- Baison, J.; Zhou, L.; Forsberg, N.; Mörling, T.; Grahn, T.; Olsson, L.; Karlsson, B.; Wu, H.X.; Mellerowicz, E.J.; Lundqvist, S.O.; et al. Genetic control of tracheid properties in Norway spruce wood. Sci. Rep. 2020, 10, 18089. [Google Scholar] [CrossRef]
- Xu, J.M.; Lu, J.X.; Bao, F.C.; Evans, R.; Downes, G.M. Climate response of cell characteristics in tree rings of Picea crassifolia. Holzforschung 2013, 67, 217–225. [Google Scholar] [CrossRef]
- Arend, M.; Fromm, J. Seasonal change in the drought response of wood cell development in Poplar. Tree Physiol. 2007, 27, 985–992. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.T.; Zhao, C.Y.; Gao, C.C.; Xie, H.H.; Qiao, Y.; Gao, Y.F.; Yuan, L.M.; Wang, W.B.; Ge, L.J.; Zhang, G.D. Effects of environmental variables on seedling-sapling distribution of Qinghai spruce (Picea crassifolia) along altitudinal gradients. Forest Ecol. Manag. 2017, 384, 54–64. [Google Scholar] [CrossRef]
- Groover, A.; Devey, M.; Fiddler, T.; Lee, J.; Megraw, R.; Mitchel-Olds, T.; Sherman, B.; Vujcic, S.; Williams, C.; Neale, D. Identification of quantitative trait loci influencing wood specific gravity in an outbred pedigree of loblolly pine. Genetics 1994, 138, 1293–1300. [Google Scholar] [CrossRef]
- Gailing, O. QTL analysis of leaf morphological characters in a Quercus robur full-sib family (Q. robur x Q. robur ssp. slavonica). Plant Biol. [CrossRef]
- Heslot, N.; Akdemir, D.; Sorrells, M.E.; Jannink, J.L. Integrating environmental covariates and crop modeling into the genomic selection framework to predict genotype by environment interactions. Theor. Appl. Genet. 2014, 127, 463–480. [Google Scholar] [CrossRef]
- Baison, J.; Vidalis, A.; Zhou, L.; Chen, Z.Q.; Li, Z.; Sillanpää, M.J.; Bernhardsson, C.; Scofield, D.; Forsberg, N.; Grahn, T.; et al. Genome-wide association study identified novel candidate loci affecting wood formation in Norway spruce. Plant J. 2019, 100, 83–100. [Google Scholar] [CrossRef]
- Porth, I.; Klapšte, J.; Skyba, O.; Hannemann, J.; McKown, A.D.; Guy, R.D.; DiFazio, S.P.; Muchero, W.; Ranjan, P.; Tuskan, G.A.; et al. Genome-wide association mapping for wood characteristics in populus identifies an array of candidate single nucleotide polymorphisms. New Phytol. 2013, 200, 710–726. [Google Scholar] [CrossRef]
- Uchiyama, K.; Iwata, H.; Moriguchi, Y.; Ujino-Ihara, T.; Ueno, S.; Taguchi, Y.; Tsubomura, M.; Mishima, K.; Iki, T.; Watanabe, A.; et al. Demonstration of genome-wide association studies for identifying markers for wood property and male strobili traits in Cryptomeria japonica. PLoS ONE 2013, 8, e79866. [Google Scholar] [CrossRef] [Green Version]
- Resende, R.T.; Resende, M.D.; Silva, F.F.; Azevedo, C.F.; Takahashi, E.K.; Silva-Junior, O.B.; Grattapaglia, D. Regional heritability mapping and genome-wide association identify loci for complex growth, wood and disease resistance traits in eucalyptus. New Phytol. 2017, 213, 1287–1300. [Google Scholar] [CrossRef] [Green Version]
- Lamara, M.; Raherison, E.; Lenz, P.; Beaulieu, J.; Bousquet, J.; MacKay, J. Genetic architecture of wood properties based on association analysis and co-expression networks in white spruce. New Phytol. 2016, 210, 240–255. [Google Scholar] [CrossRef] [Green Version]
- Ithnin, M.; Xu, Y.; Marjuni, M.; Mohamed, S.N.; Din, A.; Low, L.; Tan, Y.C.; Yap, S.J.; Ooi, L.C.L.; Nookiah, R.; et al. Multiple locus genome-wide association studies for important economic traits of oil palm. Tree Genet. Genomes 2017, 13, 103. [Google Scholar] [CrossRef]
- Fahrenkrog, A.M.; Neves, L.G.; Resende, M.F., Jr.; Vazquez, A.I.; de Los, C.G.; Dervinis, C.; Sykes, R.; Davis, M.; Davenport, R.; Barbazuk, W.B.; et al. Genome-wide association study reveals putative regulators of bioenergy traits in Populus deltoides. New Phytol. 2016, 213, 799–811. [Google Scholar] [CrossRef]
- Josephs, E.B.; Stinchcombe, J.R.; Wright, S.I. What can genome-wide association studies tell us about the evolutionary forces maintaining genetic variation for quantitative traits? New Phytol. 2017, 214, 21–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Hu, Y.; Wu, X.; Ma, R.; Jiang, Q.; Wang, Y. Identifying liver cancer-related enhancer SNPs by integrating GWAS and histone modification ChIP-seq data. Biomed. Res. Int. 2016, 2395341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Liu, D.; Zhang, X.; Li, W.; Liu, H.; Hong, W.; Jiang, C.; Guan, N.; Ma, C.; Zeng, H.; et al. SLAF-seq: An efficient method of large-scale de novo SNP discovery and genotyping using high-throughput sequencing. PLoS ONE 2013, 8, e58700. [Google Scholar] [CrossRef] [PubMed]
- Geng, X.; Jiang, C.; Yang, J.; Wang, L.; Wu, X.; Wei, W. Rapid identification of candidate genes for seed weight using the SLAF-seq method in brassica napus. PLoS ONE 2016, 11, e0147580. [Google Scholar] [CrossRef]
- Zhang, J.F.; Stewart, J.M. Economical and Rapid Method for Extracting Cotton Genomic DNA. J. Cotton Sci. 2000, 4, 193–201. [Google Scholar]
- Li, H.; Durbin, R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The genome analysis toolkit: A mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Price, A.L.; Patterson, N.J.; Plenge, R.M.; Weinblatt, M.E.; Shadick, N.A.; Reich, D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006, 38, 904–909. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2010, 88, 76–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardy, O.J.; Vekemans, X. SPAGeDi: A versatile computer program to analyze spatial genetic structure at the individual or population levels. Mol. Ecol. Notes. 2002, 2, 618–620. [Google Scholar] [CrossRef] [Green Version]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; De Pristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef]
- Lippert, C.; Listgarten, J.; Liu, Y.; Kadie, C.M.; Davidson, R.I.; Heckerman, D. FaST linear mixed models for genome-wide association studies. Nat. Methods 2011, 8, 833–835. [Google Scholar] [CrossRef]
- Kang, H.M.; Sul, J.H.; Service, S.K.; Zaitlen, N.A.; Kong, S.Y.; Freimer, N.B.; Sabatti, C.; Eskin, E. Variance component model to account for sample structure in genome-wide association studies. Nat. Genet. 2010, 42, 348–354. [Google Scholar] [CrossRef] [Green Version]
- Neale, D.B.; Savolainen, O. Association genetics of complex traits in conifers. Trends Plant Sci. 2004, 9, 325–330. [Google Scholar] [CrossRef]
- Naoki, T.; Mohammad, N.; Widiyatno, S.I.; Kentaro, U.; Rempei, S.; Kevin, K.S.N.; Soon, L.L.; Yoshihiko, T. Potential of genome-wide association studies and genomic selection to improve productivity and quality of commercial timber species in tropical rainforest, a Case Study of Shorea platyclados. Forests 2020, 11, 239. [Google Scholar] [CrossRef] [Green Version]
- Nystedt, B.; Street, N.R.; Wetterbom, A.; Zuccolo, A.; Lin, Y.C.; Scofield, D.G.; Vezzi, F.; Delhomme, N.; et al.; Giacomello, S.; Alexeyenko, A.; et al. The Norway spruce genome sequence and conifer genome evolution. Nature 2013, 497, 579–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohorn, B.; Kohorn, S. The cell wall-associated kinases, waks, as pectin receptors. Front. Plant Sci. 2012, 3, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majda, M.; Robert, S. The role of auxin in cell wall expansion. Int. J. Mol. Sci. 2018, 19, 951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schröder, F.; Lisso, J.; Lange, P.; Müssig, C. The extracellular EXO protein mediates cell expansion in Arabidopsis leaves. BMC Plant Biol. 2009, 9, 20. [Google Scholar] [CrossRef] [Green Version]
- Tibbs, C.L.; Zhang, Z.; Yu, J. Status and prospects of genome-wide association studies in plants. Plant Genome 2021, 14, e20077. [Google Scholar] [CrossRef]
- Allwright, M.R.; Payne, A.; Emiliani, G.; Milner, S.; Viger, M.; Rouse, F.; Keurentjes, J.J.B.; Bérard, A.; Wildhagen, H.; Faivre-Rampant, P.; et al. Biomass traits and candidate genes for bioenergy revealed through association genetics in coppiced european Populus nigra (L.). Biotech. Biofuels 2016, 9, 195. [Google Scholar] [CrossRef] [Green Version]
- Cappa, E.P.; El-Kassaby, Y.A.; Garcia, M.N.; Acuña, C.; Borralho, N.M.; Grattapaglia, D.; Marcucci-Poltri, S.N. Impacts of population structure and analytical models in genome-wide association studies of complex traits in forest trees: A case study in Eucalyptus globulus. PLoS ONE 2013, 8, e81267. [Google Scholar] [CrossRef] [Green Version]
- Lipka, A.E.; Kandianis, C.B.; Hudson, M.E.; Yu, J.; Drnevich, J.; Bradbury, P.J.; Gore, M.A. From association to prediction: Statistical methods for the dissection and selection of complex traits in plants. Curr. Opin. Plant Biol. 2015, 24, 110–118. [Google Scholar] [CrossRef]
- Visscher, P.M.; Wray, N.R.; Zhang, Q.; Sklar, P.; McCarthy, M.I.; Brown, M.A.; Yang, J. 10 years of GWAS discovery: Biology, function, and translation. Am. J. Hum. Genet. 2017, 101, 5–22. [Google Scholar] [CrossRef] [Green Version]
- Thistlethwaite, F.R.; Gamal El-Dien, O.; Ratcliffe, B.; Klápště, J.; Porth, I.; Chen, C.; Stoehr, M.U.; Ingvarsson, P.K.; El-Kassaby, Y.A. Linkage disequilibrium vs. pedigree: Genomic selection prediction accuracy in conifer species. PLoS ONE 2020, 15, e0232201. [Google Scholar] [CrossRef]
- Parchman, T.L.; Gompert, Z.; Mudge, J.; Schilkey, F.D.; Benkman, C.W.; Buerkle, C.A. Genome-wide association genetics of an adaptive trait in lodgepole pine. Mol. Ecol. 2012, 21, 2991–3005. [Google Scholar] [CrossRef] [PubMed]
- De La Torre, A.R.; Wilhite, B.; Puiu, D.; St Clair, J.B.; Crepeau, M.W.; Salzberg, S.L.; Langley, C.H.; Allen, B.; Neale, D.B. Dissecting the polygenic basis of cold adaptation using genome-wide association of traits and environmental data in Douglas-fir. Genes 2021, 12, 110. [Google Scholar] [CrossRef]
- Plomion, C.; Chancerel, E.; Endelman, J.; Lamy, J.B.; Mandrou, E.; Lesur, I.; Ehrenmann, F.; Isik, F.; Bink, M.C.; van Heerwaarden, J.; et al. Genome-wide distribution of genetic diversity and linkage disequilibrium in a mass-selected population of maritime pine. BMC Genomics 2014, 15, 171. [Google Scholar] [CrossRef] [PubMed]
- Ta, F.; Liu, X.D.; Huang, D.L.; Wang, L.; Liu, R.H.; Zhao, W.J.; Jing, W.M. Quantitative dynamics of Picea crassifolia population in Dayekou basin of Qianlian mountains. Acta Ecol. Sin. 2021, 41, 6871–6882. (In Chinese) [Google Scholar]
- Birol, I.; Raymond, A.; Jackman, S.D.; Pleasance, S.; Coope, R.; Taylor, G.A.; Yuen, M.M.; Keeling, C.I.; Brand, D.; Vandervalk, B.P.; et al. Assembling the 20-Gb white spruce (Picea glauca) genome from whole-genome Shotgun sequencing data. Bioinformatics 2013, 29, 1492–1497. [Google Scholar] [CrossRef] [PubMed]
- Zimin, A.; Stevens, K.A.; Crepeau, M.W.; Holtz-Morris, A.; Koriabine, M.; Marçais, G.; Puiu, D.; Roberts, M.; Wegrzyn, J.L.; de Jong, P.J.; et al. Sequencing and assembly of the 22-Gb loblolly pine genome. Genetics 2014, 196, 875–890. [Google Scholar] [CrossRef] [Green Version]
- Gu, X.; Feng, C.; Ma, L.; Song, C.; Wang, Y.; Da, Y.; Li, H.; Chen, K.; Ye, S.; Ge, C.; et al. Genome-wide Association Study of Body Weight in Chicken F2 Resource Population. PLoS ONE 2011, 6, e21872. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Sun, Y.; Zhao, G.; Wang, F.; Wu, D.; Zheng, M.; Chen, J.; Zhang, L.; Hu, Y.; Wen, J. Genome-wide association study identifies loci and candidate genes for body composition and meat quality traits in Beijing-You chickens. PLoS ONE 2013, 8, e61172. [Google Scholar] [CrossRef]
- Bai, Q.; Cai, Y.; He, B.; Liu, W.; Pan, Q.; Zhang, Q. Core set construction and association analysis of Pinus massoniana from Guangdong province in southern China using SLAF-seq. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef]
- Yang, Y.; Xuan, L.; Yu, C.; Wang, Z.; Xu, J.; Fan, W.; Guo, J.; Yin, Y. High-density genetic map construction and quantitative trait loci identification for growth traits in (Taxodium distichum var. distichum × T. mucronatum) × T. mucronatum. BMC Plant Biol. 2018, 18, 263. [Google Scholar] [CrossRef]
- Huang, X.; Wei, X.; Sang, T.; Zhao, Q.; Feng, Q.; Zhao, Y.; Li, C.; Zhu, C.; Lu, T.; Zhang, Z.; et al. Genome-wide association studies of 14 agronomic traits in rice landraces. Nat. Genet. 2010, 42, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yan, J.; Shah, T.; Warburton, M.L.; Li, Q.; Li, L.; Gao, Y.; Chai, Y.; Fu, Z.; Zhou, Y.; et al. Genetic analysis and characterization of a new maize association mapping panel for quantitative trait loci dissection. Theor. Appl. Genet. 2010, 121, 417–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Ersoz, E.; Lai, C.Q.; Todhunter, R.J.; Tiwari, H.K.; Gore, M.A.; Bradbury, P.J.; Yu, J.; Arnett, D.K.; Ordovas, J.M.; et al. Mixed linear model approach adapted for genome-wide association studies. Nat. Genet. 2010, 42, 355–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Huang, M.; Fan, B.; Buckler, E.S.; Zhang, Z. Iterative usage of fixed and random effect models for powerful and efficient genome-wide association studies. PLoS Genet. 2016, 12, e1005767. [Google Scholar] [CrossRef] [PubMed]
- Dhanapal, A.P.; Crisosto, C.H. Association genetics of chilling injury susceptibility in peach (Prunus persica (L.) Batsch) across multiple years. 3 Biotech 2013, 3, 481–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hecht, B.C.; Campbell, N.R.; Holecek, D.E.; Narum, S.R. Genome-wide association reveals genetic basis for the propensity to migrate in wild populations of rainbow and steelhead trout. Mol. Ecol. 2013, 22, 3061–3076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Fan, X.C.; Zhang, Y.; Jiang, J.F.; Liu, C.H. Identification of favorable SNP alleles and candidate genes for seedlessness in Vitis vinifera L. using genome-wide association mapping. Euphytica 2017, 213, 136. [Google Scholar] [CrossRef]
- Kudo, K.; Nabeshima, E.; Begum, S.; Yamagishi, Y.; Nakaba, S.; Oribe, Y.; Yasue, K.; Funada, R. Formation of new networks of earlywood vessels in seedlings of the deciduous ring-porous hardwood Quercus Serrata in springtime. Trees Struct. Funct. 2018, 32, 725–734. [Google Scholar] [CrossRef]
- Martin, H.; Hanuš, V. Comparison of earlywood vessel variables in the wood of Quercus robur L. and Quercus petraea (Mattuschka) Liebl. growing at the same site. Dendrochronologia 2014, 32, 284–289. [Google Scholar] [CrossRef]
- Tsai, D.M.; Yang, C.H. A quantile–quantile plot based pattern matching for defect detection. Pattern Recognit. Lett. 2005, 26, 1948–1962. [Google Scholar] [CrossRef]
- Li, L.; Li, K.; Ali, A.; Guo, Y. AtWAKL10, a cell wall associated receptor-like kinase, negatively regulates leaf senescence in Arabidopsis thaliana. Int. J. Mol. Sci. 2021, 22, 4885. [Google Scholar] [CrossRef] [PubMed]
- Daniel, J.C. Plant cell walls: Wall-associated kinases and cell expansion. Curr. Biol. 2001, 11, R558–R559. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phenotype | Abbreviation | Unit |
|---|---|---|
| Earlywood tracheid length | EL | μm |
| Earlywood tracheid diameter | ED | μm |
| Earlywood tracheid lumen diameter | ELD | μm |
| Earlywood tracheid wall thickness | EWT | μm |
| Latewood tracheid length | LL | μm |
| Latewood tracheid diameter | LD | μm |
| Latewood tracheid lumen diameter | LLD | μm |
| Latewood tracheid wall thickness | LWT | μm |
| Earlywood tracheid lumen–diameter ratio | ELDR | |
| Earlywood tracheid wall–lumen ratio | EWLR | |
| Earlywood tracheid length–width ratio | ELWR | |
| Latewood tracheid lumen–diameter ratio | LLDR | |
| Latewood tracheid wall–lumen ratio | LWLR | |
| Latewood tracheid length–width ratio | LLWR |
| Trait | Min | Max | Mean | SD | CV (%) | Skewness | Kurtosis |
|---|---|---|---|---|---|---|---|
| EL | 2303.61 | 4131.40 | 3237.20 | 348.74 | 10.77 | −0.42 | −0.01 |
| ED | 44.37 | 66.46 | 54.14 | 5.26 | 9.71 | −0.40 | 0.36 |
| ELD | 20.17 | 59.62 | 46.78 | 5.97 | 12.76 | 2.71 | −0.42 |
| EWT | 5.28 | 30.29 | 7.36 | 2.40 | 32.64 | 80.35 | 8.41 |
| ELDR | 43.44 | 76.76 | 60.98 | 6.99 | 11.47 | −0.38 | −0.03 |
| EWLR | 0.41 | 0.91 | 0.86 | 0.05 | 5.62 | 69.75 | −7.56 |
| EDLR | 0.10 | 1.74 | 0.17 | 0.16 | 89.47 | 98.73 | 9.77 |
| LL | 2434.23 | 4317.94 | 3505.45 | 339.43 | 9.68 | 0.28 | −0.21 |
| LD | 34.20 | 47.83 | 40.45 | 2.57 | 6.34 | 0.39 | 0.07 |
| LLD | 14.82 | 26.64 | 20.83 | 2.45 | 11.76 | −0.33 | −0.01 |
| LWT | 15.21 | 23.69 | 19.62 | 1.90 | 9.66 | −0.29 | −0.26 |
| LLDR | 67.79 | 101.16 | 87.21 | 6.59 | 7.56 | 0.13 | −0.44 |
| LWLR | 0.40 | 0.62 | 0.51 | 0.04 | 8.71 | 0.10 | −0.04 |
| LDLR | 0.63 | 1.67 | 1.02 | 0.20 | 19.47 | 0.75 | 0.72 |
| Trait | Gene ID | Loci | Gene-start | Gene-end | SNP location | Allele | Annotation |
|---|---|---|---|---|---|---|---|
| ELDR, EWLR, EWT | MA_10430313g0010 | MA_10428216 | 15911 | 16486 | 8522 | T/A | Serine/threonine-protein kinase |
| MA_10429843g0020 | MA_10429843 | 27084 | 33339 | 14836 | G/T | Aminodeoxychorismate synthase | |
| MA_10434936g0010 | MA_10434936 | 3785 | 12707 | 16639 | T/C | Poly (ADP-ribose) glycohydrolase 1 | |
| MA_11137g0010 | MA_11137 | 25232 | 25570 | 17526 | G/A | Histone H1 | |
| MA_119933g0010 | MA_119933 | 7320 | 8147 | 15072 | C/A | Wall-associated receptor kinase | |
| MA_17692g0010 | MA_17692 | 82061 | 82593 | 87850 | G/A | Serine carboxypeptidase | |
| MA_19953g0020 | MA_19953 | 81799 | 91867 | 38054 | C/T | Pathogenesis-related protein 5 | |
| EWLR, EWT | MA_10303051g0010 | MA_10303051 | 23536 | 23793 | 2008 | G/A | Multicopper oxidase |
| MA_10883g0010 | MA_10883 | 53514 | 53744 | 17064 | G/A | EXORDIUM-like 2 | |
| EWLR | MA_101170g0020 | MA_101170 | 24857 | 25888 | 32445 | C/T | Carboxylesterase 17 |
| MA_10427040g0010 | MA_10427040 | 22428 | 23383 | 2504 | A/G | Heat shock 22 kDa protein | |
| MA_10427391g0010 | MA_10427391 | 21043 | 42330 | 58343 | G/A | MAG2 | |
| MA_10431643g0010 | MA_10431643 | 1 | 11637 | 12312 | A/G | Proteasome subunit alpha type-5 | |
| MA_10432762g0010 | MA_10432762 | 17888 | 18253 | 1144 | G/A | Telomerase reverse transcriptase | |
| MA_10433801g0010 | MA_10433801 | 551 | 15049 | 64877 | C/T | E3 ubiquitin-protein ligase | |
| MA_10437109g0030 | MA_10437109 | 94725 | 96103 | 91706 | G/C | ABC transporter D family member 1 | |
| MA_10437270g0010 | MA_10437270 | 42477 | 60307 | 1265 | G/T | IPG1 | |
| MA_114300g0010 | MA_114300 | 1314 | 3462 | 4171 | A/G | STRUBBELIG-RECEPTOR FAMILY 8 | |
| MA_174203g0010 | MA_174203 | 12346 | 13811 | 5106 | A/G | Sodium/metabolite cotransporter BASS1 | |
| MA_17795g0010 | MA_17795 | 42316 | 42738 | 57812 | T/A | Histone H2B.1 | |
| MA_182831g0010 | MA_182831 | 20010 | 21544 | 31133 | C/T | Hyoscyamine 6-dioxygenase | |
| MA_20154g0020 | MA_20154 | 67781 | 78419 | 57944 | C/T | Phosphoenolpyruvate carboxylase | |
| MA_31g0010 | MA_31 | 21393 | 40460 | 18526 | A/G | Ubiquitin carboxyl-terminal hydrolase 12 | |
| MA_34680g0010 | MA_34680 | 1 | 23548 | 847 | G/A | Peroxiredoxin-2F | |
| MA_39532g0010 | MA_39532 | 4950 | 6096 | 29400 | A/G | Copper transport protein CCH | |
| MA_417207g0010 | MA_417207 | 3307 | 5128 | 5643 | G/T | Purple acid phosphatase 3 | |
| MA_482451g0010 | MA_482451 | 1 | 1106 | 5136 | C/T | Ribulose-1,5 bisphosphate carboxylase | |
| MA_5201g0010 | MA_5201 | 4440 | 9434 | 12271 | G/A | Proline transporter 2 | |
| MA_588g0010 | MA_588 | 18671 | 26200 | 22351 | T/A | Abscisic acid hydroxylase 1 | |
| MA_74441g0010 | MA_74441 | 1775 | 11928 | 1579 | A/T | Aldo-keto reductase family 4 member C9 | |
| MA_80965g0010 | MA_80965 | 17882 | 22398 | 39315 | C/G | Flavin-containing monooxygenase 1 | |
| MA_951514g0010 | MA_951514 | 262 | 1867 | 3347 | A/G | 2-alkenal reductase | |
| MA_958g0010 | MA_958 | 40918 | 71141 | 41171 | G/T | MODIFIER OF SNC1 1 | |
| MA_99302g0010 | MA_99302 | 18687 | 19646 | 28542 | C/T | Thioredoxin-like protein CDSP32 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, C.; Guo, Y.; Chen, Y.; Zhang, H.; El-Kassaby, Y.A.; Li, W. Genome Wide Association Study Identifies Candidate Genes Related to the Earlywood Tracheid Properties in Picea crassifolia Kom. Forests 2022, 13, 332. https://0-doi-org.brum.beds.ac.uk/10.3390/f13020332
Zhou C, Guo Y, Chen Y, Zhang H, El-Kassaby YA, Li W. Genome Wide Association Study Identifies Candidate Genes Related to the Earlywood Tracheid Properties in Picea crassifolia Kom. Forests. 2022; 13(2):332. https://0-doi-org.brum.beds.ac.uk/10.3390/f13020332
Chicago/Turabian StyleZhou, Chengcheng, Yingtian Guo, Yali Chen, Hongbin Zhang, Yousry A. El-Kassaby, and Wei Li. 2022. "Genome Wide Association Study Identifies Candidate Genes Related to the Earlywood Tracheid Properties in Picea crassifolia Kom." Forests 13, no. 2: 332. https://0-doi-org.brum.beds.ac.uk/10.3390/f13020332