
Genome-Wide Investigation of the MiR166 Family Provides New Insights into Its Involvement in the Drought Stress Responses of Tea Plants (Camellia sinensis (L.) O. Kuntze)

 and
and 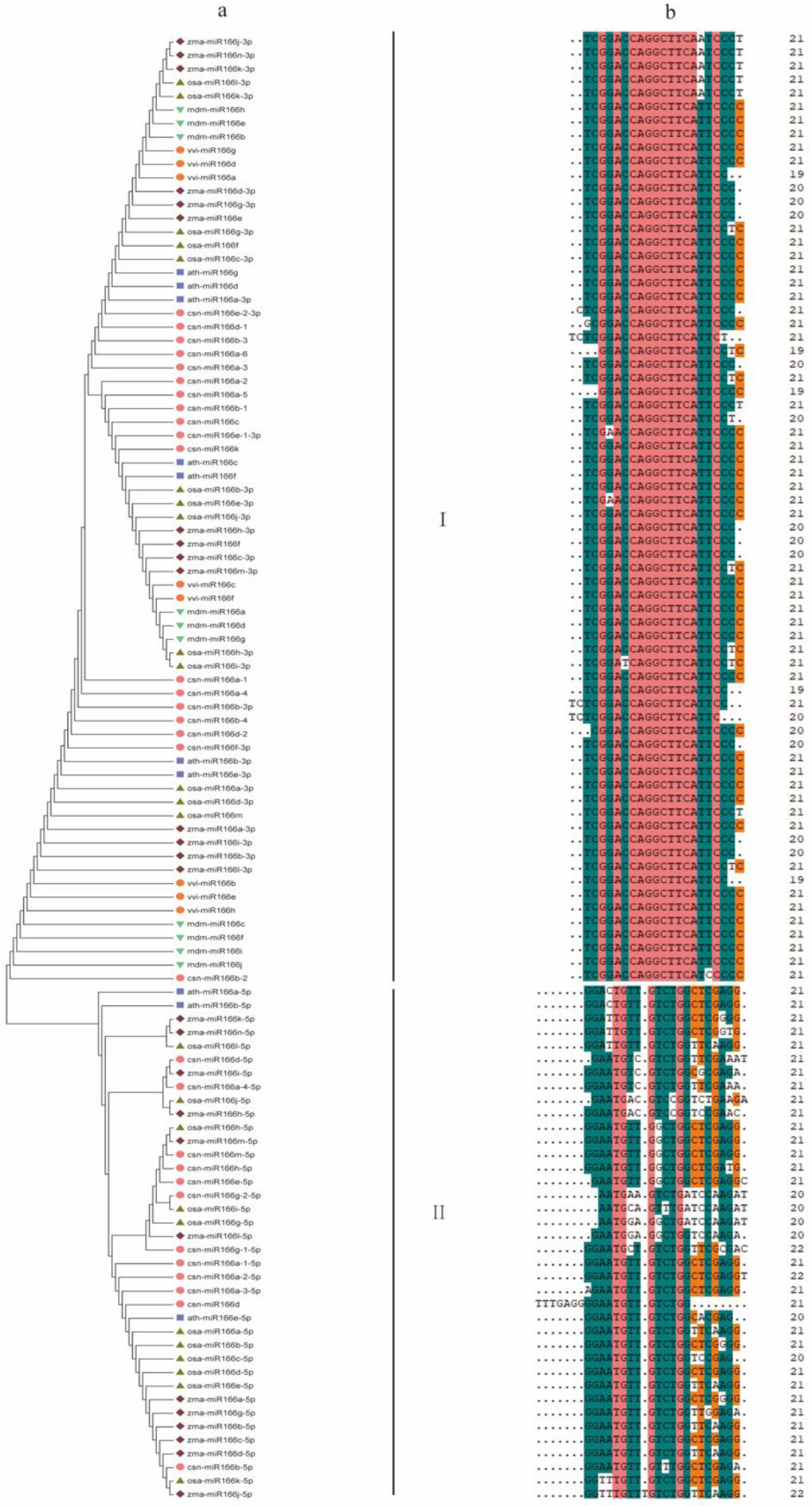{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Treatments
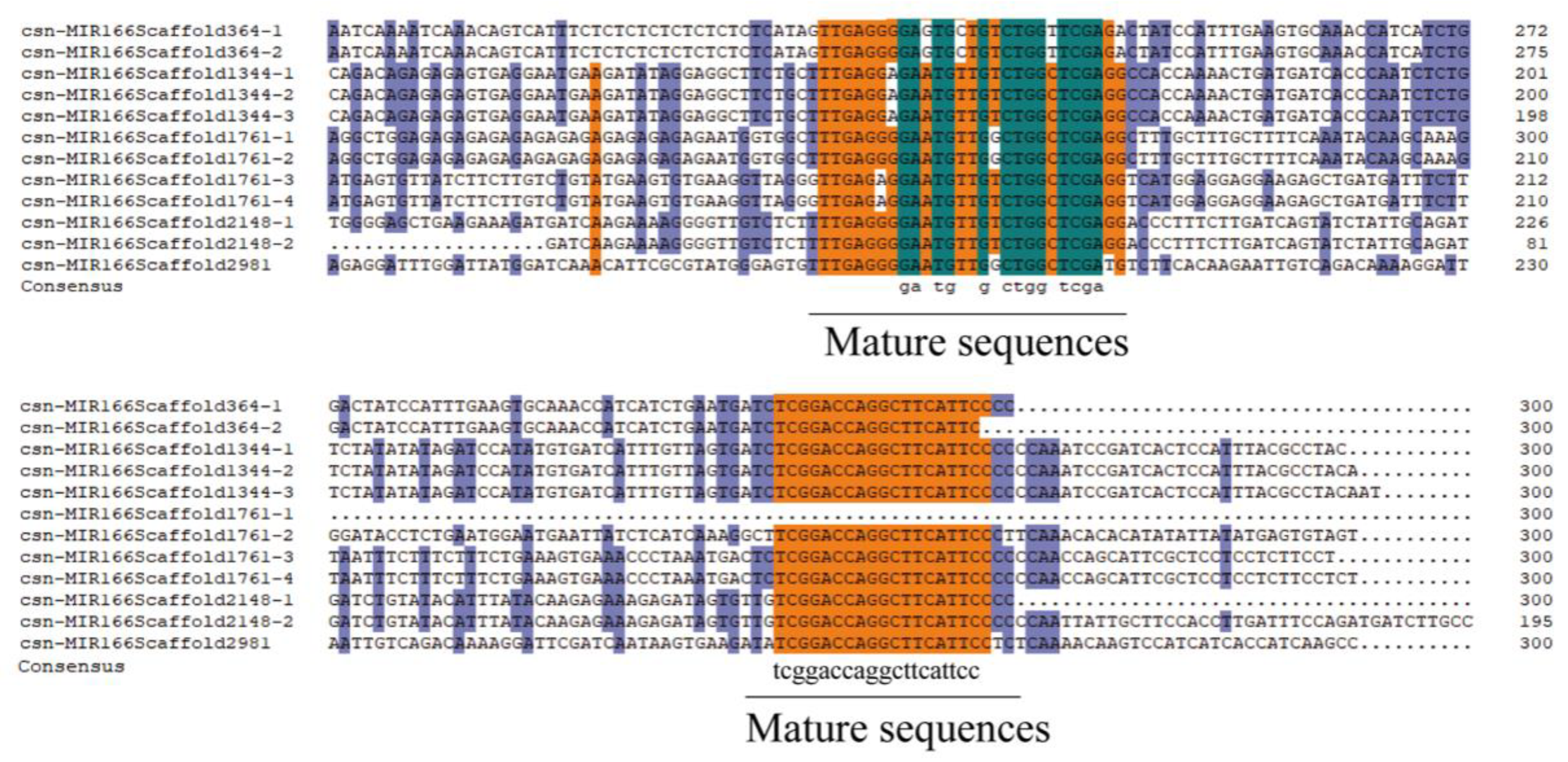
2.2. Identification of Csn-MiR166s in Tea Plant’s Genome and Acquisition of Plant MiR166 Sequences
2.3. Bioinformatic Analyses of Csn-MiR166
2.4. Physiological Characterization of the Tea Plant under Different Drought Degrees
2.5. Total RNA Isolation and cDNA Synthesis
2.6. Reverse Transcription Quantitative Polymerase Chain Reaction (RT-qPCR) Analyses
2.7. Cleavage Site Identification on the Basis of a Modified 5′ RNA Ligase-Mediated Rapid Amplification of cDNA Ends (RLM-RACE)
2.8. Statistical Analysis
3. Results
3.1. Analysis of MiR166 Sequences in Plants
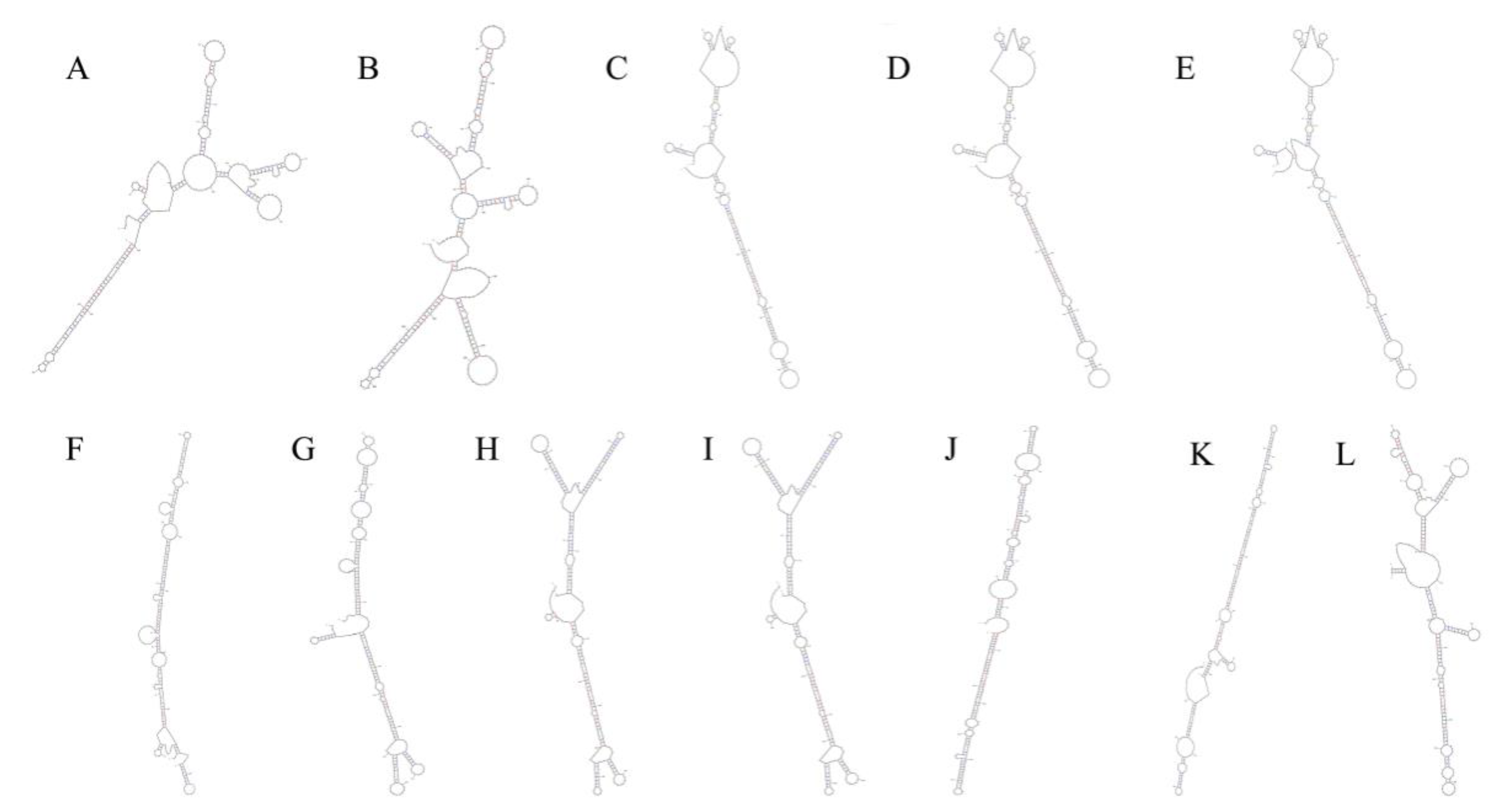
3.2. Sequence Analysis and Predicted Secondary Structure Analysis of Pre-MiR166s in Tea Genome
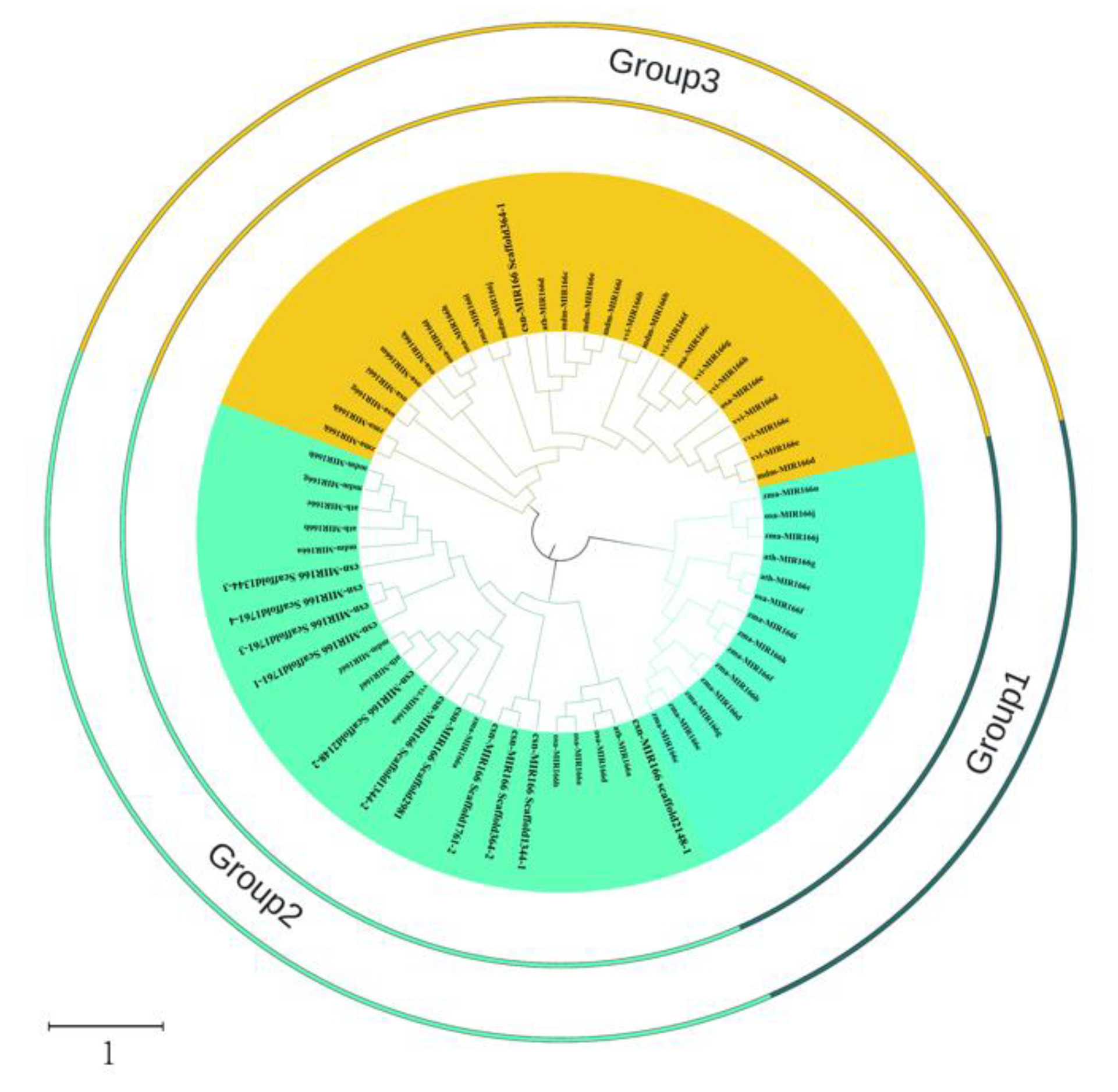
3.3. Evolutionary Relationships among Pre-MiR166s in Plants
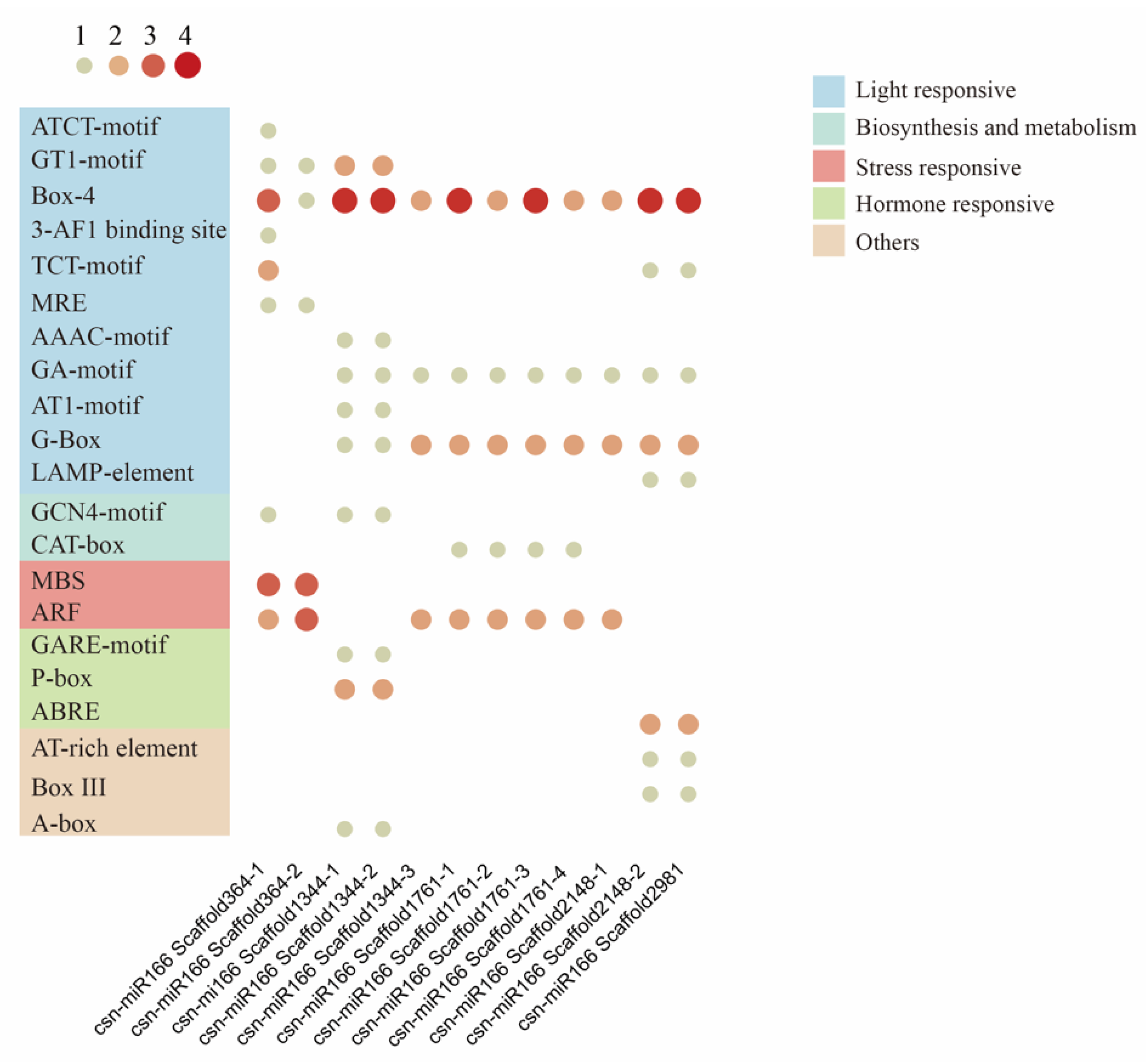
3.4. Analysis of Cis-Acting Elements in CsMIR166 Promoter Regions
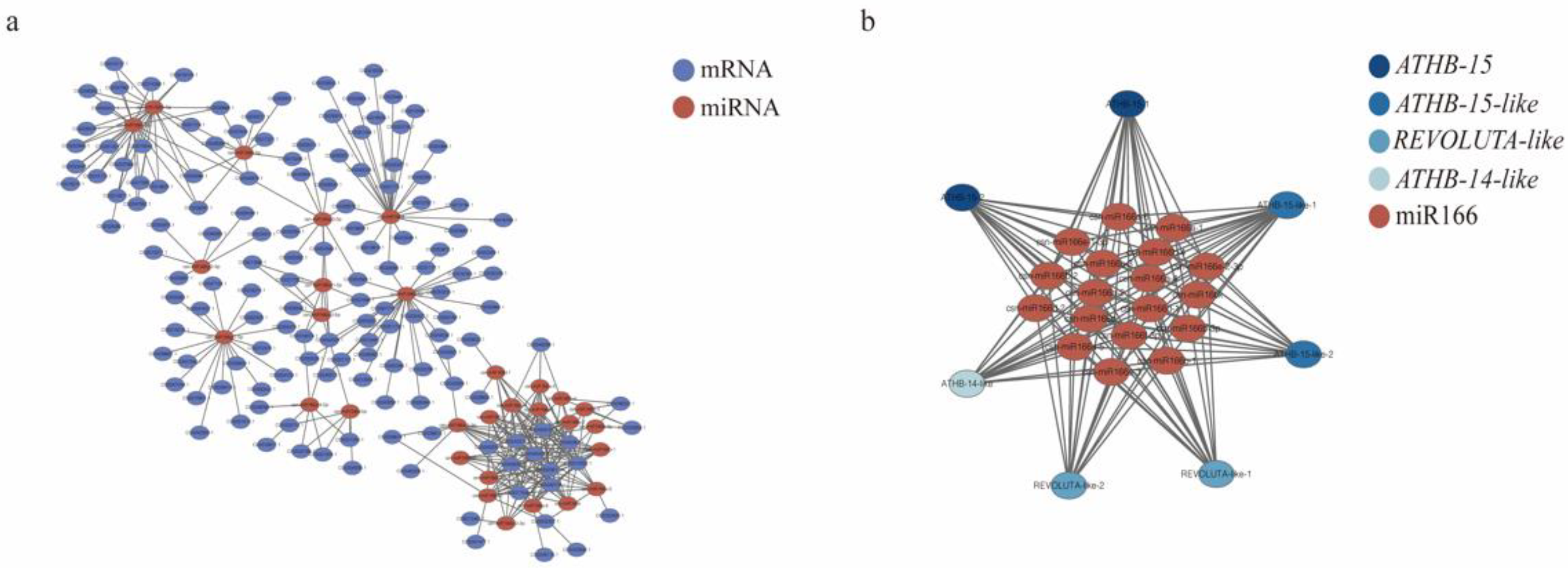
3.5. Prediction of csn-miR166 targets
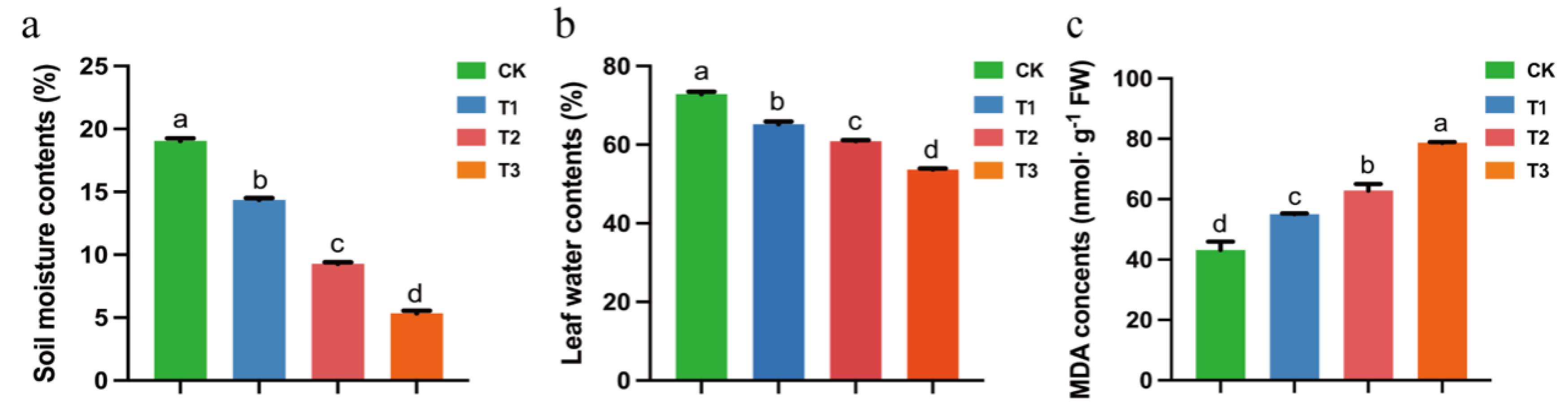
3.6. Analysis of Tea Plants Relative Water Content and Malondialdehyde Content under Drought Stress
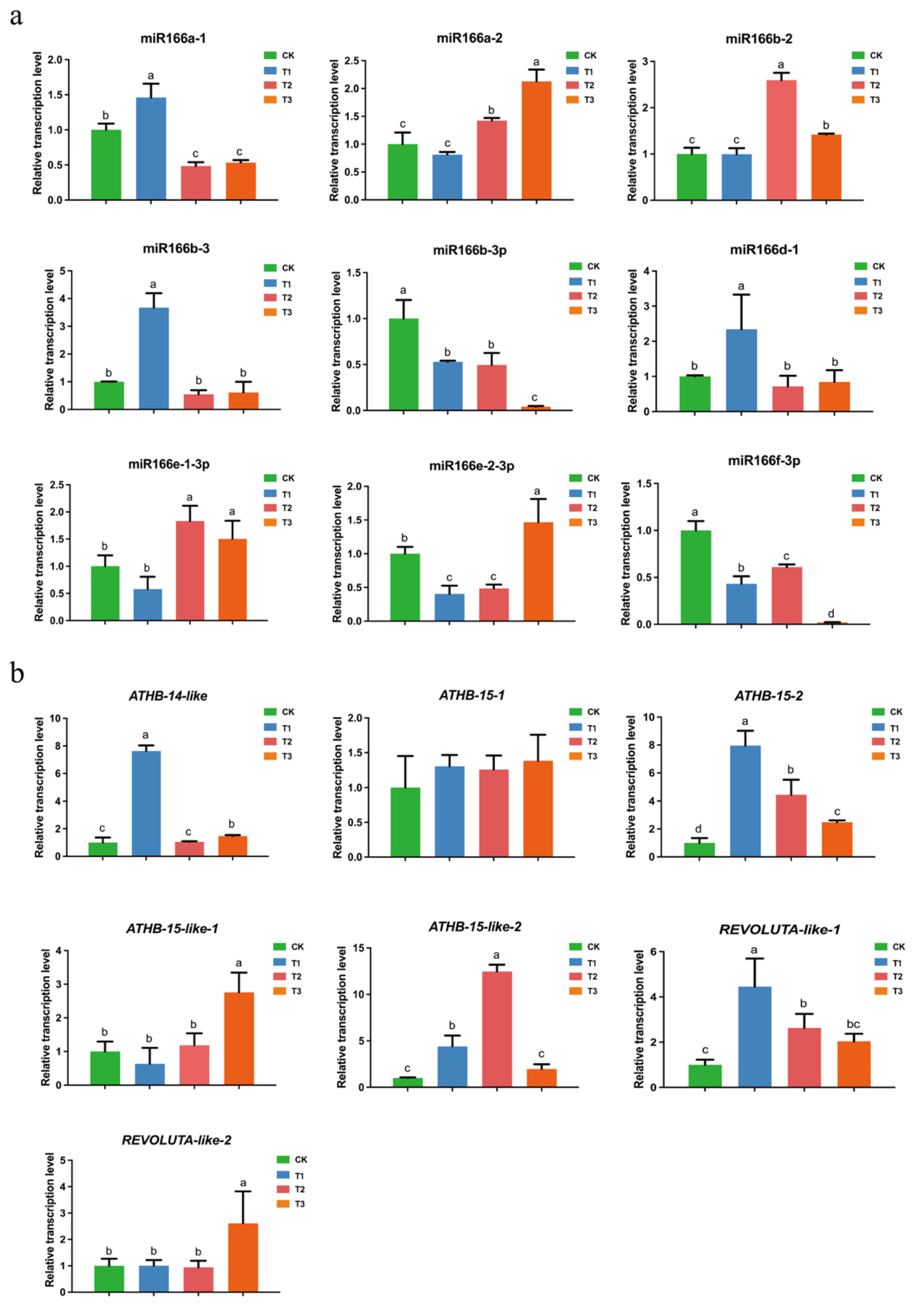
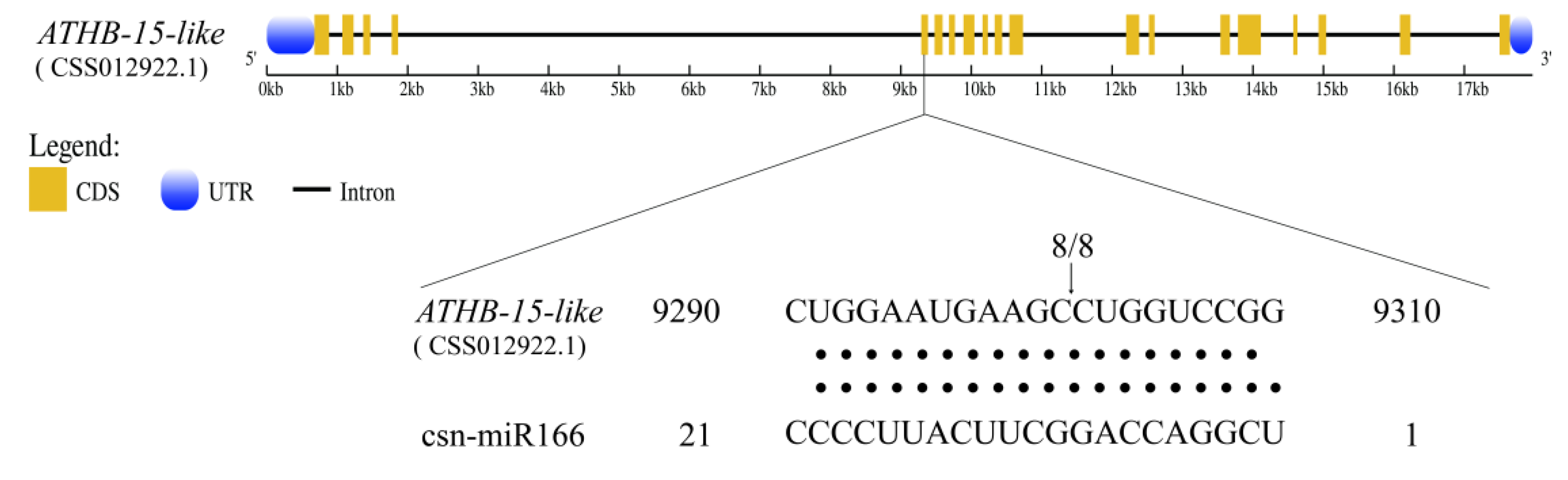
3.7. Transcription Profiling and Cleavage Site Identification of Csn-MiR166s and Their Targets under Different Drought Degrees
4. Discussion
4.1. Evolutionary Characteristics of Csn-MiR166s
4.2. Cleavage of HD-ZIP III Transcripts by Csn-MiR166s Is a Crucial Mechanism Underlying Tea Plant Drought Tolerance
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cheruiyot, E.K.; Mumera, L.M.; Ng’Etich, W.K.; Hassanali, A.; Wachira, F.N. High fertilizer rates increase susceptibility of tea to water stress. J. Plant Nutr. 2010, 33, 29–115. [Google Scholar] [CrossRef]
- Guo, Y.Q.; Zhao, S.S.; Zhu, C.; Chang, X.J.; Yue, C.; Wang, Z.; Lin, Y.L.; Lai, Z.X. Identification of drought-responsive miRNAs and physiological characterization of tea plant (Camellia sinensis L.) under drought stress. BMC Plant Biol. 2017, 17, 211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.C.; Yao, M.Z.; Jin, J.Q.; Ma, J.Q.; Li, C.F.; Chen, L. Physiological changes and differential gene expression of tea plant under dehydration and rehydration conditions. Sci. Hortic. 2015, 184, 129–141. [Google Scholar] [CrossRef]
- Maritim, T.K.; Kamunya, S.M.; Mireji, P.; Mwendia, C.; Muoki, R.C.; Cheruiyot, E.K.; Wachira, F.N. Physiological and biochemical response of tea [Camellia sinensis (L.) O. Kuntze] to water-deficit stress. J. Hortic. Sci. Biotechnol. 2015, 90, 395–400. [Google Scholar] [CrossRef]
- Li, H.; Teng, R.M.; Liu, J.X.; Yang, Y.Z.; Lin, S.J.; Han, M.H.; Liu, J.Y.; Zhuang, J. Identification and Analysis of Genes Involved in Auxin, Abscisic Acid, Gibberellin, and Brassinosteroid Metabolisms Under Drought Stress in Tender Shoots of Tea Plants. DNA Cell Biol. 2019, 38, 1292–1302. [Google Scholar] [CrossRef] [PubMed]
- Song, X.W.; Li, Y.; Cao, X.F.; Qi, Y.J. MicroRNAs and Their Regulatory Roles in Plant-Environment Interactions. Annu. Rev. Plant Biol. 2019, 70, 489–525. [Google Scholar] [CrossRef]
- Reinhart, B.J.; Weinstein, E.G.; Rhoades, M.W.; Bartel, B.; Bartel, D.P. MicroRNAs in plants. Genes Dev. 2002, 13, 1616–32132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones Rhoades, M.W.; Bartel, D.P.; Bartel, B. MicroRNAs and Their Regulatory Roles in Plants. Annu. Rev. Plant Biol. 2006, 57, 19–53. [Google Scholar] [CrossRef]
- Zhang, B.H.; Wang, Q.L. MicroRNA-based biotechnology for plant improvement. J. Cell. Physiol. 2015, 230, 1–15. [Google Scholar] [CrossRef]
- Arshad, M.; Feyissa, B.A.; Amyot, L.; Aung, B.; Hannoufa, A. MicroRNA156 improves drought stress tolerance in alfalfa (Medicago sativa) by silencing SPL13. Plant Sci. 2017, 258, 122–136. [Google Scholar] [CrossRef]
- Zhang, J.H.; Zhang, H.; Srivastava, A.K.; Pan, Y.J.; Bai, J.J.; Fang, J.J.; Shi, H.Z.; Zhu, J.K. Knockdown of Rice MicroRNA166 Confers Drought Resistance by Causing Leaf Rolling and Altering Stem Xylem Development. Plant Physiol. 2018, 176, 2082–2094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, K.F.; Wang, R.; Ou, X.J.; Fang, Z.M.; Tian, C.E.; Duan, J.; Wang, Y.Q.; Zhang, M.Y. OsTIR1 and OsAFB2 downregulation via OsmiR393 overexpression leads to more tillers, early flowering and less tolerance to salt and drought in rice. PLoS ONE 2012, 7, e30039. [Google Scholar] [CrossRef] [PubMed]
- Baek, D.; Chun, H.J.; Kang, S.; Shin, G.; Park, S.J.; Hong, H.; Kim, C.; Kim, D.H.; Lee, S.Y.; Kim, M.C.; et al. A Role for Arabidopsis miR399f in Salt, Drought, and ABA Signaling. Mol. Cells 2016, 39, 111–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, J.; Park, C. MIR166/165 genes exhibit dynamic expression patterns in regulating shoot apical meristem and floral development in Arabidopsis. Planta 2007, 225, 1327–1338. [Google Scholar] [CrossRef]
- Zhu, H.L.; Hu, F.Q.; Wang, R.H.; Zhou, X.; Sze, S.H.; Liou, L.W.; Barefoot, A.; Dickman, M.; Zhang, X.R. Arabidopsis Argonaute10 Specifically Sequesters miR166/165 to Regulate Shoot Apical Meristem Development. Cell 2011, 145, 242–256. [Google Scholar] [CrossRef] [Green Version]
- Akdogan, G.; Tufekci, E.D.; Uranbey, S.; Unver, T. miRNA-based drought regulation in wheat. Funct. Integr. Genom. 2016, 16, 221–233. [Google Scholar] [CrossRef]
- Aravind, J.; Rinku, S.; Pooja, B.; Shikha, M.; Kaliyugam, S.; Mallikarjuna, M.G.; Kumar, A.; Rao, A.R.; Nepolean, T. Identification, Characterization, and Functional Validation of Drought-responsive MicroRNAs in Subtropical Maize Inbreds. Front. Plant Sci. 2017, 8, 2–14. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.S.; Li, M.; Zhang, Z.C.; Tie, W.W.; Chen, X.; Jin, L.F.; Zhai, N.; Zheng, Q.X.; Zhang, J.F.; Wang, R.; et al. Integrated mRNA and microRNA analysis identifies genes and small miRNA molecules associated with transcriptional and post-transcriptional-level responses to both drought stress and re-watering treatment in tobacco. BMC Genom. 2017, 18, 62–73. [Google Scholar] [CrossRef] [Green Version]
- Yadav, A.; Kumar, S.; Verma, R.; Lata, C.; Sanyal, I.; Rai, S.P. microRNA 166: An evolutionarily conserved stress biomarker in land plants targeting HD-ZIP family. Physiol. Mol. Biol. Plants 2021, 27, 2471–2485. [Google Scholar] [CrossRef]
- Hamza, N.B.; Sharma, N.; Tripathi, A.; Sanan-Mishra, N. MicroRNA expression profiles in response to drought stress in Sorghum bicolor. Gene Expr. Patterns 2016, 20, 88–98. [Google Scholar] [CrossRef]
- Singh, A.; Singh, S.; Panigrahi, K.; Reski, R.; Sarkar, A.K. Balanced activity of microRNA166/165 and its target transcripts from the class III homeodomain-leucine zipper family regulates root growth in Arabidopsis thaliana. Plant Cell Rep. 2014, 33, 945–953. [Google Scholar] [CrossRef] [PubMed]
- Rong, F.; Chen, F.; Huang, L.; Zhang, J.; Zhang, C.; Hou, D.; Cheng, Z.; Weng, Y.; Chen, P.; Li, Y. A mutation in class III homeodomain-leucine zipper (HD-ZIP III) transcription factor results in curly leaf (cul) in cucumber (Cucumis sativus L.). Theor. Appl. Genet. 2019, 132, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.X.; Wang, Y.Y.; Teotia, S.C.; Wang, Z.H.; Shi, C.N.; Sun, H.W.; Gu, Y.Y.; Zhang, Z.H.; Tang, G.L. The interaction between miR160 and miR165/166 in the control of leaf development and drought tolerance in Arabidopsis. Sci. Rep. 2019, 9, 2832. [Google Scholar] [CrossRef] [PubMed]
- Li, R.X. Identification and Functional Characterization of Drought-Resistance Related Micro RNAs and Their Targets in Mulberry. Ph.D. Thesis, Jiangsu University of Science and Technology, Zhenjiang, China, 2018. [Google Scholar]
- Li, X.Y.; Xie, X.; Li, J.; Cui, Y.H.; Hou, Y.M.; Zhai, L.L.; Wang, X.; Fu, Y.L.; Liu, R.R.; Bian, S.M. Conservation and diversification of the miR166 family in soybean and potential roles of newly identified miR166s. BMC Plant Biol. 2017, 17, 32–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.L.; Zhang, Q.L.; Zeng, Y.J.; Chen, X.H.; Zhang, Z.H.; Chen, Y.K.; Lai, Z.X. Analysis on Evolutionary Characteristics and the Temporal and Spatial Expression Patterns of miR166 Gene Family in Dimocarpus longan. Acta Hortic. Sin. 2017, 44, 2285–2295. [Google Scholar] [CrossRef]
- Xia, E.H.; Zhang, H.B.; Sheng, J.; Li, K.; Zhang, Q.J.; Kim, C.H.; Zhang, Y.; Liu, Y.; Zhu, T.; Li, W.; et al. The Tea Tree Genome Provides Insights into Tea Flavor and Independent Evolution of Caffeine Biosynthesis. Mol. Plant 2017, 10, 866–877. [Google Scholar] [CrossRef] [Green Version]
- Wei, C.L.; Yang, H.; Wang, S.B.; Zhao, J.; Liu, C.; Gao, L.P.; Xia, E.H.; Lu, Y.; Tai, Y.L.; She, G.B.; et al. Draft genome sequence of Camellia sinensis var. sinensis provides insights into the evolution of the tea genome and tea quality. Proc. Natl. Acad. Sci. USA 2018, 115, E4151–E4158. [Google Scholar] [CrossRef] [Green Version]
- Xia, E.H.; Tong, W.; Hou, Y.; An, Y.L.; Chen, L.B.; Wu, Q.; Liu, Y.L.; Yu, J.; Li, F.D.; Li, R.P.; et al. The Reference Genome of Tea Plant and Resequencing of 81 Diverse Accessions Provide Insights into Its Genome Evolution and Adaptation. Mol. Plant 2020, 13, 1013–1026. [Google Scholar] [CrossRef]
- Chen, J.D.; Zheng, C.; Ma, J.Q.; Jiang, C.K.; Ercisli, S.; Yao, M.Z.; Chen, L. The chromosome-scale genome reveals the evolution and diversification after the recent tetraploidization event in tea plant. Hortic. Res. 2020, 7, 63–74. [Google Scholar] [CrossRef]
- Zhang, Q.J.; Li, W.; Li, K.; Nan, H.; Shi, C.; Zhang, Y.; Dai, Z.Y.; Lin, Y.L.; Yang, X.L.; Tong, Y.; et al. The Chromosome-Level Reference Genome of Tea Tree Unveils Recent Bursts of Non-autonomous LTR Retrotransposons in Driving Genome Size Evolution. Mol. Plant 2020, 13, 935–938. [Google Scholar] [CrossRef]
- Wang, X.C.; Feng, H.; Chang, Y.X.; Ma, C.L.; Wang, L.Y.; Hao, X.Y.; Li, A.L.; Cheng, H.; Wang, L.; Cui, P.; et al. Population sequencing enhances understanding of tea plant evolution. Nat. Commun. 2020, 11, 4447. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.Y.; Zhang, Y.J.; Qiu, H.J.; Guo, Y.F.; Wan, H.L.; Zhang, X.L.; Scossa, F.; Alseekh, S.; Zhang, Q.H.; Wang, P.; et al. Genome assembly of wild tea tree DASZ reveals pedigree and selection history of tea varieties. Nat. Commun. 2020, 11, 3719. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.J.; Yu, J.X.; Jin, S.; Chen, S.; Yue, C.; Wang, W.L.; Gao, S.L.; Cao, H.L.; Zheng, Y.C.; Gu, M.Y.; et al. Genetic basis of high aroma and stress tolerance in the oolong tea cultivar genome. Hortic. Res. 2021, 8, 107. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Du, G.Y.; Li, X.H.; Zhang, C.Y.; Guo, J.K. A major locus controlling malondialdehyde content under water stress is associated with Fusarium crown rot resistant in wheat. Mol. Genet. Genom. 2015, 290, 1955–1962. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.L.; Cai, H.; Zhai, H.; Luo, X.; Wang, Z.Y.; Cui, L.; Bai, X. Over-expression of Glycine soja WRKY20 enhances both drought and salt tolerance in transgenic alfalfa (Medicago sativa L.). Plant Cell. Tissue Organ Cult. 2014, 118, 1–10. [Google Scholar] [CrossRef]
- Sheng, L. A Literature-Curated Datebase for miRNA Molecular in Plant Response to Abiotic Stress and Application in Tea Plant (Camellia sinensis). Master’s Thesis, Anhui Agricultural University, Hefei, China, 2013. [Google Scholar]
- Zhang, Y.; Zhu, X.J.; Chen, X.; Song, C.N.; Zou, Z.W.; Wang, Y.H.; Wang, M.L.; Fang, W.P.; Li, X.H. Identification and characterization of cold-responsive microRNAs in tea plant (Camellia sinensis) and their targets using high-throughput sequencing and degradome analysis. BMC Plant Biol. 2014, 14, 271. [Google Scholar] [CrossRef] [Green Version]
- Anburaj, J.; Xiao, Z.; Yan, H.; Mingzhu, S.; Prabu, G.; Yun, L.Y.; Ling, W.C. Genome-wide identification of conserved and novel microRNAs in one bud and two tender leaves of tea plant (Camellia sinensis) by small RNA sequencing, microarray-based hybridization and genome survey scaffold sequences. BMC Plant Biol. 2017, 17, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.L.; Lai, Z.X. Reference gene selection for qPCR analysis during somatic embryogenesis in longan tree. Plant Sci. 2010, 178, 359–365. [Google Scholar] [CrossRef]
- Zhou, C.; Zhu, C.; Fu, H.; Li, X.; Chen, L.; Lin, Y.; Lai, Z.; Guo, Y. Genome-wide investigation of superoxide dismutase (SOD) gene family and their regulatory miRNAs reveal the involvement in abiotic stress and hormone response in tea plant (Camellia sinensis). PLoS ONE 2019, 14, e223609. [Google Scholar] [CrossRef]
- Thakur, V.; Wanchana, S.; Xu, M.; Bruskiewich, R.; Quick, W.P.; Mosig, A.; Zhu, X.G. Characterization of statistical features for plant microRNA prediction. BMC Genom. 2011, 12, 108. [Google Scholar] [CrossRef] [Green Version]
- Griffiths Jones, S. The microRNA Registry. Nucleic Acids Res. 2004, 32, 109D–111D. [Google Scholar] [CrossRef] [PubMed]
- Jones Rhoades, M.W. Conservation and divergence in plant microRNAs. Plant Mol. Biol. 2012, 80, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.H.; Tian, X.; Li, Y.J.; Wu, C.A.; Zheng, C.C. Microarray-based analysis of stress-regulated microRNAs in Arabidopsis thaliana. RNA 2008, 14, 836–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferdous, J.; Hussain, S.S.; Shi, B.J. Role of microRNAs in plant drought tolerance. Plant Biotechnol. J. 2015, 13, 293–305. [Google Scholar] [CrossRef]
- Prigge, M.J.; Otsuga, D.; Alonso, J.M.; Ecker, J.R.; Drews, G.N.; Clark, S.E. Class III Homeodomain-Leucine Zipper Gene Family Members Have Overlapping, Antagonistic, and Distinct Roles in Arabidopsis Development. Plant Cell 2005, 17, 61–76. [Google Scholar] [CrossRef] [Green Version]
- Simon, T.; Patrick, G.; David, B. Tracheary Element Differentiation. Annu. Rev. Plant Biol. 2007, 58, 407–433. [Google Scholar] [CrossRef] [Green Version]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, C.; Zhou, C.; Zhu, C.; Chen, L.; Shi, B.; Lin, Y.; Lai, Z.; Guo, Y. Genome-Wide Investigation of the MiR166 Family Provides New Insights into Its Involvement in the Drought Stress Responses of Tea Plants (Camellia sinensis (L.) O. Kuntze). Forests 2022, 13, 628. https://0-doi-org.brum.beds.ac.uk/10.3390/f13040628
Tian C, Zhou C, Zhu C, Chen L, Shi B, Lin Y, Lai Z, Guo Y. Genome-Wide Investigation of the MiR166 Family Provides New Insights into Its Involvement in the Drought Stress Responses of Tea Plants (Camellia sinensis (L.) O. Kuntze). Forests. 2022; 13(4):628. https://0-doi-org.brum.beds.ac.uk/10.3390/f13040628
Chicago/Turabian StyleTian, Caiyun, Chengzhe Zhou, Chen Zhu, Lan Chen, Biying Shi, Yuling Lin, Zhongxiong Lai, and Yuqiong Guo. 2022. "Genome-Wide Investigation of the MiR166 Family Provides New Insights into Its Involvement in the Drought Stress Responses of Tea Plants (Camellia sinensis (L.) O. Kuntze)" Forests 13, no. 4: 628. https://0-doi-org.brum.beds.ac.uk/10.3390/f13040628