
Protein Markers for the Identification of Cork Oak Plants Infected with Phytophthora cinnamomi by Applying an (α, β)-k-Feature Set Approach


Abstract
:1. Introduction
2. Materials and Methods
2.1. Public Cork Oak Protein Dataset
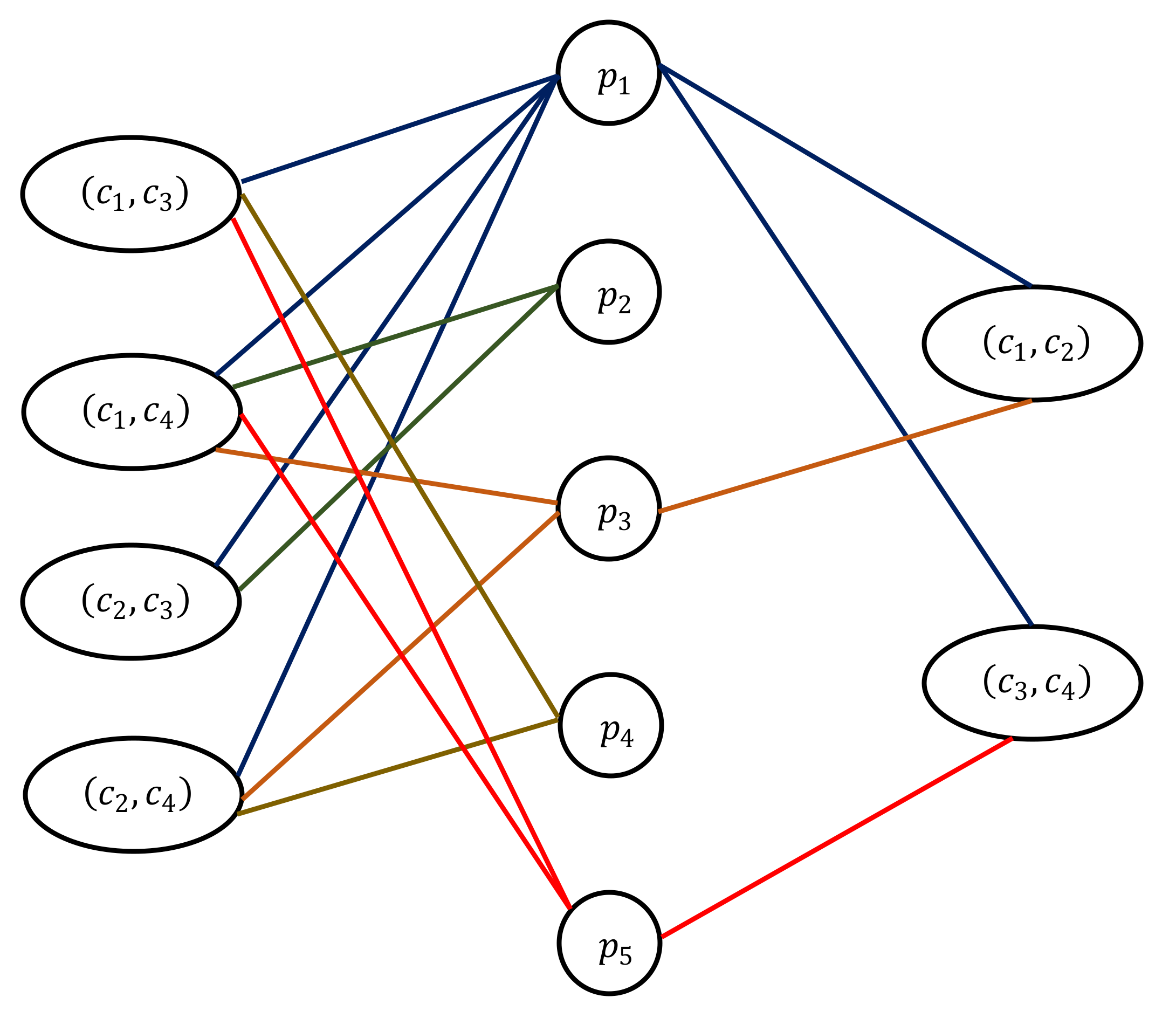
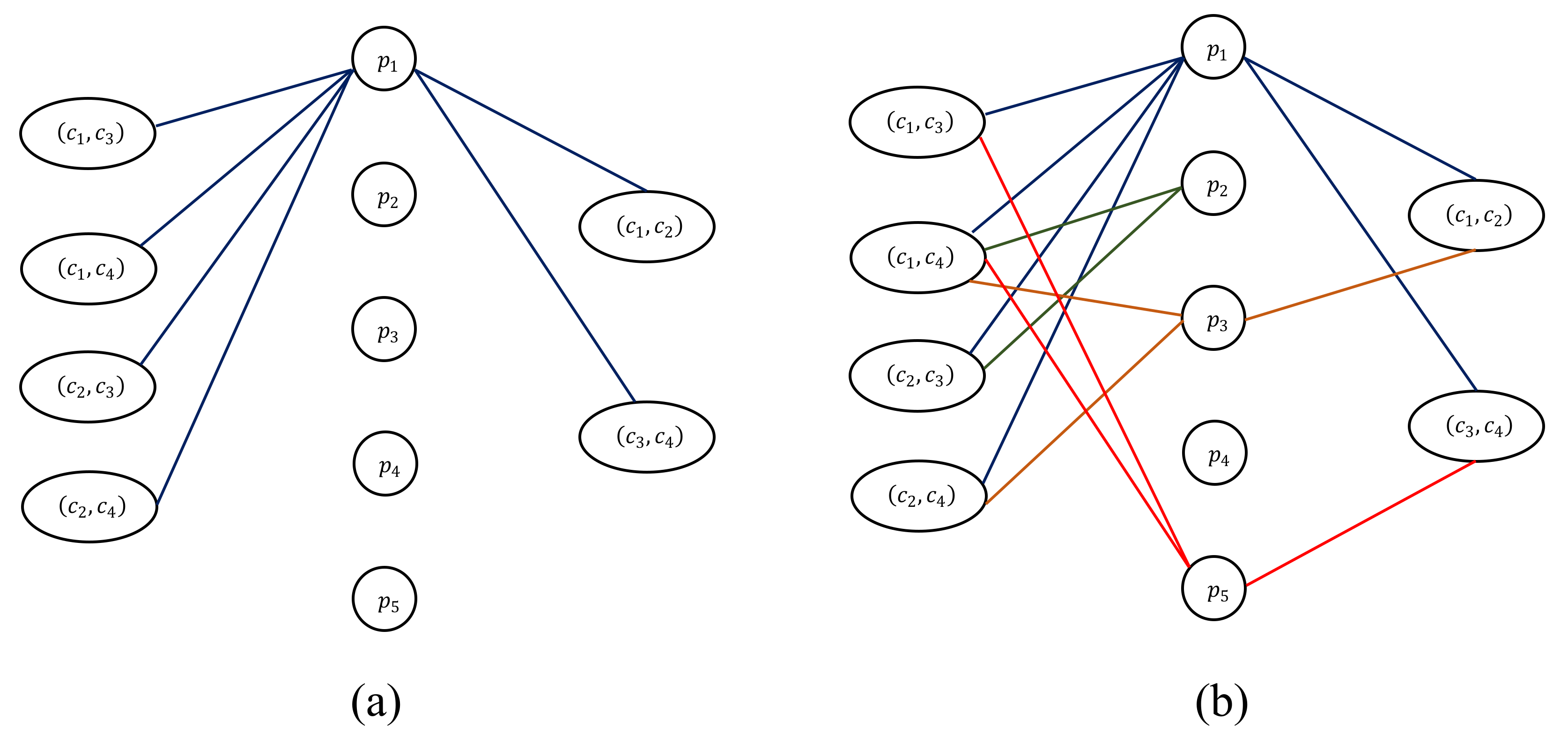
2.2. Modeling the Cork Oak Protein Dataset as an (α, β)-k-Feature Set Problem
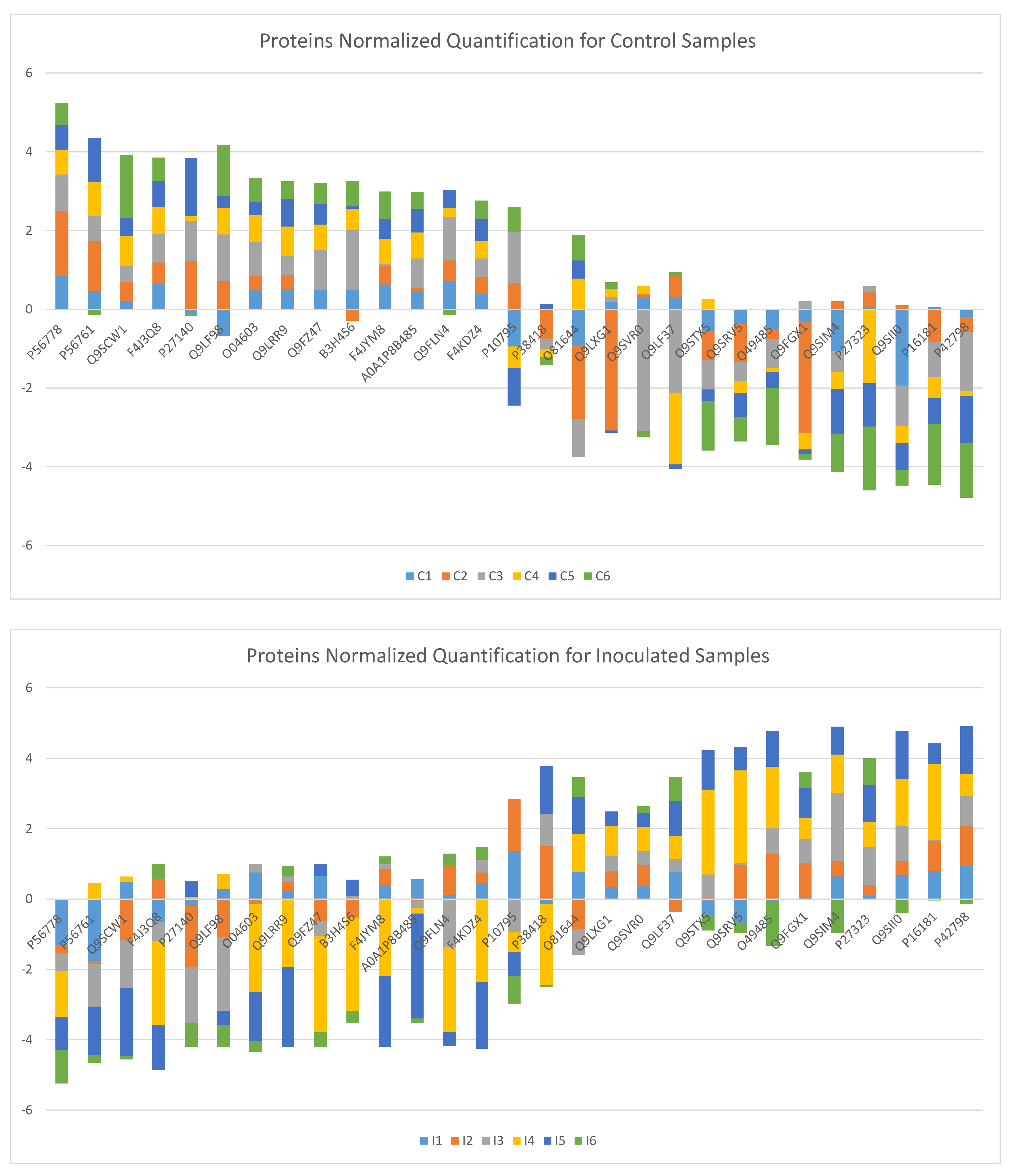
3. Results and Discussion
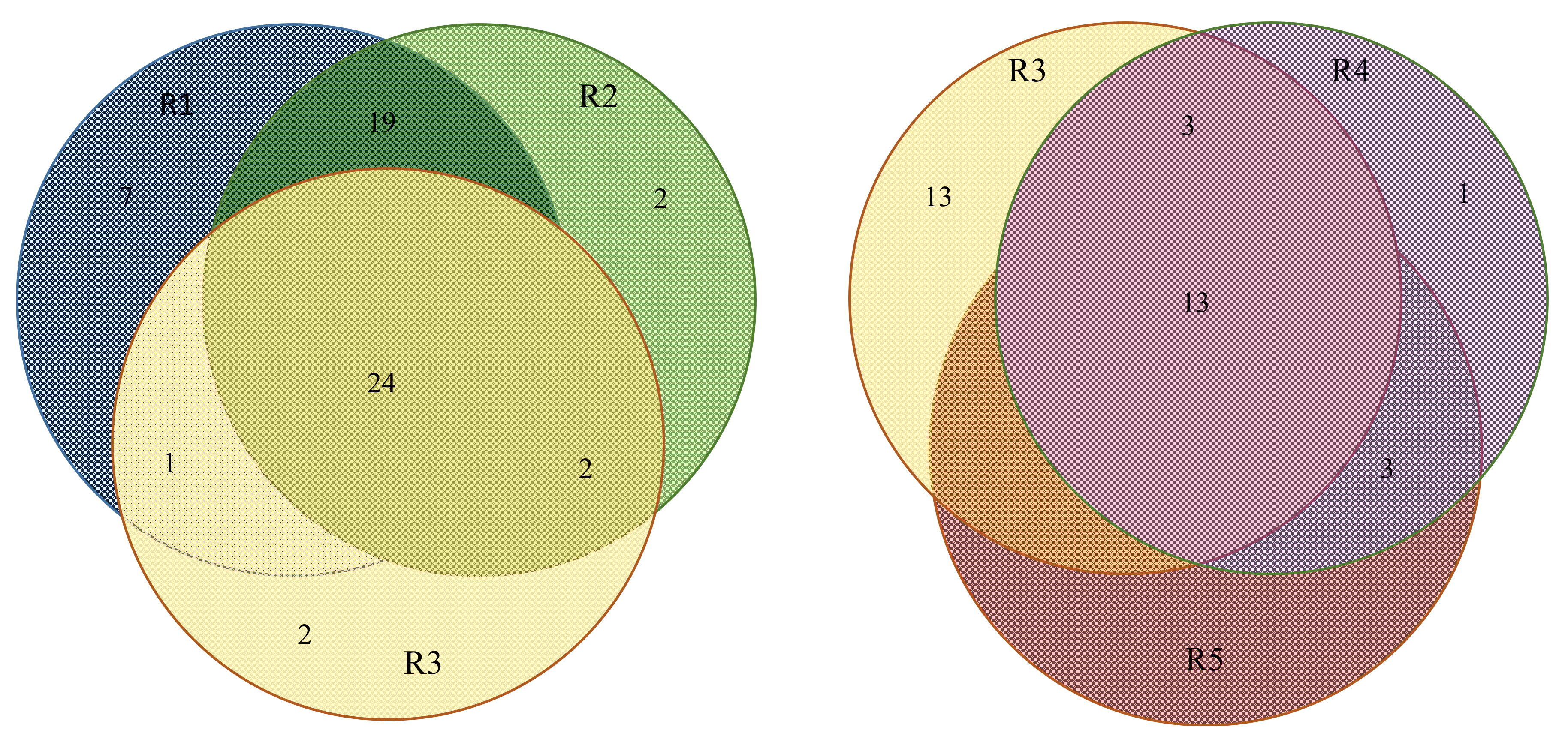
3.1. (α, β)-k-Feature Set Problem
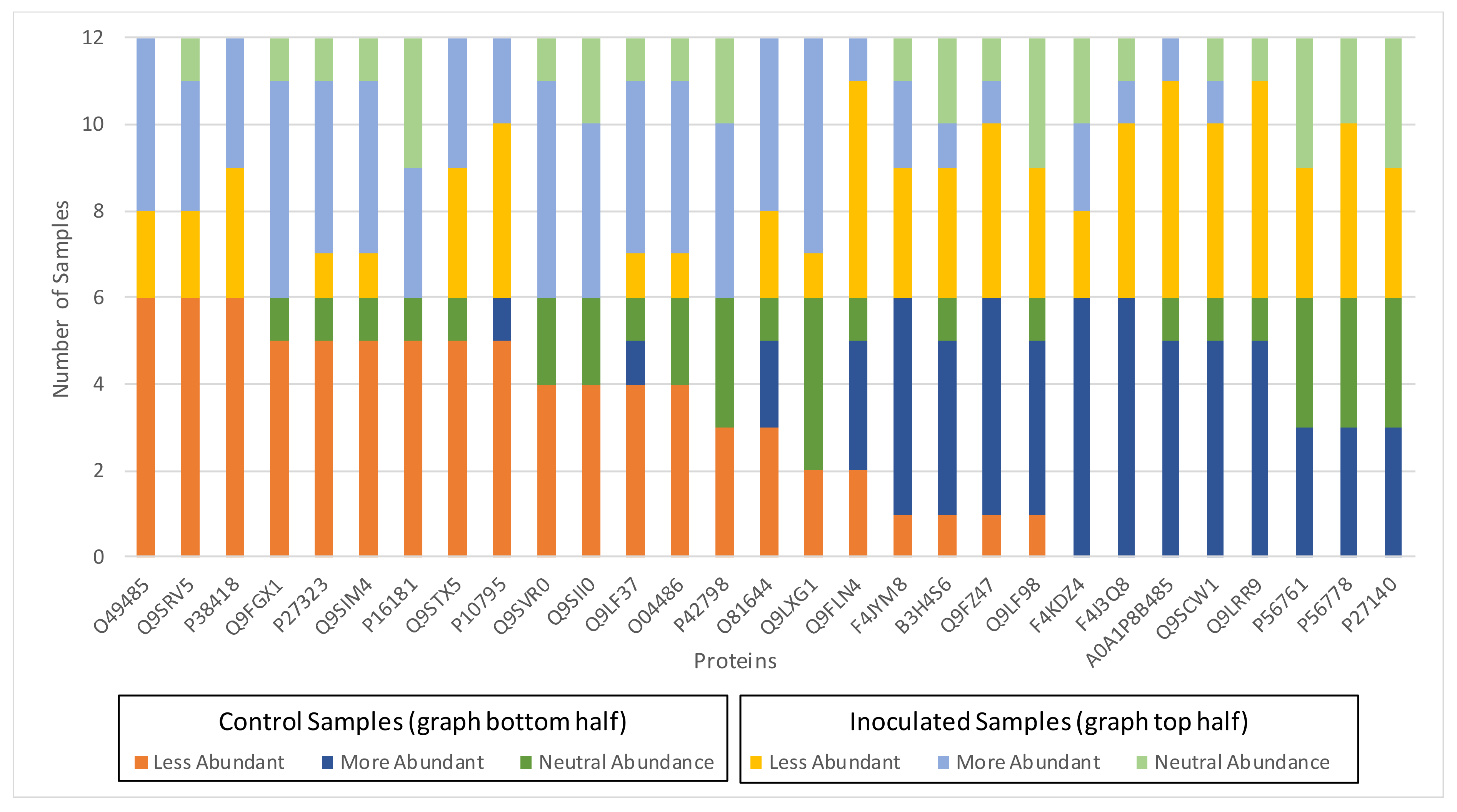
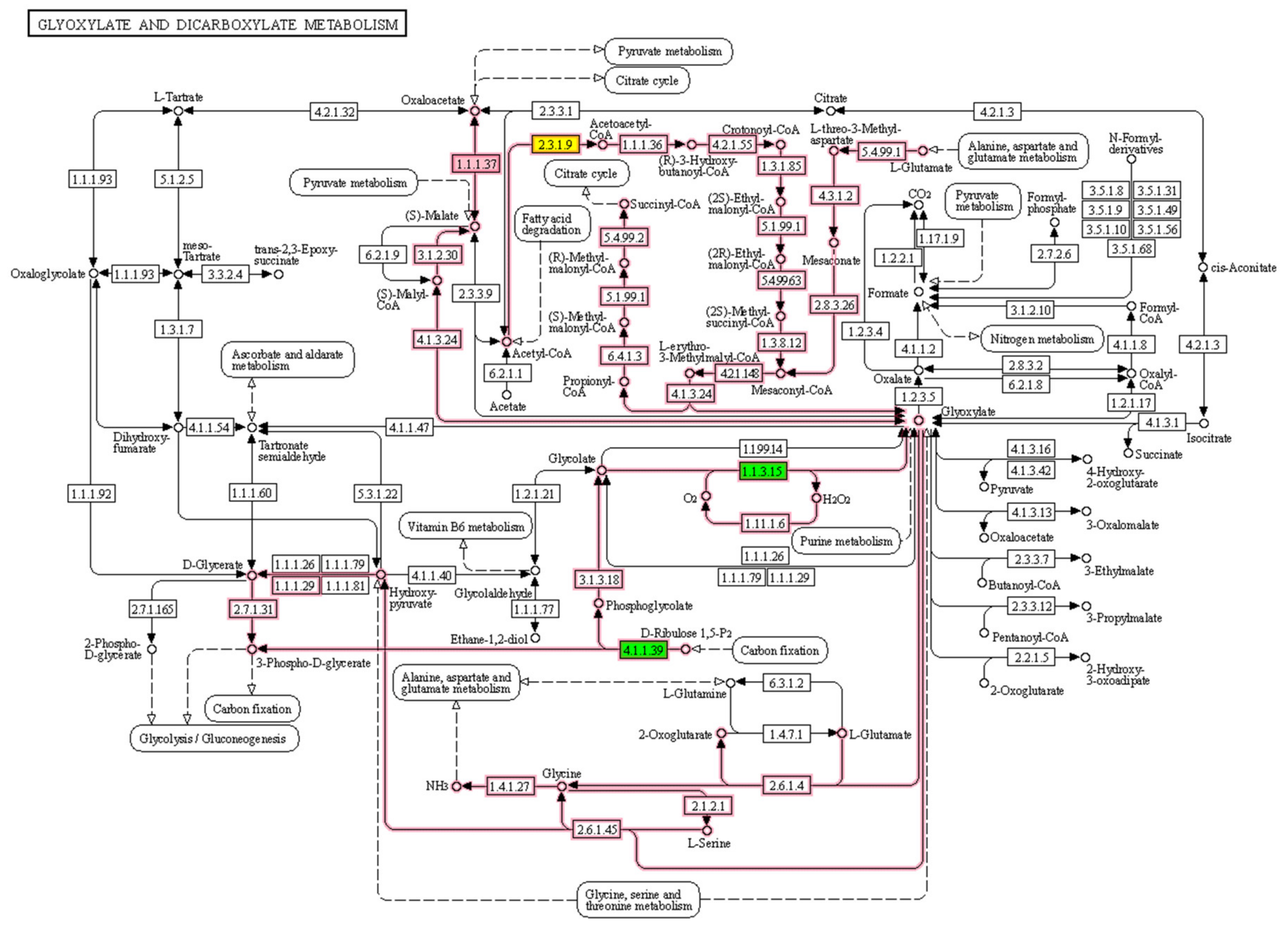
3.2. Biological Relevance of the S-R3 Protein Set
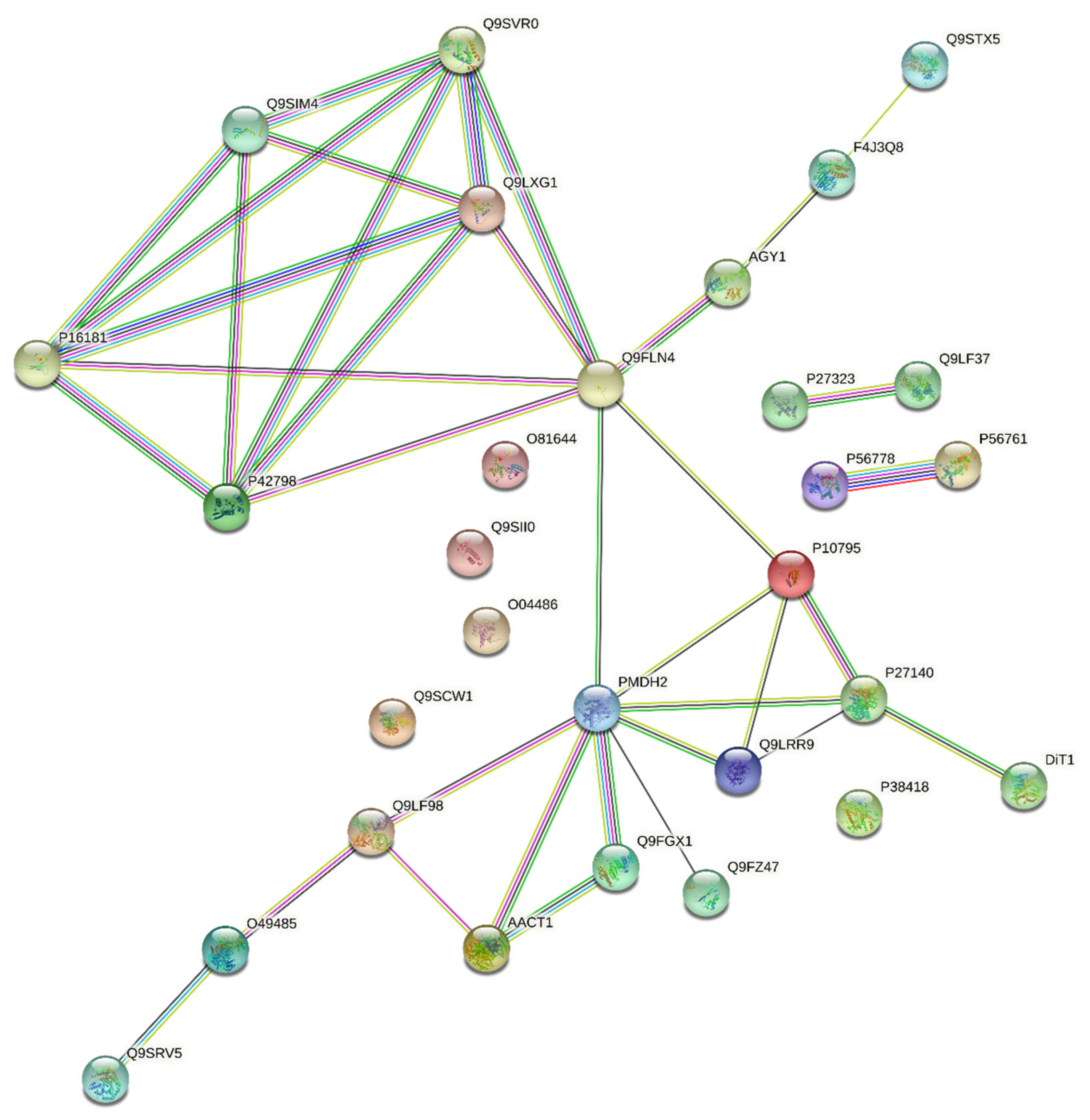
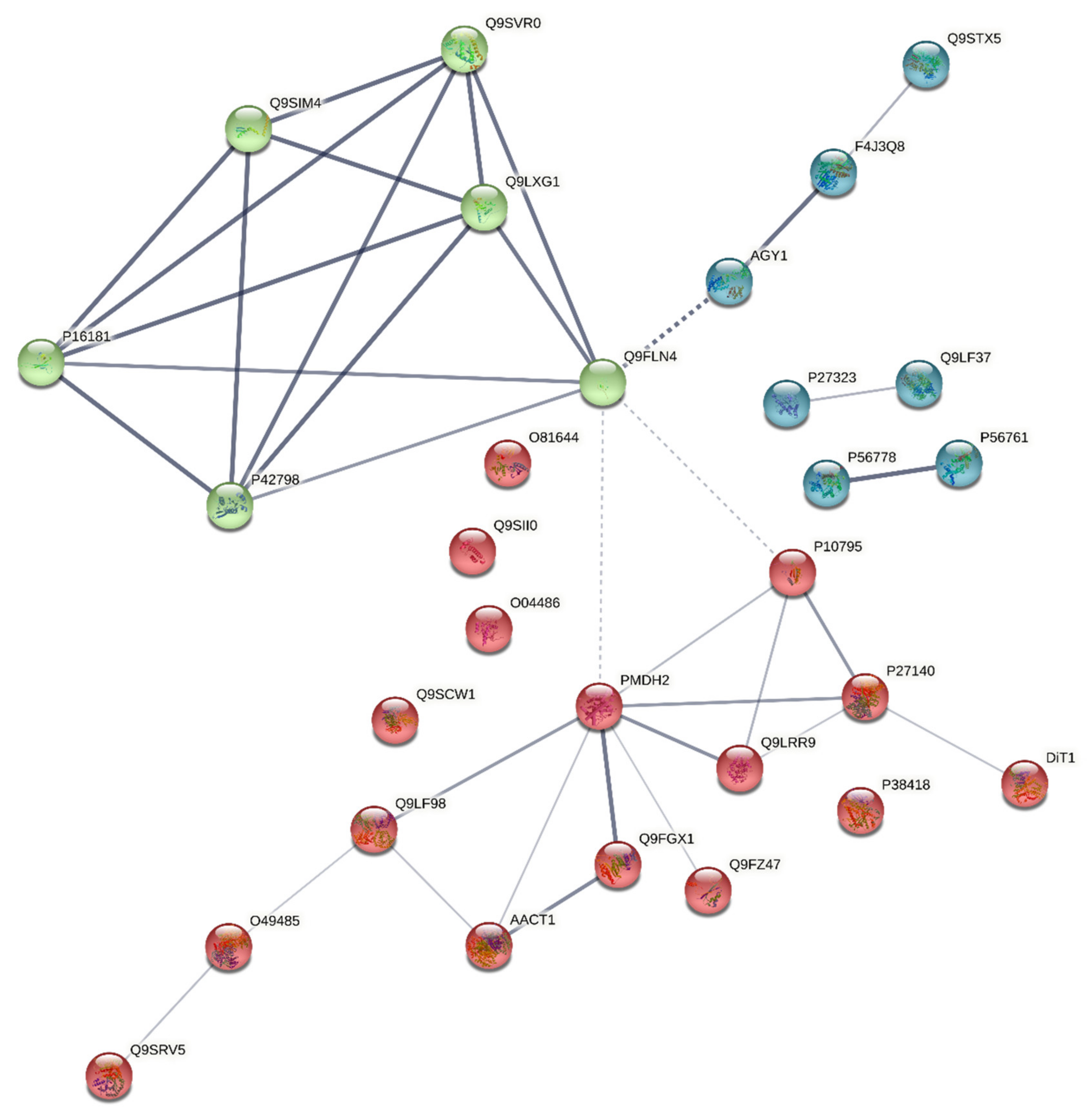
3.3. Harmonisation between the PPI Network and the Coverage Problem Approach
- –
- A more or neutral abundant level of the proteins P16181, P42798, Q9SIM4, Q9SVR0, Q9LXG1, and Q9FLN4;
- –
- A less or neutral abundant level of the proteins P56761 and P56778;
- –
- A less or neutral abundant level of the proteins PMDH2, AACT1, Q9LRR9, and P10795.
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Ávila, J.M.; Linares, J.C.; García-Nogales, A.; Sánchez, M.E.; Gomez-Aparicio, L. Across-scale patterning of plant–soil–pathogen interactions in Quercus suber decline. Eur. J. For. Res. 2017, 136, 677–688. [Google Scholar] [CrossRef]
- Camilo-Alves, C.D.E.P.; da Clara, M.I.E.; Ribeiro, N.M.C.D. Decline of Mediterranean oak trees and its association with Phytophthora cinnamomi: A review. Eur. J. For. Res. 2013, 132, 411–432. [Google Scholar] [CrossRef]
- Lopéz-Tirado, J.; Hidalgo, P.J. Predictive modelling of climax oak trees in southern Spain: Insights in a scenario of global change. Plant Ecol. 2016, 217, 451–463. [Google Scholar] [CrossRef]
- Acha, A.; Newing, H.S. Cork Oak Landscapes, Promised or Compromised Lands? A Case Study of a Traditional Cultural Landscape in Southern Spain. Hum. Ecol. 2015, 43, 601–611. [Google Scholar] [CrossRef]
- Acácio, V.; Dias, F.; Catry, F.X.; Rocha, M.; Moreira, F. Landscape dynamics in Mediterranean oak forests under global change: Understanding the role of anthropogenic and environmental drivers across forest types. Glob. Change Biol. 2017, 23, 1199–1217. [Google Scholar] [CrossRef] [PubMed]
- Duque-Lazo, L.; Navarro-Cerrillo, R.M.; Ruíz-Gómez, F.J. Assessment of the future stability of cork oak (Quercus suber L.) afforestation under climate change scenarios in Southwest Spain. For. Ecol. Manag. 2018, 409, 444–456. [Google Scholar] [CrossRef]
- Ibáñez, B.; Gómez-Aparicio, L.; Ávila, J.M.; Pérez-Ramos, I.M.; Marañón, T. Effects of Quercus suber Decline on woody Plant Regeneration: Potential Implications for Successional Dynamics in Mediterranean Forests. Ecosystems 2017, 20, 630–644. [Google Scholar] [CrossRef] [Green Version]
- Sánchez, M.E.; Caetano, P.; Ferraz, J.; Trapero, A. Phytophthora disease of Quercus ilex in southwestern Spain. For. Pathol. 2002, 32, 5–18. [Google Scholar] [CrossRef]
- Cardillo, E.; Abad, E.; Meyer, S. Iberian oak decline caused by Phytophthora cinnamomi: A spatiotemporal analysis incorporating the effect of host heterogeneities at landscape scale. For. Pathol. 2021, 00, e12667. [Google Scholar] [CrossRef]
- Serrano, M.S.; Rios, P.; Gonzalez, M.; Sánchez, M.E. Experimental minimum threshold for Phytophthora cinnamomi root disease expression on Quercus suber. Phytopathol. Mediterr. 2015, 54, 461–464. [Google Scholar] [CrossRef]
- Sghaier-Hammami, B.; Valero-Galvàn, J.; Romero-Rodríguez, M.C.; Navarro-Cerrillo, R.M.; Abdelly, C.; Jorrín-Novo, J. Physiological and proteomics analyses of Holm oak (Quercus ilex subsp. ballota [Desf.] Samp.) responses to Phytophthora cinnamomi. Plant Physiol. Biochem. 2013, 71, 191–202. [Google Scholar] [CrossRef]
- Coelho, A.C.; Pires, R.; Schütz, G.; Santa, C.; Manadas, B.; Pinto, P. Disclosing proteins in the leaves of cork oak plants associated with the immune response to Phytophthora cinnamomi inoculation in the roots: A long-term proteomics approach. PLoS ONE 2021, 16, e0245148. [Google Scholar] [CrossRef]
- Gómez-Aparicio, L.; Domínguez-Begines, J.; Kardol, P.; Ávila, J.M.; Ibáñez, B.; Garcia, L.V. Plant-soil feedbacks in declining forests: Implications for species coexistence. Ecology 2017, 98, 1908–1921. [Google Scholar] [CrossRef] [Green Version]
- Costa, C.; Madeira, M.; Plieninger, T. Cork oak woodlands patchiness: A signature of imminent deforestation? Appl. Geogr. 2014, 54, 18–26. [Google Scholar] [CrossRef]
- Vanhove, M.; Pina-Martins, F.; Coelho, A.C.; Branquinho, C.; Costa, A.; Batista, D.; Silva, A.; Sousa, P.; Marques, I.; Belkadi, B.; et al. Using Gradient Forest to predict climate response and adaptation in Cork Oak. J. Evol. Biol. 2021, 34, 910–923. [Google Scholar] [CrossRef]
- Ramírez-Valiente, J.A.; Valladares, F.; Aranda, I. Exploring the impact of neutral evolution on intrapopulation genetic differentiation in functional traits in a long-lived plant. Tree Genet. Genomes 2014, 10, 1181–1190. [Google Scholar] [CrossRef]
- Inventário Florestal Nacional (6º IFN); ICNF—Instituto de Conservação da Natureza e das Florestas: Lisboa, Portugal, 2015.
- Lancia, G.; Mathieson, L.; Moscato, P. Separating sets of strings by finding matching patterns is almost always hard. Theor. Comput. Sci. 2017, 665, 73–86. [Google Scholar] [CrossRef] [Green Version]
- Rocha de Paula, M.; Berretta, R.; Moscato, P. A fast meta-heuristic approach for the (α, β)-k-feature set problem. J. Heuristics 2016, 22, 199–220. [Google Scholar] [CrossRef]
- Berretta, R.; Mendes, A.; Moscato, P. Selection of discriminative genes in microarray experiments using mathematical programming. J. Res. Pract. Inf. Technol. 2007, 39, 287–299. [Google Scholar]
- Mendes, A.; Scott, R.J.; Moscato, P. Microarrays-identifying molecular portraits for prostate tumors with different Gleason patterns. In Clinical Bioinformatics Methods in Molecular Medicine; Trent, R.J., Ed.; Humana Press: Totowa, NJ, USA, 2008; Volume 141, pp. 131–151. [Google Scholar] [CrossRef]
- Arefin, A.; Inostroza-Ponta, M.; Mathieson, L.; Berretta, R.; Moscato, P. Clustering nodes in large-scale biological networks using external memory algorithms. In Algorithms and Architectures for Parallel Processing; Springer: Berlin/Heidelberg, Germany, 2011. [Google Scholar] [CrossRef]
- Gómez Ravetti, M.; Moscato, P. Identification of a 5-protein biomarker molecular signature for predicting Alzheimer’s disease. PLoS ONE 2008, 3, e3111. [Google Scholar] [CrossRef] [Green Version]
- Gómez Ravetti, M.; Rosso, O.A.; Berretta, R.; Moscato, P. Uncovering molecular biomarkers that correlate cognitive decline with the changes of hippocampus’ gene expression profiles in Alzheimer’s disease. PLoS ONE 2010, 5, e10153. [Google Scholar] [CrossRef] [Green Version]
- Berretta, R.; Costa, W.; Moscato, P. Combinatorial optimization models for finding genetic signatures from gene expression datasets. Methods Mol. Biol. 2008, 453, 363–377. [Google Scholar] [CrossRef]
- Rosso, O.A.; Mendes, A.; Berretta, R.; Rostas, J.A.; Hunter, M.; Moscato, P. Distinguishing childhood absence epilepsy patients from controls by the analysis of their background brain electrical activity (II): A combinatorial optimization approach for electrode selection. J. Neurosci. Methods 2009, 181, 257–267. [Google Scholar] [CrossRef]
- Cotta, C.; Sloper, C.; Moscato, P. Evolutionary search of thresholds for robust feature set selection: Application to the analysis of microarray data. In Applications of Evolutionary Computing; EvoWorkshops, Raidl, G.R., Cagnoni, S., Branke, J., Wolfe Corne, D., Drechsler, R., Jin, Y., Johnson, C.G., Machado., O., Marchiori, E., et al., Eds.; Springer: Berlin/Heidelberg, Germany, 2004; Volume 3005, pp. 21–30. [Google Scholar]
- Davies, S.; Russell, S. NP-completeness of searches for smallest possible feature sets. In Proceedings of the AAAI Symposium on Relevance; Greiner, R., Subramanian, D., Eds.; AAAI Press: New Orleans, CA, USA, 1994; pp. 41–43. [Google Scholar]
- Richard, M.K. Reducibility among combinatorial problems. In Complexity of Computer Computations; Miller, R.E., Thatcher, J.W., Eds.; Plenum: New York, NY, USA, 1972; pp. 85–103. [Google Scholar]
- Vizcaíno, J.A.; Csordas, A.; Del-Toro, N.; Dianes, J.A.; Griss, J.; Lavidas, I.; Mayer, G.; Perez-Riverol, Y.; Reisinger, F.; Ternent, T.; et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016, 44, D447–D456. [Google Scholar] [CrossRef]
- STRING: Functional Protein Associations Networks. Available online: http://string-db.org (accessed on 28 February 2022).
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- KEGG: Kyoto Encyclopedia of Genes and Genomes. Available online: http://www.kegg.jp (accessed on 3 May 2022).
- Romani, I.; Tadini, L.; Rossi, F.; Masiero, S.; Pribil, M.; Jahns, P.; Kater, M.; Leister, D.; Pesaresi, P. Versatile roles of Arabidopsis plastid ribosomal proteins in plant growth and development. Plant J. 2012, 72, 922–934. [Google Scholar] [CrossRef]
- Liu, D.; Gong, Q.; Ma, Y.; Li, P.; Li, J.; Yang, S.; Yuan, L.; Yu, Y.; Pan, D.; Xu, F.; et al. cpSecA, a thylakoid protein translocase subunit, is essential for photosynthetic development in Arabidopsis. J. Exp. Bot. 2010, 61, 1655–1669. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.; Henry, R.; McCaffery, M.; Cline, K. SecA homolog in protein transport within chloroplasts: Evidence for endosymbiont-derived sorting. Science 1994, 266, 796–798. [Google Scholar] [CrossRef]
- Pracharoenwattana, I.; Cornah, J.E.; Smith, S.M. Arabidopsis peroxisomal malate dehydrogenase functions in b-oxidation but not in the glyoxylate cycle. Plant J. 2007, 50, 381–390. [Google Scholar] [CrossRef]
- Hu, J.; Baker, A.; Bartel, B.; Linka, N.; Mullen, R.T.; Reumann, S.; Zolman, B.K. Plant Peroxisomes: Biogenesis and Function. Plant Cell 2012, 24, 2279–2303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortega-Galisteo, A.P.; Rodríguez-Serrano, M.; Pazmiño, D.M.; Gupta, D.K.; Sandalio, L.M.; Romero-Puertas, M.C. S-Nitrosylated proteins in pea (Pisum sativum L.) leaf peroxisomes: Changes under abiotic stress. J. Exp. Bot. 2012, 63, 2089–2103. [Google Scholar] [CrossRef] [PubMed]
- Torres-Zabala, M.; Littlejohn, G.; Jayaraman, S.; Studholme, D.; Bailey, T.; Lawson, T.; Tillich, M.; Licht, D.; Bölter, B.; Delfino, L.; et al. Chloroplasts play a central role in plant defence and are targeted by pathogen effectors. Nat. Plants 2015, 1, 15074. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| D | Proteins (P) | ||||||
|---|---|---|---|---|---|---|---|
| T | |||||||
| Samples (C) | 0 | 0 | 1 | 1 | 0 | Control | |
| 0 | 1 | 1 | 0 | 1 | Control | ||
| 1 | 0 | 1 | 0 | 1 | Inoculated | ||
| 1 | 1 | 0 | 1 | 1 | Inoculated | ||
| Proteins (P) | ||||||
|---|---|---|---|---|---|---|
| Sample pairs | A | |||||
| 1 | 0 | 0 | 1 | 1 | ||
| 1 | 1 | 1 | 0 | 1 | ||
| 1 | 1 | 0 | 0 | 0 | ||
| 1 | 0 | 1 | 1 | 0 | ||
| B | ||||||
| 1 | 0 | 1 | 0 | 0 | ||
| 1 | 0 | 0 | 0 | 1 | ||
| R1 | R2 | R3 | R4 | R5 | |
|---|---|---|---|---|---|
| μ ± 0.05 μ | μ ± 0.075 μ | μ ± 0.1 μ | μ ± 0.125 μ | μ ± 0.15 μ | |
| Bio/statistical significative proteins | 80 | 80 | 80 | 80 | 80 |
| Alpha (α) | 16 | 11 | 8 | 6 | 5 |
| Beta (β) | 18 | 15 | 9 | 5 | 4 |
| Optimum number of proteins (k) | 51 | 47 | 29 | 20 | 16 |
| S | Protein Information a | |
|---|---|---|
| R3 | ||
| Arabidopsis UniProt Accession | Protein Name | Initial |
| P16181 | 40S ribosomal protein S11-1 | RS111 |
| P42798 | 40S ribosomal protein S15a-1 | R15A1 |
| Q9STX5 | Endoplasmin homolog | ENPL |
| P10795 | Ribulose bisphosphate carboxylase small chain 1A | RBS1A/RBCS1A |
| Q9SIM4 | 60S ribosomal protein L14-1 | RL141 |
| Q9LF37 | Chaperone protein | ClpB3 |
| F4J3Q8 | P-loop containing nucleoside triphosphate hydrolases superfamily | F4J3Q8 |
| Q9SII0 | Probable histone H2A variant 2 | H2AV2 |
| Q9LRR9 | (S)-2-hydroxy-acid oxidase GLO1 | GLO1/GOX1 |
| Q9LXG1 | 40S ribosomal protein S9-1 | RS91 |
| Q9SVR0 | 60S ribosomal protein L13a-3 | R13A3 |
| P56761 | Photosystem II D2 | PSBD |
| F4JYM8 | Thiolase family protein | F4JYM8/AACT1 |
| O04486 | Ras-related protein | RABA2a |
| Q9FZ47 | ACT domain-containing protein ACR11 | ACR11 |
| P38418 | Lipoxygenase 2 | LOX 2 |
| Q9FLN4 | 50S ribosomal protein L27 | RK27 |
| Q9FGX1 | ATP-citrate synthase beta chain protein 2 | ACLB2 |
| Q9SRV5 | 5-methyltetrahydropteroyltriglutamate-homocysteine methyltransferase 2 | METE2 |
| P27140 | Beta carbonic anhydrase 1 | BCA1 |
| Q9SCW1 | Beta-galactosidase 1 | BGAL1 |
| B3H4S6 | Dicarboxylate transporter 1 | DiT1 |
| O49485 | D-3-phosphoglycerate dehydrogenase 1 | SERA1 |
| Q9LF98 | Fructose-bisphosphate aldolase 8 | ALFC8/FBA8 |
| P27323 | Heat shock protein 90-1 | HSP90-1 |
| F4KDZ4 | Malate dehydrogenase | FAKDZ4/PMDH2 |
| P56778 | Photosystem II CP43 reaction center protein | PSBC |
| A0A1P8B485 | Protein translocase subunit Sec A | AGY1 |
| O81644 | Villin-2 | VILI2 |
| Biological Process | Arabidopsis UniProt Accession | Number (N) and Relative Frequency (f) of Abundance Quantifications | ||||||
|---|---|---|---|---|---|---|---|---|
| Control Samples | Inoculated Samples | |||||||
| Less | More | Neutral | Less | More | Neutral | |||
| Protein synthesis | P16181; P42798 Q9SIM4; Q9SVR0 Q9LXG1; Q9FLN4 | N f | 21 0.58 | 3 0.08 | 12 0.33 | 7 0.19 | 22 0.61 | 7 0.19 |
| Photosynthesis | P56778; P56761 | N f | 0 0.0 | 6 0.5 | 6 0.5 | 7 0.58 | 0 0.0 | 5 0.42 |
| Glyoxylate and dicarboxylate metabolism | PMDH2; AACT1; Q9LRR9; P10795 | N f | 6 0.25 | 17 0.71 | 1 0.04 | 14 0.58 | 6 0.25 | 4 0.17 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coelho, A.C.; Schütz, G. Protein Markers for the Identification of Cork Oak Plants Infected with Phytophthora cinnamomi by Applying an (α, β)-k-Feature Set Approach. Forests 2022, 13, 940. https://0-doi-org.brum.beds.ac.uk/10.3390/f13060940
Coelho AC, Schütz G. Protein Markers for the Identification of Cork Oak Plants Infected with Phytophthora cinnamomi by Applying an (α, β)-k-Feature Set Approach. Forests. 2022; 13(6):940. https://0-doi-org.brum.beds.ac.uk/10.3390/f13060940
Chicago/Turabian StyleCoelho, Ana Cristina, and Gabriela Schütz. 2022. "Protein Markers for the Identification of Cork Oak Plants Infected with Phytophthora cinnamomi by Applying an (α, β)-k-Feature Set Approach" Forests 13, no. 6: 940. https://0-doi-org.brum.beds.ac.uk/10.3390/f13060940