Rotavirus Antagonism of the Innate Immune Response

Abstract
:1. Introduction
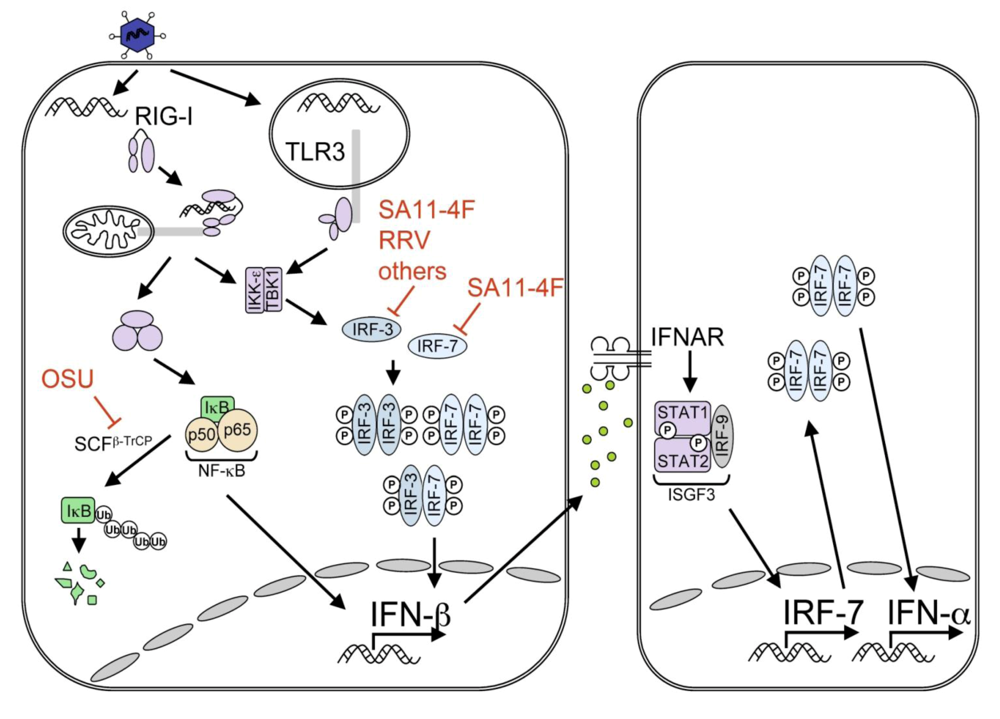
IFN Response to Viral Infection

2. Rotavirus Biology
2.1. Replication
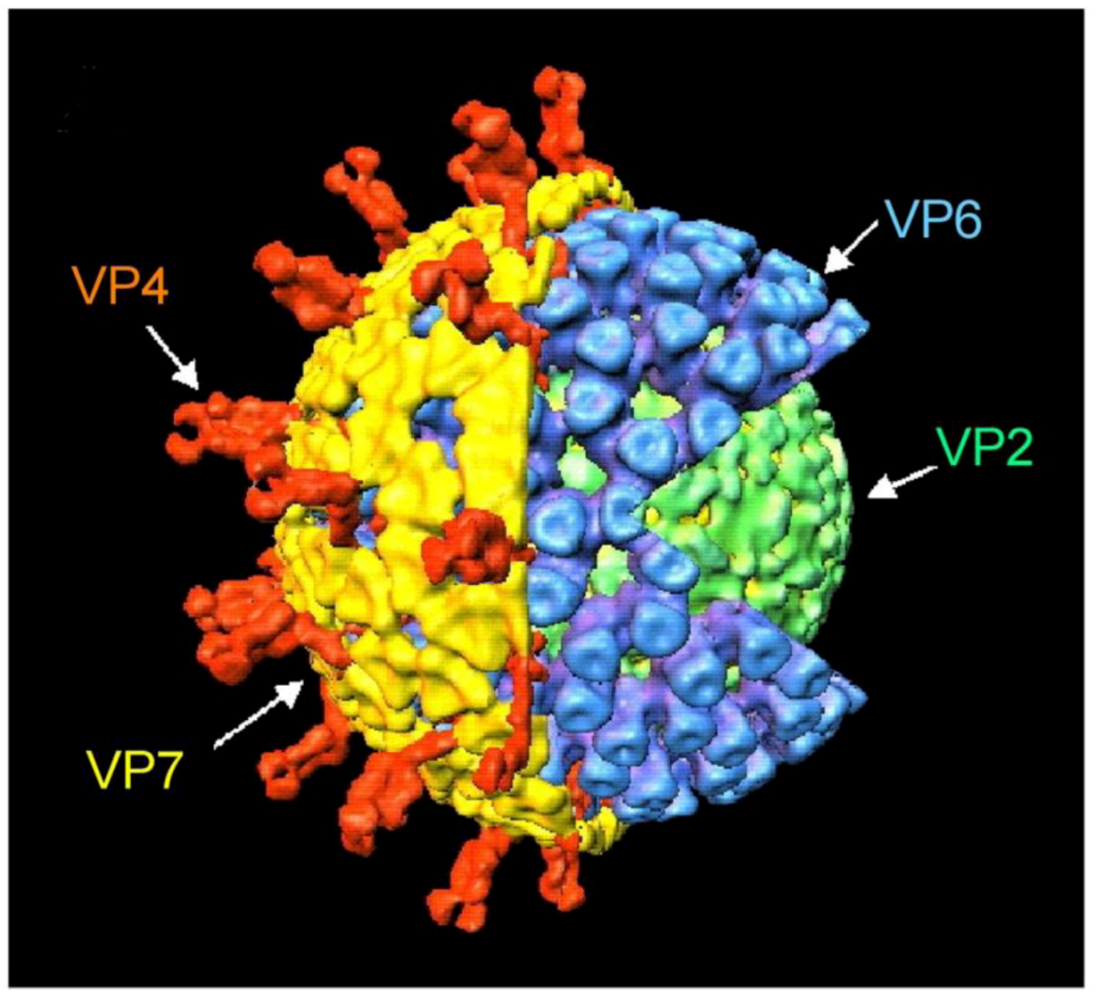
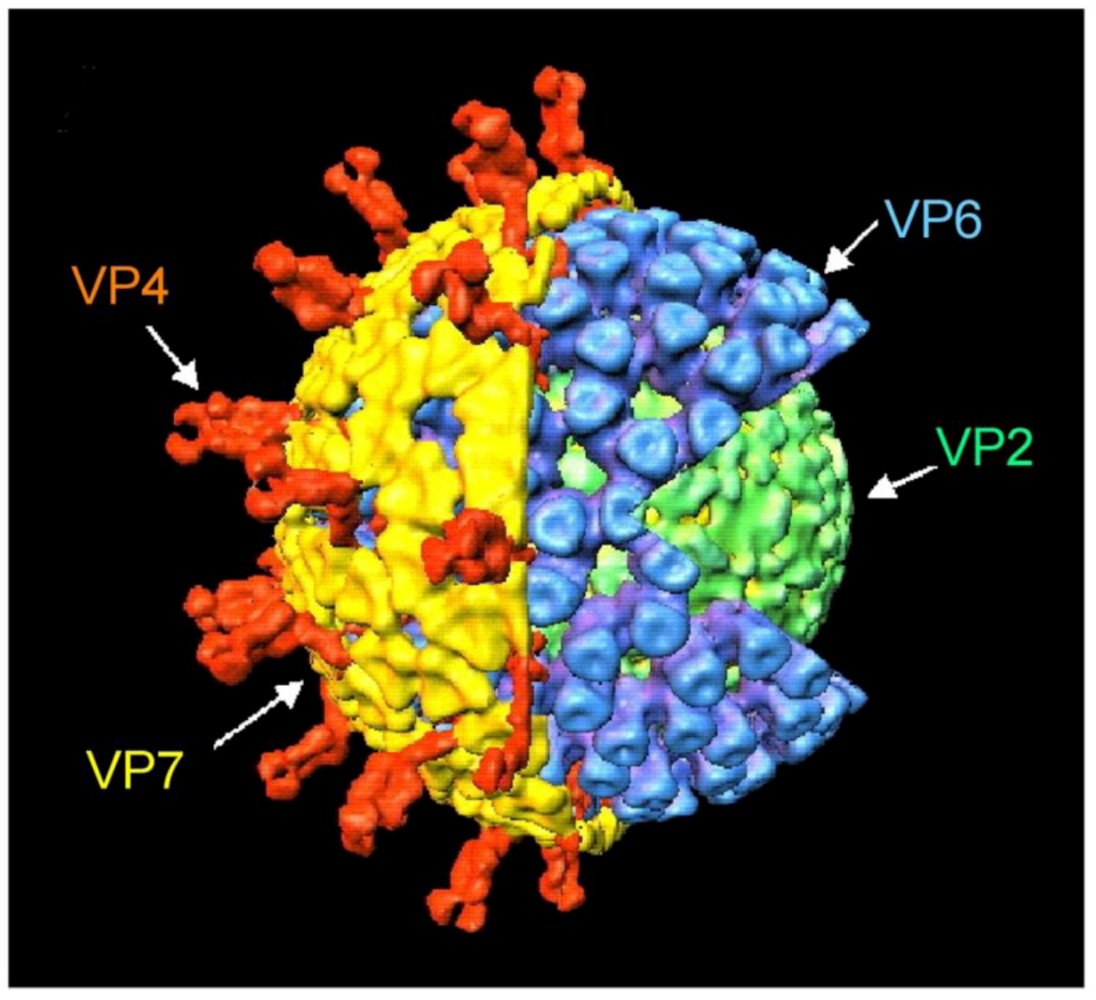
2.2. Classification
2.3. Pathogenesis
2.4. RV Host Range Restriction
2.5. Role of IFN in RV infection
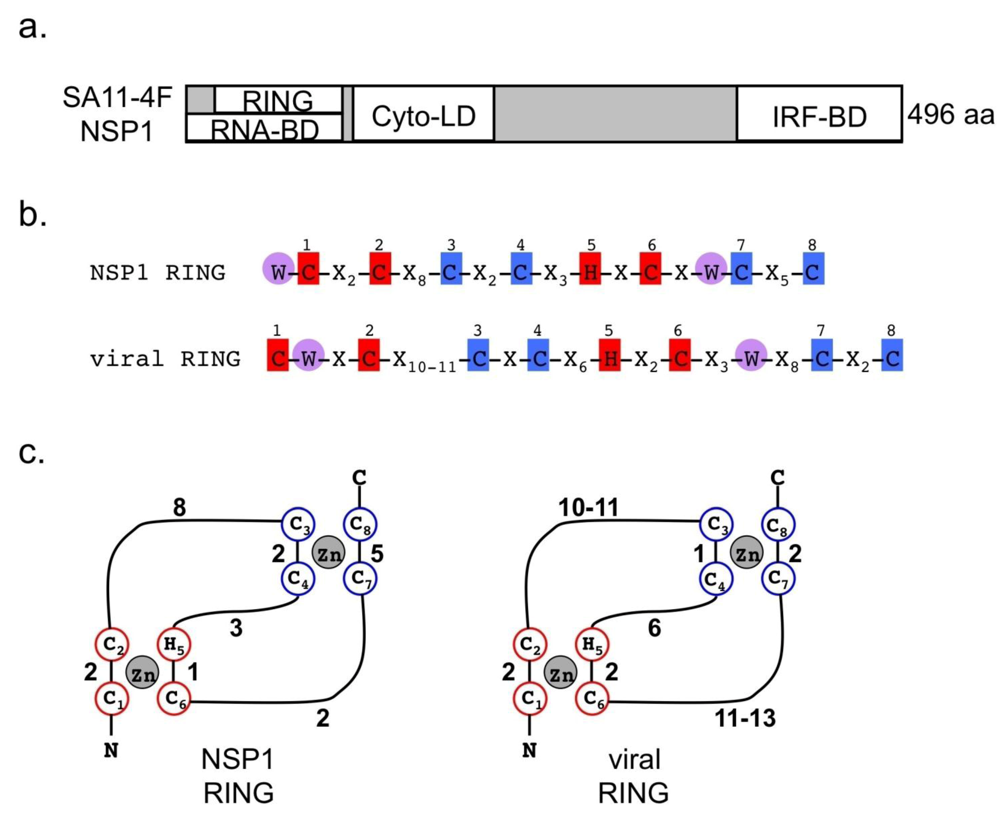
2.6. Properties of NSP1

3. Interaction of NSP1 with Transcription Factors of the IFN Induction Pathway
3.1. NSP1-Induced Degradation of IRF Proteins

{kind=link}
{kind=link}
{kind=link}
| RV Strain | Host Species | IRF3 Degradation (Cell Type Tested) | Reference |
|---|---|---|---|
| SA11-4F | Simian | + (Caco-2, FRhL2, MA104) | [86,89,90] |
| 30-19 | Simian | + (FRhL2) | [89] |
| RRV | Simian | + (Cos7, MEF) | [91,92] |
| UK | Bovine | + (Cos7) - (MEF) | [91,92] |
| B641 | Bovine | + (MA104) | [90] |
| NCDV | Bovine | + (MA104, HEK293) - (MEF) | [90,92] |
| OSU | Porcine | - (MA104, HEK293) | [90,92] |
| ETD | Murine | + (Cos7, MEF) | [91,92] |
| EW | Murine | + (MEF) | [91] |
3.2. NSP1 Inhibition of the NF-κB Pathway
3.3. Host Cell Dependence of NSP1 Activity
3.4. NSP1 as an E3 Ubiquitin Ligase
3.5. NSP1 Similarities to Other Viral Proteins
4. Conclusions and Perspectives
Acknowledgments
References
- Bonjardim, C.A.; Ferreira, P.C.; Kroon, E.G. Interferons: signaling, antiviral and viral evasion. Immunol. Lett. 2009, 122, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Der, S.D.; Zhou, A.; Williams, B.R.; Silverman, R.H. Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc. Natl. Acad. Sci. U S A 1998, 95, 15623–15628. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed]
- Paun, A.; Pitha, P.M. The IRF family, revisited. Biochimie 2007, 89, 744–753. [Google Scholar] [CrossRef] [PubMed]
- Takaoka, A.; Wang, Z.; Choi, M.K.; Yanai, H.; Negishi, H.; Ban, T.; Lu, Y.; Miyagishi, M.; Kodama, T.; Honda, K.; Ohba, Y.; Taniguchi, T. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 2007, 448, 501–505. [Google Scholar] [CrossRef] [PubMed]
- Maniatis, T.; Falvo, J.V.; Kim, T.H.; Kim, T.K.; Lin, C.H.; Parekh, B.S.; Wathelet, M.G. Structure and function of the interferon-beta enhanceosome. Cold Spring Harb. Symp. Quant. Biol. 1998, 63, 609–620. [Google Scholar] [PubMed]
- Honda, K.; Taniguchi, T. IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat. Rev. Immunol. 2006, 6, 644–658. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Pagano, J.S. IRF-7, a new interferon regulatory factor associated with Epstein-Barr virus latency. Mol. Cell. Biol. 1997, 17, 5748–5757. [Google Scholar] [PubMed]
- Fitzgerald, K.A.; McWhirter, S.M.; Faia, K.L.; Rowe, D.C.; Latz, E.; Golenbock, D.T.; Coyle, A. J.; Liao, S.M.; Maniatis, T. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 2003, 4, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; tenOever, B.R.; Grandvaux, N.; Zhou, G.P.; Lin, R.; Hiscott, J. Triggering the interferon antiviral response through an IKK-related pathway. Science 2003, 300, 1148–1151. [Google Scholar] [CrossRef] [PubMed]
- Barnes, B.; Lubyova, B.; Pitha, P.M. On the role of IRF in host defense. J. Interferon Cytokine Res. 2002, 22, 59–71. [Google Scholar] [PubMed]
- Lin, R.; Mamane, Y.; Hiscott, J. Multiple regulatory domains control IRF-7 activity in response to virus infection. J. Biol. Chem. 2000, 275, 34320–34327. [Google Scholar] [CrossRef] [PubMed]
- Marie, I.; Durbin, J.E.; Levy, D.E. Differential viral induction of distinct interferon-alpha genes by positive feedback through interferon regulatory factor-7. EMBO J. 1998, 17, 6660–6669. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Hata, N.; Asagiri, M.; Nakaya, T.; Taniguchi, T.; Tanaka, N. Positive feedback regulation of type I IFN genes by the IFN-inducible transcription factor IRF-7. FEBS Lett. 1998, 441, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Suemori, H.; Hata, N.; Asagiri, M.; Ogasawara, K.; Nakao, K.; Nakaya, T.; Katsuki, M.; Noguchi, S.; Tanaka, N.; Taniguchi, T. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity 2000, 13, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M.; Trinchieri, G.; Liu, Y.J. Plasmacytoid dendritic cells in immunity. Nat. Immunol. 2004, 5, 1219–1226. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Yanai, H.; Negishi, H.; Asagiri, M.; Sato, M.; Mizutani, T.; Shimada, N.; Ohba, Y.; Takaoka, A.; Yoshida, N.; Taniguchi, T. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 2005, 434, 772–777. [Google Scholar] [CrossRef] [PubMed]
- Yeow, W.S.; Au, W.C.; Juang, Y.T.; Fields, C.D.; Dent, C.L.; Gewert, D.R.; Pitha, P.M. Reconstitution of virus-mediated expression of interferon alpha genes in human fibroblast cells by ectopic interferon regulatory factor-7. J. Biol. Chem. 2000, 275, 6313–6320. [Google Scholar] [CrossRef] [PubMed]
- Kroll, M.; Margottin, F.; Kohl, A.; Renard, P.; Durand, H.; Concordet, J.P.; Bachelerie, F.; Arenzana-Seisdedos, F.; Benarous, R. Inducible degradation of IkappaBalpha by the proteasome requires interaction with the F-box protein h-betaTrCP. J. Biol. Chem. 1999, 274, 7941–7945. [Google Scholar] [CrossRef] [PubMed]
- Parashar, U.D.; Gibson, C.J.; Bresse, J.S.; Glass, R.I. Rotavirus and severe childhood diarrhea. Emerg. Infect. Dis. 2006, 12, 304–306. [Google Scholar] [PubMed]
- Greenberg, H.B.; Estes, M.K. Rotaviruses: from pathogenesis to vaccination. Gastroenterology 2009, 136, 1939–1951. [Google Scholar] [CrossRef] [PubMed]
- Prasad, B.V.; Wang, G.J.; Clerx, J.P.; Chiu, W. Three-dimensional structure of rotavirus. J. Mol. Biol. 1988, 199, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Yeager, M.; Dryden, K.A.; Olson, N.H.; Greenberg, H.B.; Baker, T.S. Three-dimensional structure of rhesus rotavirus by cryoelectron microscopy and image reconstruction. J. Cell Biol. 1990, 110, 2133–2144. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Luongo, C.L.; Nibert, M.L.; Patton, J.T. Rotavirus open cores catalyze 5'-capping and methylation of exogenous RNA: evidence that VP3 is a methyltransferase. Virology 1999, 265, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Lawton, J.A.; Estes, M.K.; Prasad, B.V. Three-dimensional visualization of mRNA release from actively transcribing rotavirus particles. Nat. Struct. Biol. 1997, 4, 118–121. [Google Scholar] [CrossRef]
- Patton, J.T.; Jones, M.T.; Kalbach, A.N.; He, Y.W.; Xiaobo, J. Rotavirus RNA polymerase requires the core shell protein to synthesize the double-stranded RNA genome. J. Virol. 1997, 71, 9618–9626. [Google Scholar] [PubMed]
- Bican, P.; Cohen, J.; Charpilienne, A.; Scherrer, R. Purification and characterization of bovine rotavirus cores. J. Virol. 1982, 43, 1113–1117. [Google Scholar] [PubMed]
- Spencer, E.; Arias, M.L. In vitro transcription catalyzed by heat-treated human rotavirus. J. Virol. 1981, 40, 1–10. [Google Scholar] [PubMed]
- Santos, N.; Hoshino, Y. Global distribution of rotavirus serotypes/genotypes and its implication for the development and implementation of an effective rotavirus vaccine. Rev. Med. Virol. 2005, 15, 29–56. [Google Scholar] [CrossRef] [PubMed]
- Prasad, B.V.; Rothnagel, R.; Zeng, C.Q.; Jakana, J.; Lawton, J.A.; Chiu, W.; Estes, M.K. Visualization of ordered genomic RNA and localization of transcriptional complexes in rotavirus. Nature 1996, 382, 471–473. [Google Scholar] [CrossRef] [PubMed]
- Imai, M.; Akatani, K.; Ikegami, N.; Furuichi, Y. Capped and conserved terminal structures in human rotavirus genome double-stranded RNA segments. J. Virol. 1983, 47, 125–136. [Google Scholar] [PubMed]
- McCrae, M.A.; McCorquodale, J.G. Molecular biology of rotaviruses V. Terminal structure of viral RNA species. Virology 1983, 126, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Zeng, C.Q.; Wentz, M.J.; Gorziglia, M.; Estes, M.K.; Ramig, R.F. Template-dependent, in vitro replication of rotavirus RNA. J. Virol. 1994, 68, 7030–7039. [Google Scholar] [PubMed]
- Patton, J.T.; Spencer, E. Genome replication and packaging of segmented double-stranded RNA viruses. Virology 2000, 277, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, L.S.; Taraporewala, Z.F.; Patton, J.T. Rotavirus replication: plus-sense templates for double-stranded RNA synthesis are made in viroplasms. J. Virol. 2004, 78, 7763–7774. [Google Scholar] [CrossRef] [PubMed]
- Estes, M.K.; Kang, G.; Zeng, C.Q.; Crawford, S.E.; Ciarlet, M. Pathogenesis of rotavirus gastroenteritis. Novartis Found. Symp. 2001, 238, 82–100. [Google Scholar] [PubMed]
- Delmas, O.; Durand-Schneider, A.M.; Cohen, J.; Colard, O.; Trugnan, G. Spike protein VP4 assembly with maturing rotavirus requires a postendoplasmic reticulum event in polarized caco-2 cells. J. Virol. 2004, 78, 10987–10994. [Google Scholar] [CrossRef] [PubMed]
- Jourdan, N.; Maurice, M.; Delautier, D.; Quero, A.M.; Servin, A.L.; Trugnan, G. Rotavirus is released from the apical surface of cultured human intestinal cells through nonconventional vesicular transport that bypasses the Golgi apparatus. J. Virol. 1997, 71, 8268–8278. [Google Scholar] [PubMed]
- Taniguchi, K.; Urasawa, S. Diversity in rotavirus genomes. Seminars in Virology 1995, 6, 123–131. [Google Scholar] [CrossRef]
- Nakagomi, O.; Nakagomi, T. Genomic relationships among rotaviruses recovered from various animal species as revealed by RNA-RNA hybridization assays. Res. Vet. Sci. 2002, 73, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Matthijnssens, J.; Ciarlet, M.; Heiman, E.; Arijs, I.; Delbeke, T.; McDonald, S.M.; Palombo, E.A.; Iturriza-Gomara, M.; Maes, P.; Patton, J.T.; Rahman, M.; Van Ranst, M. Full genome-based classification of rotaviruses reveals a common origin between human Wa-Like and porcine rotavirus strains and human DS-1-like and bovine rotavirus strains. J. Virol. 2008, 82, 3204–3219. [Google Scholar] [CrossRef] [PubMed]
- Nakagomi, O.; Nakagomi, T. Genetic diversity and similarity among mammalian rotaviruses in relation to interspecies transmission of rotavirus. Arch. Virol. 1991, 120, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, Y.; Kapikian, A.Z. Classification of rotavirus VP4 and VP7 serotypes. Arch. Virol. Suppl. 1996, 12, 99–111. [Google Scholar] [PubMed]
- Matthijnssens, J.; Potgieter, C.A.; Ciarlet, M.; Parreno, V.; Martella, V.; Banyai, K.; Garaicoechea, L.; Palombo, E.A.; Novo, L.; Zeller, M.; Arista, S.; Gerna, G.; Rahman, M.; Van Ranst, M. Are human P[14] rotavirus strains the result of interspecies transmissions from sheep or other ungulates that belong to the mammalian order Artiodactyla? J. Virol. 2009, 83, 2917–2929. [Google Scholar] [CrossRef] [PubMed]
- Schumann, T.; Hotzel, H.; Otto, P.; Johne, R. Evidence of interspecies transmission and reassortment among avian group A rotaviruses. Virology 2009, 386, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Solberg, O.D.; Hasing, M.E.; Trueba, G.; Eisenberg, J.N. Characterization of novel VP7, VP4, and VP6 genotypes of a previously untypeable group A rotavirus. Virology 2009, 385, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Trojnar, E.; Otto, P.; Johne, R. The first complete genome sequence of a chicken group A rotavirus indicates independent evolution of mammalian and avian strains. Virology 2009, 386, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Bass, E.S.; Pappano, D.A.; Humiston, S.G. Rotavirus. Pediatr. Rev. 2007, 28, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Ramig, R.F. Systemic rotavirus infection. Expert Rev. Anti. Infect. Ther. 2007, 5, 591–612. [Google Scholar] [CrossRef] [PubMed]
- Crawford, S.E.; Patel, D.G.; Cheng, E.; Berkova, Z.; Hyser, J.M.; Ciarlet, M.; Finegold, M.J.; Conner, M.E.; Estes, M.K. Rotavirus viremia and extraintestinal viral infection in the neonatal rat model. J. Virol. 2006, 80, 4820–4832. [Google Scholar] [CrossRef] [PubMed]
- Blutt, S.E.; Conner, M.E. Rotavirus: to the gut and beyond! Curr Opin. Gastroenterol. 2007, 23, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Fenaux, M.; Cuadras, M.A.; Feng, N.; Jaimes, M.; Greenberg, H.B. Extraintestinal spread and replication of a homologous EC rotavirus strain and a heterologous rhesus rotavirus in BALB/c mice. J. Virol. 2006, 80, 5219–5232. [Google Scholar] [CrossRef] [PubMed]
- Mossel, E.C.; Ramig, R.F. Rotavirus genome segment 7 (NSP3) is a determinant of extraintestinal spread in the neonatal mouse. J. Virol. 2002, 76, 6502–6509. [Google Scholar] [CrossRef] [PubMed]
- Mossel, E.C.; Ramig, R.F. A lymphatic mechanism of rotavirus extraintestinal spread in the neonatal mouse. J. Virol. 2003, 77, 12352–12356. [Google Scholar] [CrossRef] [PubMed]
- Broome, R.L.; Vo, P.T.; Ward, R.L.; Clark, H.F.; Greenberg, H.B. Murine rotavirus genes encoding outer capsid proteins VP4 and VP7 are not major determinants of host range restriction and virulence. J. Virol. 1993, 67, 2448–2455. [Google Scholar] [PubMed]
- Ciarlet, M.; Estes, M.K.; Barone, C.; Ramig, R.F.; Conner, M.E. Analysis of host range restriction determinants in the rabbit model: comparison of homologous and heterologous rotavirus infections. J. Virol. 1998, 72, 2341–2351. [Google Scholar] [PubMed]
- Hoshino, Y.; Saif, L.J.; Kang, S.Y.; Sereno, M.M.; Chen, W.K.; Kapikian, A.Z. Identification of group A rotavirus genes associated with virulence of a porcine rotavirus and host range restriction of a human rotavirus in the gnotobiotic piglet model. Virology 1995, 209, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Azim, T.; Ahmad, S.M.; Sefat, E.K.; Sarker, M.S.; Unicomb, L.E.; De, S.; Hamadani, J.D.; Salam, M.A.; Wahed, M.A.; Albert, M.J. Immune response of children who develop persistent diarrhea following rotavirus infection. Clin. Diagn. Lab. Immunol. 1999, 6, 690–695. [Google Scholar] [PubMed]
- De Boissieu, D.; Lebon, P.; Badoual, J.; Bompard, Y.; Dupont, C. Rotavirus induces alpha-interferon release in children with gastroenteritis. J. Pediatr. Gastroenterol. Nutr. 1993, 16, 29–32. [Google Scholar] [PubMed]
- Wang, Y.; Dennehy, P.H.; Keyserling, H.L.; Tang, K.; Gentsch, J.R.; Glass, R.I.; Jiang, B. Rotavirus infection alters peripheral T-cell homeostasis in children with acute diarrhea. J. Virol. 2007, 81, 3904–3912. [Google Scholar] [CrossRef] [PubMed]
- Chaplin, P.J.; Entrican, G.; Gelder, K.I.; Collins, R.A. Cloning and biologic activities of a bovine interferon-alpha isolated from the epithelium of a rotavirus-infected calf. J. Interferon Cytokine Res. 1996, 16, 25–30. [Google Scholar] [CrossRef] [PubMed]
- La Bonnardiere, C.; Cohen, J.; Contrepois, M. Interferon activity in rotavirus infected newborn calves. Ann. Rech. Vet. 1981, 12, 85–91. [Google Scholar] [PubMed]
- Lecce, J.G.; Cummins, J.M.; Richards, A.B. Treatment of rotavirus infection in neonate and weanling pigs using natural human interferon alpha. Mol. Biother. 1990, 2, 211–216. [Google Scholar] [PubMed]
- Schwers, A.; Vanden Broecke, C.; Maenhoudt, M.; Beduin, J.M.; Werenne, J.; Pastoret, P.P. Experimental rotavirus diarrhoea in colostrum-deprived newborn calves: assay of treatment by administration of bacterially produced human interferon (Hu-IFN alpha 2). Ann. Rech. Vet. 1985, 16, 213–218. [Google Scholar] [PubMed]
- Angel, J.; Franco, M.A.; Greenberg, H.B.; Bass, D. Lack of a role for type I and type II interferons in the resolution of rotavirus-induced diarrhea and infection in mice. J. Interferon Cytokine Res. 1999, 19, 655–659. [Google Scholar] [PubMed]
- Feng, N.; Kim, B.; Fenaux, M.; Nguyen, H.; Vo, P.; Omary, M.B.; Greenberg, H.B. Role of interferon in homologous and heterologous rotavirus infection in the intestines and extraintestinal organs of suckling mice. J. Virol. 2008, 82, 7578–7590. [Google Scholar] [CrossRef] [PubMed]
- Dagenais, L.; Pastoret, P.P.; Van den Broecke, C.; Werenne, J. Susceptibility of bovine rotavirus to interferon. Brief report. Arch. Virol. 1981, 70, 377–379. [Google Scholar] [CrossRef] [PubMed]
- La Bonnardiere, C.; de Vaureix, C.; L'Haridon, R.; Scherrer, R. Weak susceptibility of rotavirus to bovine interferon in calf kidney cells. Arch. Virol. 1980, 64, 167–170. [Google Scholar] [CrossRef] [PubMed]
- Bass, D.M. Interferon gamma and interleukin 1, but not interferon alfa, inhibit rotavirus entry into human intestinal cell lines. Gastroenterology 1997, 113, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Casola, A.; Estes, M.K.; Crawford, S.E.; Ogra, P.L.; Ernst, P.B.; Garofalo, R.P.; Crowe, S.E. Rotavirus infection of cultured intestinal epithelial cells induces secretion of CXC and CC chemokines. Gastroenterology 1998, 114, 947–955. [Google Scholar] [CrossRef] [PubMed]
- Casola, A.; Garofalo, R.P.; Crawford, S.E.; Estes, M.K.; Mercurio, F.; Crowe, S.E.; Brasier, A.R. Interleukin-8 gene regulation in intestinal epithelial cells infected with rotavirus: role of viral-induced IkappaB kinase activation. Virology 2002, 298, 8–19. [Google Scholar] [CrossRef]
- Rollo, E.E.; Kumar, K.P.; Reich, N.C.; Cohen, J.; Angel, J.; Greenberg, H.B.; Sheth, R.; Anderson, J.; Oh, B.; Hempson, S.J.; Mackow, E.R.; Shaw, R.D. The epithelial cell response to rotavirus infection. J. Immunol. 1999, 163, 4442–4452. [Google Scholar] [PubMed]
- Johnson, M.A.; McCrae, M.A. Molecular biology of rotaviruses VIII. Quantitative analysis of regulation of gene expression during virus replication. J. Virol. 1989, 63, 2048–2055. [Google Scholar] [PubMed]
- Hua, J.; Chen, X.; Patton, J.T. Deletion mapping of the rotavirus metalloprotein NS53 (NSP1): the conserved cysteine-rich region is essential for virus-specific RNA binding. J. Virol. 1994, 68, 3990–4000. [Google Scholar] [PubMed]
- Hua, J.; Mansell, E.A.; Patton, J.T. Comparative analysis of the rotavirus NS53 gene: conservation of basic and cysteine-rich regions in the protein and possible stem-loop structures in the RNA. Virology 1993, 196, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Dunn, S.J.; Cross, T.L.; Greenberg, H.B. Comparison of the rotavirus nonstructural protein NSP1 (NS53) from different species by sequence analysis and northern blot hybridization. Virology 1994, 203, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Kojima, K.; Taniguchi, K.; Kobayashi, N. Species-specific and interspecies relatedness of NSP1 sequences in human, porcine, bovine, feline, and equine rotavirus strains. Arch. Virol. 1996, 141, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Tian, Y.; Tarlow, O.; Harbour, D.; McCrae, M.A. Molecular biology of rotaviruses IX. Conservation and divergence in genome segment 5. J. Gen. Virol. 1994, 75, 3413–3421. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, D.B.; Both, G.W. Conservation of a potential metal binding motif despite extensive sequence diversity in the rotavirus nonstructural protein NS53. Virology 1990, 174, 618–621. [Google Scholar] [CrossRef] [PubMed]
- Pina-Vazquez, C.; De Nova-Ocampo, M.; Guzman-Leon, S.; Padilla-Noriega, L. Post-translational regulation of rotavirus protein NSP1 expression in mammalian cells. Arch. Virol. 2007, 152, 345–368. [Google Scholar] [CrossRef] [PubMed]
- Brottier, P.; Nandi, P.; Bremont, M.; Cohen, J. Bovine rotavirus segment 5 protein expressed in the baculovirus system interacts with zinc and RNA. J. Gen. Virol. 1992, 73. [Google Scholar] [CrossRef]
- Hua, J.; Patton, J.T. The carboxyl-half of the rotavirus nonstructural protein NS53 (NSP1) is not required for virus replication. Virology 1994, 198, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Okada, J.; Kobayashi, N.; Taniguchi, K.; Shiomi, H. Functional analysis of the heterologous NSP1 genes in the genetic background of simian rotavirus SA11. Arch. Virol. 1999, 144, 1439–1449. [Google Scholar] [CrossRef] [PubMed]
- Patton, J.T.; Taraporewala, Z.; Chen, D.; Chizhikov, V.; Jones, M.; Elhelu, A.; Collins, M.; Kearney, K.; Wagner, M.; Hoshino, Y.; Gouvea, V. Effect of intragenic rearrangement and changes in the 3' consensus sequence on NSP1 expression and rotavirus replication. J. Virol. 2001, 75, 2076–2086. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Kojima, K.; Urasawa, S. Nondefective rotavirus mutants with an NSP1 gene which has a deletion of 500 nucleotides, including a cysteine-rich zinc finger motif-encoding region (nucleotides 156 to 248), or which has a nonsense codon at nucleotides 153-155. J. Virol. 1996, 70, 4125–4130. [Google Scholar] [PubMed]
- Barro, M.; Patton, J.T. Rotavirus NSP1 inhibits expression of type I interferon by antagonizing the function of interferon regulatory factors IRF3, IRF5, and IRF7. J. Virol. 2007, 81, 4473–4481. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Tarlow, O.; Ballard, A.; Desselberger, U.; McCrae, M.A. Genomic concatemerization/deletion in rotaviruses: a new mechanism for generating rapid genetic change of potential epidemiological importance. J. Virol. 1993, 67, 6625–6632. [Google Scholar] [PubMed]
- Graff, J.W.; Mitzel, D.N.; Weisend, C.M.; Flenniken, M.L.; Hardy, M.E. Interferon regulatory factor 3 is a cellular partner of rotavirus NSP1. J. Virol. 2002, 76, 9545–9550. [Google Scholar] [CrossRef] [PubMed]
- Barro, M.; Patton, J.T. Rotavirus nonstructural protein 1 subverts innate immune response by inducing degradation of IFN regulatory factor 3. Proc. Natl. Acad. Sci. U S A 2005, 102, 4114–4119. [Google Scholar] [CrossRef] [PubMed]
- Graff, J.W.; Ewen, J.; Ettayebi, K.; Hardy, M.E. Zinc-binding domain of rotavirus NSP1 is required for proteasome-dependent degradation of IRF3 and autoregulatory NSP1 stability. J. Gen. Virol. 2007, 88, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Feng, N.; Sen, A.; Nguyen, H.; Vo, P.; Hoshino, Y.; Deal, E.M.; Greenberg, H.B. Variation in antagonism of the interferon response to rotavirus NSP1 results in differential infectivity in mouse embryonic fibroblasts. J. Virol. 2009, 83, 6987–6994. [Google Scholar] [CrossRef] [PubMed]
- Sen, A.; Feng, N.; Ettayebi, K.; Hardy, M.E.; Greenberg, H.B. IRF3 Inhibition by Rotavirus NSP1 is Host cell and Viral Strain Dependent but Independent of NSP1 Proteasomal Degradation. J. Virol. 2009. [Google Scholar] [PubMed]
- Mesa, M.C.; Rodriguez, L.S.; Franco, M.A.; Angel, J. Interaction of rotavirus with human peripheral blood mononuclear cells: plasmacytoid dendritic cells play a role in stimulating memory rotavirus specific T cells in vitro. Virology 2007, 366, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Barnes, B.J.; Moore, P.A.; Pitha, P.M. Virus-specific activation of a novel interferon regulatory factor, IRF-5, results in the induction of distinct interferon alpha genes. J. Biol. Chem. 2001, 276, 23382–23390. [Google Scholar] [CrossRef] [PubMed]
- Douagi, I.; McInerney, G.M.; Hidmark, A.S.; Miriallis, V.; Johansen, K.; Svensson, L.; Karlsson Hedestam, G.B. Role of interferon regulatory factor 3 in type I interferon responses in rotavirus-infected dendritic cells and fibroblasts. J. Virol. 2007, 81, 2758–2768. [Google Scholar] [CrossRef] [PubMed]
- Graff, J.W.; Ettayebi, K.; Hardy, M.E. Rotavirus NSP1 inhibits NFkappaB activation by inducing proteasome-dependent degradation of beta-TrCP: a novel mechanism of IFN antagonism. PLoS Pathog. 2009, 5, e1000280. [Google Scholar] [CrossRef] [PubMed]
- Holloway, G.; Truong, T.T.; Coulson, B.S. Rotavirus antagonizes cellular antiviral responses by inhibiting the nuclear accumulation of STAT1, STAT2, and NF-kappaB. J. Virol. 2009, 83, 4942–4951. [Google Scholar] [CrossRef] [PubMed]
- Vancott, J.L.; McNeal, M.M.; Choi, A.H.; Ward, R.L. The role of interferons in rotavirus infections and protection. J. Interferon Cytokine Res. 2003, 23, 163–170. [Google Scholar] [PubMed]
- Aravind, L.; Iyer, L.M.; Koonin, E.V. Scores of RINGS but no PHDs in ubiquitin signaling. Cell Cycle 2003, 2, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Dodd, R.B.; Allen, M.D.; Brown, S.E.; Sanderson, C.M.; Duncan, L.M.; Lehner, P.J.; Bycroft, M.; Read, R.J. Solution structure of the Kaposi's sarcoma-associated herpesvirus K3 N-terminal domain reveals a Novel E2-binding C4HC3-type RING domain. J. Biol. Chem. 2004, 279, 53840–53847. [Google Scholar] [CrossRef] [PubMed]
- Weber, F.; Kochs, G.; Haller, O. Inverse interference: how viruses fight the interferon system. Viral Immunol. 2004, 17, 498–515. [Google Scholar] [PubMed]
- Katze, M.G.; He, Y.; Gale Jr., M. Viruses and interferon: a fight for supremacy. Nat. Rev. Immunol. 2002, 2, 675–687. [Google Scholar] [CrossRef] [PubMed]
- Ronco, L.V.; Karpova, A.Y.; Vidal, M.; Howley, P.M. Human papillomavirus 16 E6 oncoprotein binds to interferon regulatory factor-3 and inhibits its transcriptional activity. Genes Dev. 1998, 12, 2061–2072. [Google Scholar] [CrossRef] [PubMed]
- La Rocca, S.A.; Herbert, R.J.; Crooke, H.; Drew, T.W.; Wileman, T.E.; Powell, P.P. Loss of interferon regulatory factor 3 in cells infected with classical swine fever virus involves the N-terminal protease, Npro. J. Virol. 2005, 79, 7239–7247. [Google Scholar] [CrossRef] [PubMed]
- Bauhofer, O.; Summerfield, A.; Sakoda, Y.; Tratschin, J.D.; Hofmann, M.A.; Ruggli, N. Classical swine fever virus Npro interacts with interferon regulatory factor 3 and induces its proteasomal degradation. J. Virol. 2007, 81, 3087–3096. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Rijnbrand, R.; Jangra, R.K.; Devaraj, S.G.; Qu, L.; Ma, Y.; Lemon, S.M.; Li, K. Ubiquitination and proteasomal degradation of interferon regulatory factor-3 induced by Npro from a cytopathic bovine viral diarrhea virus. Virology 2007, 366, 277–292. [Google Scholar] [CrossRef] [PubMed]
- Hilton, L.; Moganeradj, K.; Zhang, G.; Chen, Y.H.; Randall, R.E.; McCauley, J.W.; Goodbourn, S. The NPro product of bovine viral diarrhea virus inhibits DNA binding by interferon regulatory factor 3 and targets it for proteasomal degradation. J. Virol. 2006, 80, 11723–11732. [Google Scholar] [CrossRef] [PubMed]
- Szymanski, M.R.; Fiebach, A.R.; Tratschin, J.D.; Gut, M.; Ramanujam, V.M.; Gottipati, K.; Patel, P.; Ye, M.; Ruggli, N.; Choi, K.H. Zinc binding in pestivirus N(pro) is required for interferon regulatory factor 3 interaction and degradation. J. Mol. Biol. 2009, 391, 438–449. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Chen, L.M.; Zeng, H.; Gomez, J.A.; Plowden, J.; Fujita, T.; Katz, J.M.; Donis, R.O.; Sambhara, S. NS1 protein of influenza A virus inhibits the function of intracytoplasmic pathogen sensor, RIG-I. Am. J. Respir. Cell. Mol. Biol. 2007, 36, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Mibayashi, M.; Martinez-Sobrido, L.; Loo, Y.M.; Cardenas, W.B.; Gale Jr., M.; Garcia-Sastre, A. Inhibition of retinoic acid-inducible gene I-mediated induction of beta interferon by the NS1 protein of influenza A virus. J. Virol. 2007, 81, 514–524. [Google Scholar] [CrossRef] [PubMed]
- Pichlmair, A.; Schulz, O.; Tan, C.P.; Naslund, T.I.; Liljestrom, P.; Weber, F.; Reis e Sousa, C. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5'-phosphates. Science 2006, 314, 997–1001. [Google Scholar] [CrossRef] [PubMed]
- Hirata, Y.; Broquet, A.H.; Menchen, L.; Kagnoff, M.F. Activation of innate immune defense mechanisms by signaling through RIG-I/IPS-1 in intestinal epithelial cells. J. Immunol. 2007, 179, 5425–5432. [Google Scholar] [PubMed]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Arnold, M.M.; Patton, J.T. Rotavirus Antagonism of the Innate Immune Response. Viruses 2009, 1, 1035-1056. https://0-doi-org.brum.beds.ac.uk/10.3390/v1031035
Arnold MM, Patton JT. Rotavirus Antagonism of the Innate Immune Response. Viruses. 2009; 1(3):1035-1056. https://0-doi-org.brum.beds.ac.uk/10.3390/v1031035
Chicago/Turabian StyleArnold, Michelle M., and John T. Patton. 2009. "Rotavirus Antagonism of the Innate Immune Response" Viruses 1, no. 3: 1035-1056. https://0-doi-org.brum.beds.ac.uk/10.3390/v1031035